
Abstract
Originally identified as the enzymes responsible for catalysing the oxidation of specific, conserved proline residues within hypoxia-inducible factor-1α (HIF-1α), the additional roles for the prolyl hydroxylase domain (PHD) proteins have remained elusive. Of the four identified PHD enzymes, PHD2 is considered to be the key oxygen sensor, as knockdown of PHD2 results in elevated HIF protein. Several recent studies have highlighted the importance of PHD2 in tumourigenesis. However, there is conflicting evidence as to the exact role of PHD2 in tumour angiogenesis. The divergence seems to be because of the contribution of stromal-derived PHD2, and in particular the involvement of endothelial cells, vs tumour-derived PHD2. This review summarises our current understanding of PHD2 and tumour angiogenesis, focusing on the influences of PHD2 on vascular normalisation and neovascularisation.
Similar content being viewed by others

Main
The rapid growth of tumours results in their surpassing the capacity of the established circulatory system to supply the developing tumour with oxygen and nutrients as well as remove waste products. For a tumour to expand, it must respond to these assaults. Thus, a necessary step in the progression of any solid tumour is adaptation to low oxygen or hypoxic conditions. Tumour cells can respond to hypoxia by increasing oxygen delivery or acclimating to decreased oxygen availability. The hypoxia-inducible factor (HIF) family of transcription factors mediates both the systemic and cellular response to hypoxia.
As master regulators of oxygen homeostasis, HIF-1 and HIF-2 must be tightly controlled to prevent the inappropriate expression of hypoxia-induced genes. HIF-1 and HIF-2 are heterodimeric transcription factors that are regulated at the post-translational level. They are composed of a constitutive β-subunit (also known as ARNT) and an oxygen-sensitive α-subunit. The α-subunit is hydroxylated on two conserved proline residues by a family of prolyl hydroxylases (Ivan et al, 2001; Jaakkola et al, 2001; Yu et al, 2001). The hydroxylation of the α-subunits allows the von Hippel-Lindau (VHL) E3 ubiquitin ligase to interact, adding ubiquitin ladders and targeting the α-subunit to the proteasome for degradation (Maxwell et al, 1999). HIF transactivation is also regulated by a third hydroxylation. This post-translational modification of a conserved asparagine residue (Asp 803 of HIF-1α and Asp 831 of HIF-2α) prevents the interaction of HIF with p300, a transcriptional coactivator (Mahon et al, 2001; Lando et al, 2002a, ). Hydroxylation of the α-subunits of HIF-1 and HIF-2 have critical roles in both the stability and activity of HIF.
Hypoxia-inducible factors and cancer
Under pathological conditions, strikingly in cancer, HIF becomes deregulated and has a key role in the development of tumours. Clinically, HIF is overexpressed in a variety of cancers, predicting poorer prognosis (Zhong et al, 1999; Semenza, 2002). Experimentally, HIF is required for the growth of solid tumours (Ryan et al, 1998, 2000). Deletion of Hif-1α in teratoma tumour models resulted in smaller tumours compared with teratomas containing wild-type Hif-1α. Using transgenic mice, Hif-1α has been shown to be an important factor in the development and metastatic spread of a variety of cancers, including those of the breast and brain (Tang et al, 2004; Liao et al, 2007; Du et al, 2008). Conversely, in embryonic stem cells, deletion of Hif-1 reduced hypoxia-induced apoptosis while increasing proliferation, resulting in larger tumours (Carmeliet et al, 1998). The role of HIF in tumourigenesis is further complicated by disparate findings for HIF-1 and HIF-2 (Acker et al, 2005; Gordan et al, 2008). However, tumour promoting or activating mutations in the HIF sequence have not been identified. A key upstream regulator of HIF is the VHL E3 ubiquitin ligase. VHL is a well-known tumour suppressor gene and VHL disease is characterised by a distinct subset of highly vascular tumours, notably renal carcinoma, angiomas, and hemangioblastomas (Kaelin, 2007). Mutation or silencing of VHL accounts for >80% of all renal carcinomas, the most common form of kidney cancer (Gnarra et al, 1993, 1994; Nickerson et al, 2008). Nevertheless, inactivation of VHL by itself cannot explain the broader variety of tumour types that have elevated HIF protein.
Another key regulator of HIF is the family of prolyl hydroxylases (PHD1, PHD2, and PHD3) that modify HIF to be recognised by VHL. The prolyl hydroxylases require oxygen and 2-oxyglutarate as substrates and Fe(II) and ascorbate as cofactors, resulting in oxidation of two highly conserved proline residues (Figure 1). Recently, a paper by Ladroue et al (2008) identified a patient who had a single amino acid substitution in the iron-binding pocket of PHD2. The patient presented with congenital erythrocytosis and multiple paragangliomas. Furthermore, Kato et al (2006) reported a high mutation rate of PHD2 in endometrial cancers. Mutations in succinate dehydrogenase and fumarate hydratase, both components of the citric acid cycle, have also been linked to tumour-prone syndromes, resulting in part because of inhibition of the PHDs (Isaacs et al, 2005; Ricketts et al, 2009). Overexpression of PHD1 has been shown to inhibit tumour growth (Erez et al, 2003), whereas PHD3 is downregulated in colorectal cancers (Xue et al, 2009). The complexity of interactions between HIFs and the PHDs were highlighted by Henze et al (2010) who identified a negative feedback loop in gliomas. Taken together, these findings suggest that other negative regulators of HIF, such as the prolyl hydroxylases, may contribute to tumourigenesis. However, the role of PHDs in tumourigenesis is poorly defined.

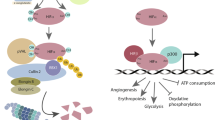
In the presence of oxygen, a family of prolyl hydroxylases oxidises HIF-1α, leading to interaction with VHL and subsequent degradation. Conversely, when the prolyl hydroxylases are not active, HIF-1α is stabilised, interacts with HIF-1β, and is transcriptionally active.
The role of PHD2 in tumour angiogenesis
Of the identified HIF prolyl hydroxylases, PHD2 is thought to be the key oxygen sensor regulating HIF (Berra et al, 2003). Silencing PHD2 through RNA interference increased HIF-1α levels under normoxic conditions. This effect was not observed with either PHD1 or PHD3. Furthermore, several groups independently generated genetic knockout mice of the different Phds (Takeda et al, 2006; Aragones et al, 2008; Minamishima et al, 2008). Phd1 and Phd3 homozygous knockout mice appeared phenotypically normal with the expected Mendelian ratios (Takeda et al, 2006). Meanwhile, homozygous knockout of Phd2 was embryonic lethal between days 12.5 and 14.5, because of vascular defects of the placenta and the heart. Moreover, conditional knockout of Phd2 increased vascular and capillary density, vessel branching, and recruitment of vascular smooth muscle cells, whereas conditional knockout of Phd1 and Phd3 did not have these vascular effects (Takeda et al, 2007). Taken together, these data suggest that PHD2 may have an important role in regulating HIF and angiogenesis.
We recently identified PHD2 as a mediator of potent tumour angiogenesis pathways (Chan et al, 2009) whereas Mazzone et al (2009) reported a different function of PHD2 in vessel normalisation. We began our studies by analysing mRNA and protein expression levels of PHD2 in human tumours. In colorectal carcinomas, PHD2 levels of mRNA and protein were both decreased in the tumour compared with non-involved, adjacent normal colon tissue, suggesting that loss of PHD2 may influence the tumour development. To further investigate, we used shRNA to stably silence PHD2 in several different human cell lines, including three colorectal cell lines and a pancreatic cell line. In each of these lines, knocking down PHD2 levels did not affect in vitro cell growth. It should be noted that we did not challenge these cells to hypoxic stress or additional PHD inhibition. Henze et al (2010) found that inhibiting PHD activity by hypoxia or DMOG reduced glioma tumour cell survival in vitro. However, when we implanted our cell lines as xenografts into the flanks of immunocompromised mice, tumour growth was significantly and dramatically increased compared with wild-type control cells. As the best-characterised target of PHD2 is HIF, we then investigated whether the enhanced tumour growth of PHD2 loss is dependent on HIF
Using HCT116 cells, a colon carcinoma, deleted for HIF-1α (Dang et al, 2006), we then silenced PHD2 and implanted these cells as tumours. Tumours that lacked HIF and PHD2 grew faster than control tumours that only lacked HIF. This provocative finding suggests that PHD2 has additional functions that are independent of HIF.
To determine the mechanism of why tumours with PHD2 silenced grew faster, we sectioned the tumours. TUNEL analysis showed no difference between control and knockdown tumours, showing that the wild-type tumours were not smaller because of an increase in apoptosis. However, Ki67 expression, a marker of proliferation, indicated that the PHD2-silenced tumours grew better than wild-type tumours in vivo. These results suggested that PHD2 disruption in tumour cells altered their interaction with the tumour microenvironment, allowing for more efficient growth. We further stained the tumour sections for CD31, a marker of blood vessels, which showed a three- to four-fold increase in tumour blood vessels in the tumours with PHD2 silenced. Similarly, several additional groups have also found that PHD2 can influence tumour growth through its effect on angiogenesis. Lee et al (2008) showed that a reduction in PHD2 leads to enhanced tumour growth and enhanced tumourigenesis. In reciprocal experiments, Matsumoto et al (2006, 2009) found that 2-oxogluturate, a substrate of PHD2, reduced both tumour growth and angiogenesis. Examining the effect of Phd2 deletion on endothelial cells directly, Takeda and Fong (2007) found that loss of Phd2 impaired proliferation. Thus, it seemed likely that the enhanced tumour blood supply of the PHD2 knockdown tumour was providing the necessary components to increase tumour growth.
We then investigated whether the PHD2 knockdown cells were secreting factors capable of influencing angiogenesis. Using a standard in vitro angiogenesis assay of endothelial cell tube formation, conditioned media from PHD2 knockdown HCT116 cells were able to cause primary endothelial cells plating on matrigel to aggregate, forming complex, tube-like structures. Conditioned media from control HCT116 cells, which do not have PHD2 knocked down, however, lacked the components to induce the endothelial cells to branch. Subjecting the conditioned media to an angiogenesis antibody array, we revealed that PHD2 consistently regulates angiogenin (ANG) and IL-8, two known soluble pro-angiogenic factors. Conditioned media from HCT116 cells that had PHD2 knocked down had elevated protein levels of ANG and IL-8. Notably, the levels of VEGF were unchanged by PHD2 levels. Silencing either ANG or IL-8 impaired both angiogenesis and tumourigenesis. These results suggested that PHD2 normally functions to inhibit angiogenesis and that silencing PHD2 promotes angiogenesis.
Tumour vasculature is actually regulated by two complementary processes: angiogenesis, which is local sprouting, and vasculogenesis, which is the de novo production of new blood vessels. Angiogenesis is the formation of new blood vessels from pre-existing vessels. This local angiogenesis can also send signals to the bone marrow, which can in turn release precursor or progenitor cells to induce neovascularisation. This process of blood vessel formation by de novo production of endothelial cells, or vasculogenesis, requires tumour cells to interact with stromal cells and circulating bone marrow-derived cells (BMDCs). We then investigated whether PHD2 knockdown affected recruitment of BMDCs to the growing tumour vasculature. To determine this we stained for two BMDC markers, CD11b and CD45. CD11b is a marker of myeloid–monocytic precursors, whereas CD45 is a myeloid marker. PHD2 silencing increased mobilisation of BMDCs to the growing tumour, whereas silencing of ANG and IL8 impaired this mobilisation. Thus, PHD2 functions to regulate both angiogenesis and vasculogenesis through ANG and IL-8.
The PHD2 regulation of tumour vasculature through ANG and IL-8 is independent of HIF. Cummins et al (2006) showed that PHD1 could regulate IKKβ, an inhibitor of NF-κB, which suggested that PHD2 might also regulate NF-κB. In PHD2-silenced cells, NF-κB activity was elevated and mutation of the NF-κB sites in the promoters of ANG and IL-8 impaired NF-κB activation. Furthermore, IL-8 is a well-characterised NF-κB target and NF-κB bound to the promoter of ANG, as determined by chromatin immunoprecipitation, showing that the influence of PHD2 on the tumour vasculature is mediated through NF-κB activity on ANG and IL-8, two known pro-angiogenic factors. Transient silencing of p65, an essential subunit of the NF-κB complex, impaired in vitro angiogenesis. Using breast cancer data sets, there was a strong, inverse correlation between PHD2 mRNA levels and NF-κB activity profiles as well as negative relationship between PHD2 and CD31 mRNA levels, as determined by microarray analysis. Furthermore, NF-κB activity profiles positively correlated with an increase in CD31 mRNA. Taken together, these data illustrate that PHD2 can regulate the tumour vasculature through an HIF-independent mechanism but relies in part on inhibition of NF-κB and the downstream targets of ANG and IL-8.
Stromal PHD2 contributions to metastasis and vessel normalisation
In a parallel study, Mazzone et al (2009) investigated the stromal role of PHD2 in the development of tumours. Interestingly, using genetically modified mice that were heterozygous for Phd2, they saw no difference in ectopic, primary tumour growth. In an orthotopic, pancreatic model, tumours grown in wild-type mice were more invasive and had more metastatic disease compared with Phd2+/− mice. This phenotype was attributed to higher intravasation of tumour cells into blood vessels of wild-type vs Phd2 heterozygous animals. The Phd2 heterozygous animals had higher expression of VE-cadherin and less hypoxia based on pimonodozole staining, oxymetry, and HIF protein levels. Tumours implanted into Phd2+/− mice also had less necrosis and lower metabolic rates. The same researchers showed that vessels of tumours implanted in heterozygous mice were normalised compared with those of wild-type mice. The vessel normalisation of tumours in Phd2 heterozygous mice resulted in less intravasation of tumour cells and consequently less metastasis.
In contrast to our findings, this group (Mazzone et al, 2009) saw equivalent tumour vessel density between the heterozygous Phd2 and wild-type mice. However, despite equal numbers of tumour vessels, the tumours of the Phd2+/− mice had smoother, more regular, and more mature blood vessels that were not as malformed or as leaky as tumours implanted in wild-type mice. These researchers suggested that the tumour vessels formed in the Phd2 heterozygous mice were ‘normalised,’ comparable to normal blood vessels. Examining the angiogenic profile of Phd2 heterozygous endothelial cells as well as tumour endothelial cells from tumours implanted into Phd2+/− mice, they found higher soluble Flt1 and VE-cadherin expression at the mRNA and protein levels, respectively. Soluble Flt1 and VE-cadherin are two angiogenic factors implicated in vessel normalisation and more specifically, cell motility and cytoskeletal reorganisation. Interestingly, in response to VEGF, endothelial cells heterozygous for Phd2 proliferated slower, had decreased motility, less lamellipodia formation, and less apoptosis. The effects of sFlt1 and VE-cadherin were dependent on HIF2 and not HIF1, as transient silencing of only HIF2 decreased levels of both angiogenic factors. Taken together, Mazzone et al (2009) found that stromal PHD2 functions to regulate vessel normalisation, providing tighter junctions and preventing metastatic spread.
Conclusions: vessel normalisation vs increased vasculature
The simplest explanation for these apparent discrepancies between our study and that of Mazzone et al (2009) is differences in the experimental models. We examined the influence of PHD2 loss on tumour growth, whereas they examined the influence of the host on tumour growth. We used shRNA to silence PHD2 in human cell lines, which were then implanted as xenografts into immunocompromised mice. In comparison, Carmeliet et al (1998) used syngeneic mouse cell lines inoculated into the flanks or orthotopically implanted into immunocompetent but heterozygous Phd2 mice. Our study examined the role of PHD2 in a tumour to contribute to its growth through angiogenesis and recruitment of bone marrow-derived cells (Figure 2A). The complimentary study examined the function of stromal PHD2 in regulating tumour metastasis (Figure 2B). Simultaneous investigations into the participation of both tumour and host will be necessary to further delineate the responsibilities of PHD2. Furthermore, using spontaneous tumour models as well as specific knockout in various cell types may help elucidate HIF-dependent and HIF-independent functions of PHD2.

(A) Loss of PHD2 in a tumour results in activation of NF-κB. In turn, NF-κB upregulates ANG and IL-8 that promote both angiogenesis and bone marrow-derived cell recruitment. These two complementary pathways drive tumour growth. (B) Tumours implanted into Phd2+/+ mice have irregular and leaky vessels, whereas those implanted into Phd2+/− mice have ‘normalised’ vessels.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Acker T, Diez-Juan A, Aragones J, Tjwa M, Brusselmans K, Moons L, Fukumura D, Moreno-Murciano MP, Herbert JM, Burger A, Riedel J, Elvert G, Flamme I, Maxwell PH, Collen D, Dewerchin M, Jain RK, Plate KH, Carmeliet P (2005) Genetic evidence for a tumor suppressor role of HIF-2alpha. Cancer Cell 8: 131–141
Aragones J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, Lambrechts D, Bishop T, Lafuste P, Diez-Juan A, Harten SK, Van Noten P, De Bock K, Willam C, Tjwa M, Grosfeld A, Navet R, Moons L, Vandendriessche T, Deroose C, Wijeyekoon B, Nuyts J, Jordan B, Silasi-Mansat R, Lupu F, Dewerchin M, Pugh C, Salmon P, Mortelmans L, Gallez B, Gorus F, Buyse J, Sluse F, Harris RA, Gnaiger E, Hespel P, Van Hecke P, Schuit F, Van Veldhoven P, Ratcliffe P, Baes M, Maxwell P, Carmeliet P (2008) Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet 40: 170–180
Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J (2003) HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J 22: 4082–4090
Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E, Keshet E (1998) Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394: 485–490
Chan DA, Kawahara TL, Sutphin PD, Chang HY, Chi JT, Giaccia AJ (2009) Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell 15: 527–538
Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT (2006) Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA 103: 18154–18159
Dang DT, Chen F, Gardner LB, Cummins JM, Rago C, Bunz F, Kantsevoy SV, Dang LH (2006) Hypoxia-inducible factor-1alpha promotes nonhypoxia-mediated proliferation in colon cancer cells and xenografts. Cancer Res 66: 1684–1936
Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G (2008) HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 13: 206–220
Erez N, Milyavsky M, Eilam R, Shats I, Goldfinger N, Rotter V (2003) Expression of prolyl-hydroxylase-1 (PHD1/EGLN2) suppresses hypoxia inducible factor-1alpha activation and inhibits tumor growth. Cancer Res 63: 8777–8783
Gnarra JR, Glenn GM, Latif F, Anglard P, Lerman MI, Zbar B, Linehan WM (1993) Molecular genetic studies of sporadic and familial renal cell carcinoma. Urol Clin North Am 20: 207–216
Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM, Lubensky I, Duan DR, Florence C, Pozzatti R, Walther MM, Bander NH, Grossman HB, Brauch H, Pomer S, Brooks JD, Isaacs WB, Lerman MI, Zbar B, Linehan WM (1994) Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 7: 85–90
Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, Greenberg RA, Flaherty KT, Rathmell WK, Keith B, Simon MC, Nathanson KL (2008) HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 14: 435–446
Henze AT, Riedel J, Diem T, Wenner J, Flamme I, Pouyseggur J, Plate KH, Acker T (2010) Prolyl hydroxylases 2 and 3 act in gliomas as protective negative feedback regulators of hypoxia-inducible factors. Cancer Res 70: 357–366
Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Linehan WM, Neckers L (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8: 143–153
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin Jr WG (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292: 464–468
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292: 468–472
Kaelin WG (2007) Von Hippel-Lindau disease. Annu Rev Pathol 2: 145–173
Kato H, Inoue T, Asanoma K, Nishimura C, Matsuda T, Wake N (2006) Induction of human endometrial cancer cell senescence through modulation of HIF-1alpha activity by EGLN1. Int J Cancer 118: 1144–1153
Ladroue C, Carcenac R, Leporrier M, Gad S, Le Hello C, Galateau-Salle F, Feunteun J, Pouyssegur J, Richard S, Gardie B (2008) PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med 359: 2685–2692
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK (2002a) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 16: 1466–1471
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML (2002b) Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295: 858–861
Lee KA, Lynd JD, O’Reilly S, Kiupel M, McCormick JJ, LaPres JJ (2008) The biphasic role of the hypoxia-inducible factor prolyl-4-hydroxylase, PHD2, in modulating tumor-forming potential. Mol Cancer Res 6: 829–842
Liao D, Corle C, Seagroves TN, Johnson RS (2007) Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res 67: 563–572
Mahon PC, Hirota K, Semenza GL (2001) FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 15: 2675–2686
Matsumoto K, Imagawa S, Obara N, Suzuki N, Takahashi S, Nagasawa T, Yamamoto M (2006) 2-Oxoglutarate downregulates expression of vascular endothelial growth factor and erythropoietin through decreasing hypoxia-inducible factor-1alpha and inhibits angiogenesis. J Cell Physiol 209: 333–340
Matsumoto K, Obara N, Ema M, Horie M, Naka A, Takahashi S, Imagawa S (2009) Antitumor effects of 2-oxoglutarate through inhibition of angiogenesis in a murine tumor model. Cancer Sci 100: 1639–1647
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399: 271–275
Mazzone M, Dettori D, Leite de Oliveira R, Loges S, Schmidt T, Jonckx B, Tian YM, Lanahan AA, Pollard P, Ruiz de Almodovar C, De Smet F, Vinckier S, Aragones J, Debackere K, Luttun A, Wyns S, Jordan B, Pisacane A, Gallez B, Lampugnani MG, Dejana E, Simons M, Ratcliffe P, Maxwell P, Carmeliet P (2009) Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 136 (5): 839–851
Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin Jr WG (2008) Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood 111: 3236–3244
Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, Navratilova M, Szeszenia-Dabrowska N, Mates D, Mukeria A, Holcatova I, Schmidt LS, Toro JR, Karami S, Hung R, Gerard GF, Linehan WM, Merino M, Zbar B, Boffetta P, Brennan P, Rothman N, Chow WH, Waldman FM, Moore LE (2008) Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res 14: 4726–4734
Ricketts CJ, Forman JR, Rattenberry E, Bradshaw N, Lalloo F, Izatt L, Cole TR, Armstrong R, Kumar VK, Morrison PJ, Atkinson AB, Douglas F, Ball SG, Cook J, Srirangalingam U, Killick P, Kirby G, Aylwin S, Woodward ER, Evans DG, Hodgson SV, Murday V, Chew SL, Connell JM, Blundell TL, Macdonald F, Maher ER (2009) Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat 31: 41–51
Ryan HE, Lo J, Johnson RS (1998) HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J 17: 3005–3015
Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS (2000) Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res 60: 4010–4015
Semenza GL (2002) HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med 8: S62–S67
Takeda K, Cowan A, Fong GH (2007) Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation 116: 774–781
Takeda K, Fong GH (2007) Prolyl hydroxylase domain 2 protein suppresses hypoxia-induced endothelial cell proliferation. Hypertension 49: 178–184
Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH (2006) Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol 26: 8336–8346
Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP, Ferrara N, Johnson RS (2004) Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 6: 485–495
Xue J, Li X, Jiao S, Wei Y, Wu G, Fang J (2009) Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKbeta independent of hydroxylase activity. Gastroenterology 138: 606–615
Yu F, White SB, Zhao Q, Lee FS (2001) HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci USA 98: 9630–9635
Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW (1999) Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 59: 5830–5835
Acknowledgements
This work was supported by NCI-CA-67166 (AJG), NCI-CA-88480 (AJG), and NCI-CA-123823 (DAC). We apologise to colleagues whose work we failed to cite.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Chan, D., Giaccia, A. PHD2 in tumour angiogenesis. Br J Cancer 103, 1–5 (2010). https://doi.org/10.1038/sj.bjc.6605682
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605682
Keywords
This article is cited by
-
Long-term treatment of chronic kidney disease patients with anemia using hypoxia-inducible factor prolyl hydroxylase inhibitors: potential concerns
Pediatric Nephrology (2024)
-
Prognostic value and model construction of preoperative inflammatory markers in patients with metastatic renal cell carcinoma
World Journal of Surgical Oncology (2023)
-
PHD2 attenuates high-glucose-induced blood retinal barrier breakdown in human retinal microvascular endothelial cells by regulating the Hif-1α/VEGF pathway
Inflammation Research (2022)
-
PHD-2 activation: a novel strategy to control HIF-1α and mitochondrial stress to modulate mammary gland pathophysiology in ER+ subtype
Naunyn-Schmiedeberg's Archives of Pharmacology (2019)
-
Antiangiogenic activity of phthalides-enriched Angelica Sinensis extract by suppressing WSB-1/pVHL/HIF-1α/VEGF signaling in bladder cancer
Scientific Reports (2017)
