
Abstract
Little is known about factors that stimulate transcription of the p53 tumor suppressor gene. Here, we report that the human pituitary homeobox 1 (hPitx1) transcription factor increases the expression of p53 at the mRNA and protein levels in human mammary carcinoma (MCF-7) cells. Increased p53 mRNA expression was due to activation of the p53 promoter by hPitx1. hPitx1 bound directly to the p53 promoter and functionally utilized two hPitx1 consensus elements. The predominant consensus element utilized by hPitx1 to stimulate p53 transcription was located within the first exon of the p53 gene. A hPitx1 mutant (hPitx1-R141P) acting as a dominant inhibitor repressed p53 transcription. Forced expression of hPitx1 resulted in cell-cycle arrest and p53-dependent apoptosis in p53-replete MCF-7 cells. Furthermore, hPitx1 stimulated the transcription of p53 target genes involved in cell-cycle arrest and apoptosis (p21 and PTGF-β), again in a p53-dependent manner. Depletion of endogenous hPitx1 by small interfering RNA (siRNA) in MCF-7 cells resulted in decreased basal expression of p53 and consequently of p21 and placental transforming growth factor β (PTGF-β). Depletion of p53 by siRNA dramatically attenuated hPitx1-induced apoptosis in MCF-7 cells. Thus, p53 is a direct transcriptional target gene of hPitx1. This observation is concordant with the recent identification of hPitx1 as a tumor suppressor gene.
Similar content being viewed by others


Main
Pituitary homeobox 1 (Pitx1) is a pan-pituitary homeobox transcription factor primarily expressed in the anterior pituitary.1 It first appears in Rathke's pouch during early stages of development, and its expression in the anterior pituitary persists thereafter. Pitx1 is also expressed in many anterior pituitary-derived cell lines such as the murine gonadotrope-derived cell lines αT3-1 and LβT2.2, 3 Pitx1 plays an important role in maintaining tissue-specific gene expression in differentiated pituitary cell subtypes. It has been demonstrated to increase the expression of many anterior pituitary cell type-specific genes, including α-glycoprotein subunit, pro-opiomelanocortin (POMC), GH, thyroid-stimulating hormone β and prolactin. Pitx1 also increases the expression levels of the gonadotrope-specific genes LHβ, FSHβ and GnRHR.1, 2, 3, 4 Pitx1 plays a crucial role in the development of the vertebrate hindlimb and determines the morphology of muscle, tendon and bones of the hindlimb.5 Pitx1-deficient mice exhibit severe craniofacial abnormalities, pituitary defects and neonatal lethality.4 Pitx1 has recently been identified by genetic screening as a suppressor of Ras activity and tumorigenicity.6
The tumor suppressor p53 is a transcription factor that negatively regulates cell proliferation and survival. It is involved in a number of cell effector pathways, including cell cycle, programmed cell death and DNA repair.7, 8 Inactivating mutations of the p53 gene are the most frequent genetic alteration in over 50% of human cancers, suggesting that loss of p53 function provides a selective advantage to tumor cells. Germline mutations in the p53 gene cause Li-Fraumeni syndrome (LFS), a genetic disorder characterized by spontaneous tumor formation. p53 mutants have been shown to possess gain-of-function oncogenic properties in several LFS mouse models.9, 10 p53-null mice develop spontaneous tumors, gene amplifications and polyploidy.11 Reciprocal studies in transgenic mice carrying supernumerary copies of the p53 gene demonstrate an increased DNA-damage response and tumor-resistance.12
The p53 protein is expressed at a very low level during normal cell growth through regulation of its protein stability. HDM2, the human homolog of mouse double-minute-2 (MDM2), binds to p53, blocks interaction with transcriptional co-activators and ubiquitinates p53, thus targeting p53 for proteosomal degradation.13 However, p53 expression is both induced and activated in response to DNA damage, oncogene activation, hypoxia, nutrient deprivation and other stress-related signals. For example, DNA-damage agents such as ionizing and UV radiation and the antimetabolite chemotherapeutic drug 5-fluorouracil, which suppresses DNA synthesis and repair and inhibits rRNA processing, activate p53 with consequent cell-cycle arrest or apoptosis through transcriptional activation of its target genes.11, 14 Regulation of p53 is known to be primarily controlled post-translationally by subcellular localization, phosphorylation, acetylation and degradation.15 Specifically, following genotoxic stress, p53 is activated and stabilized by the DNA-damage kinases, such as ataxia telangiectasia mutated kinase (ATM), ataxia telangiectasia RAD3-related kinase (ATR), checkpoint kinase-2 (Chk2) and checkpoint kinase-1 (Chk1). Phosphorylation of p53 at N-terminal sites stabilizes the p53 protein by disrupting MDM2 binding and promoting acetylation and phosphorylation of the C-terminus. Recently, translational regulation of p53 has also been demonstrated. Several RNA-binding proteins, including HuR (Hu antigen R), ribosomal protein L26 (RPL26) and nucleolin have recently been shown to increase p53 levels after DNA damage through promotion of p53 translation.16 However, little is known about the transcriptional regulation of the p53 gene. Nevertheless, factors controlling transcription of the p53 gene itself have been identified. HoxA5, a homeodomain containing gene, binds to HOX consensus elements in the p53 promoter and transcriptionally upregulates p53 gene transcription leading to apoptosis in human mammary carcinoma cells.17 Oncoprotein Bcl6 has been found to suppress p53 expression through binding to two specific sites within the p53 promoter region.18 By genome-wide functional analysis,19 two additional genes, NR2F2 and SFRS10, have been demonstrated to result in the transcriptional upregulation of p53. Although NR2F2 is a transcription factor, it was not demonstrated whether it binds to the p53 promoter directly or indirectly affects other regulators of p53 transcription.19 However, p53 transcription induced by DNA-damaging chemotherapeutic agents does involve transactivation of endogenous p53 expression through direct binding of p73 or p53 protein to the p53 promoter.20 In a large-scale RNA interference-based screening, another four genes, GAS41, RPS6K4, RUNDC1 and CRMP-2, have been found to be negatively involved in the regulation of p53 transcriptional activity.21 The interference with each of these four genes resulted in the upregulation of p53 transcriptional activity and, conversely, their overexpression resulted in the inhibition of p53 target promoters and p53-mediated apoptosis.
Here, we report that forced expression of hPitx1 resulted in p53-dependent apoptosis in MCF-7 cells and p53-independent apoptosis in human osteosarcoma MG-63 cells. We demonstrate that hPitx1 is able to directly activate p53 gene transcription in MCF-7 cells by binding to two hPitx1 consensus elements in the p53 promoter. hPitx1 subsequently utilized p53 to transactivate the cyclin-dependent kinase inhibitor p21 and placental transforming growth factor β (PTGFβ). Depletion of endogenous hPitx1 by small interfering RNA (siRNA) in MCF-7 cells resulted in a decrease in the basal expression of p53 and its targets p21 and PTGF-β. Furthermore, p53 depletion by siRNA dramatically attenuated hPitx1-induced apoptosis in MCF-7 cells. We have therefore identified p53 as a direct transcriptional target of the homeodomain-containing transcription factor hPitx1.
Results
Forced expression of hPitx1 results in apoptosis in MCF-7 cells
Reverse transcriptase-PCR (RT-PCR) was used to detect the expression of hPitx1 in mammary carcinoma MCF-7 cells. One specific fragment of the predicted size appropriate for hPitx1 mRNA was detected in the RNA isolated from MCF-7 cells and its identity was confirmed by restriction enzyme digestion and DNA sequencing (data not shown). To determine the role of hPitx1 in the behavior of MCF-7 cells, we transiently transfected MCF-7 cells with either a c-Myc-tagged hPitx1 expression plasmid (pUSE-hPitx1-MycII) or control empty pUSE vector. The c-Myc-tagged hPitx1 was observed in MCF-7 cells as a protein band of 47 kDa on Western blot as detected by anti-c-Myc antibody 9E10 (Figure 1a, upper left panel). In an attempt to establish MCF-7 cells stably expressing hPitx1, G-418 selection was applied. However, after 4 weeks of G-418 selection, the expression of hPitx1 could no longer be detected, indicating that forced expression of hPitx1 affected the viability of MCF-7 cells (Figure 1a, lower left panel).

Forced overexpression of hPitx1 caused apoptotic cell death in p53-replete MCF-7 cells. (a) Detection of forced hPitx1 overexpression by Western blot. Cells were transfected with plasmids expressing Myc-tagged hPitx1 (Myc-hPitx1) and Myc-tagged mutant hPitx1-R141P (Myc-hPitx1-R141P) or the empty vector plasmid as control as indicated. Cells were harvested either 24 h after transfection (transient) or after further 4 weeks of G-418 selection (stable). Soluble whole-cell extracts were run on a 12% SDS-PAGE, transferred to nitrocellulose and immunoblotted using the anti-c-Myc antibody (clone 9E10) for the detection of hPitx1-MycII or hPitx1-MycII-R141P protein. The same blots were stripped and reprobed with monoclonal antibody against β-actin to ensure equal loading of the cell lysate proteins. (b, c) hPitx1 induced apoptosis in MCF-7 cells. Cells were transfected with EGFP-tagged hPitx1 expression plasmid pEGFP-hPitx1-MycII (EGFP-hPitx1, upper panel in (b)) or empty pEGFP vector plasmid (EGFP, lower panel in (b)). On each day after transfection, cells were fixed and nuclei were stained with Hoechst 33258. Nuclei of green cells were examined with fluorescence microscopy (b), and apoptosis resulted from the transient expression of EGFP (open bar) or EGFP-hPitx1-MycII (hatched bar) was presented as percentage of cells with apoptotic nuclei out of green cells (c). For each point, 200–300 cells were examined. Data were analyzed by Student's t-test and are presented as mean±S.D. ***P<0.001, relative to vector transfection. (d) MCF-7 cells were transiently transfected with the c-Myc-tagged hPitx1 (hPitx1) and mutant hPitx1-R141P (hPitx1-R141P) or the empty vector plasmid (Vector). The expression of hPitx1 was detected as red with 9E10 by indirect immunofluorescence staining. DNA breaks were determined as green by fluorometric TUNEL assays. Scale bars=5 μm
To determine whether forced expression of hPitx1 produced apoptosis, MCF-7 cells were transiently transfected with an enhanced green fluorescent protein (EGFP)-tagged hPitx1 expression vector or pEGFP vector as control. Thus, the expression of EGFP-hPitx1 or EGFP proteins could be observed under a UV-visible fluorescence microscope. The nuclear morphology of transfected green cells was examined by DNA-specific Hoechst 33258 staining. EGFP-hPitx1 expression in MCF-7 cells indeed resulted in apoptotic cell death, with characteristic nuclear features such as condensation and fragmentation, compared with EGFP-only-expressing cells (Figure 1b). As observed in Figure 1c, forced expression of EGFP-hPitx1 caused marked apoptosis in MCF-7 cells in contrast to EGFP-transfected control cells. At 1 day after transfection, approximately 13% of EGFP-expressing cells were apoptotic, whereas 46% of EGFP-hPitx1-expressing cells displayed apoptotic characteristics. After 2 days, the percentage of apoptotic cells expressing EGFP-hPitx1 increased to 81%, whereas the percentage of apoptotic cells expressing EGFP did not change appreciably (12%). On the third day after transfection, almost all the EGFP-hPitx1-expressing cells (91%) were apoptotic in contrast to only 8% of the EGFP-expressing cells.
To further confirm the observation, TdT-mediated dUTP nick end labeling (TUNEL) analyses were performed on MCF-7 cells transiently transfected with the c-Myc-tagged hPitx1 expression plasmid (pUSE-hPitx1-MycII) or control empty pUSE vector. As shown in Figure 1d, the expression of hPitx1 was detected only in the pUSE-hPitx1-MycII-transfected but not in the vector-transfected cells. Similar apoptosis frequency was observed with the TUNEL assay. Forty-three percent of hPitx1-expressing cells were apoptotic after 24 h compared to 9% of vector-transfected cells.
hPitx1-induced apoptosis in MCF-7 cells is mediated by p53
The p53 tumor suppressor protein, a transcription factor which produces both growth arrest and apoptosis, is frequently inactivated by mutation or other mechanisms in human breast cancer.22 Among the many genes that possess a role in the regulation of apoptosis, p53 is one of the most important.8 MCF-7 cells express wild-type p53.23 We therefore first tested the effect of hPitx1 on p53 expression. MCF-7 cells were transiently transfected with the c-Myc-tagged hPitx1 plasmid or the control empty vector. Forced expression of hPitx1 in MCF-7 cells increased p53 mRNA level as detected by RT-PCR (Figure 2a). In addition, forced expression of hPitx1 increased p53 protein expression as detected by Western blot analysis using a monoclonal antibody against p53 (Figure 2b, top panel). The expression of c-Myc-hPitx1 was confirmed by Western blotting with the anti-c-Myc monoclonal antibody (clone 9E10) (Figure 2b, middle panel).

hPitx1-induced apoptosis was mediated by p53 in MCF-7 cells. (a, b) Forced expression of hPitx1 increased p53 expression. Cells were transfected with Myc-tagged hPitx1 expression plasmid pUSE-hPitx1-MycII (Myc-hPitx1) or the empty vector plasmid pUSE (vector). (a) Cells were harvested 24 h after transfection. Total RNA was isolated and the mRNA levels of p53 and β-actin were detected by RT-PCR using One-step RT-PCR kit from Qiagen. (b) Soluble whole-cell extracts were run on a 12% SDS-PAGE and transferred to nitrocellulose for Western blot analyses. Duplicated blots were immunoblotted using the mouse monoclonal antibodies anti-p53 (clone DO-1, Santa Cruz Biotechnology) and anti-c-Myc (clone 9E10) for the detection of p53 and hPitx1-MycII proteins, respectively. One of the blots was stripped and reprobed with the monoclonal antibody against β-actin to ensure equal loading of the cell lysate proteins. (c) Immunofluorescence staining of p53 in MCF-7 cells transiently expressing hPitx1. Cells were transfected with EGFP-tagged hPitx1-expressing plasmid pEGFP-hPitx1-MycII (EGFP-hPitx1, upper panel) or empty pEGFP vector plasmid (EGFP, lower panel). At 24 h after transfection, cells were fixed and p53 expression was detected with anti-p53 antibody clone DO-1 by indirect immunofluorescence staining. Nuclei of cells were visualized by Hoechst 33258 staining. Seventy percent of cells expressing EGFP-hPitx1 exhibited higher intensity nuclear p53 staining, versus 10% of cells expressing EGFP alone. (d) Transcriptional activation of p53 by hPitx1 in MCF-7 cells. Cells were co-transfected with p53 luciferase reporter plasmid pGL3-p53pro or the promoterless empty pGL3 vector plasmid in conjunction with hPitx1-expressing plasmid (hatched bar) or the control empty vector (open bar). Luciferase activity was measured using a kit (Promega). Experiments were carried out in six-well plates in three replicates. (e) p53 mutant attenuated hPitx1-induced apoptosis in MCF-7 cells. Cells were co-transfected with EGFP-tagged hPitx1 expression plasmid pEGFP-hPitx1-MycII (EGFP-hPitx1) or empty pEGFP vector (EGFP), as indicated in conjunction with plasmids expressing wild-type p53 (hatched bar) and its mutant p53-143A (solid bar) or control vector plasmid (open bar). At 24 h after transfection, apoptosis was determined using fluorescence microscopy as in Figure 1c. ***P<0.001, relative to vector as indicated. Scale bar=10 μm
p53 upregulation by hPitx1 was further evidenced by fluorescence microscopy using an antibody against p53 as shown in Figure 2c. MCF-7 cells were transiently transfected with EGFP-tagged hPitx1 expression vector or pEGFP vector as control. The intensity of nuclear p53 staining of EGFP-expressing cells remained indistinguishable from that of untransfected MCF-7 cells (Figure 2c, lower panel), EGFP-hPitx1-expressing cells exhibited higher intensity nuclear p53 labeling compared with both untransfected MCF-7 cells and EGFP-expressing cells (Figure 2c, upper panel). Seventy percent of cells expressing EGFP-hPitx1 exhibited higher intensity nuclear p53 staining versus 10% of cells expressing EGFP alone.
The hPitx1-stimulated increase in p53 mRNA expression may be due to hPitx1 transcriptional activation of the p53 promoter. We therefore constructed a p53 promoter luciferase reporter plasmid pGL3-p53pro, which contains a 433-bp genomic fragment consisting of the promoter region of wild-type p53 sequence (−302 to −1) and a partial sequence of exon I (+1 to +131) of the human p53 gene in MCF-7 cells. MCF-7 cells were co-transfected with the pGL3-p53pro or the empty pGL3 vector in combination with the hPitx1-expressing plasmid or the control empty vector. As observed in Figure 2d, whereas hPitx1 expression did not have any effect on the basal level of luciferase expression of the promoterless empty pGL3 vector, hPitx1 expression did specifically increase the luciferase expression of the reporter plasmid pGL3-p53pro by threefold relative to the control. Thus, hPitx1 was able to increase p53 transcription in MCF-7 cells.
To determine whether the hPitx1-stimulated increase in p53 was mediating the apoptotic effect of hPitx1 overexpression, MCF-7 cells were co-transfected with EGFP-tagged hPitx1-expressing vector or the empty vector pEGFP in conjunction with either wild-type or the p53-143A mutant-expressing plasmid. As observed in Figure 2e, forced expression of wild-type p53 increased apoptosis of MCF-7 cells expressing either EGFP or EGFP-hPitx1. Forced expression of p53-143A mutant preferentially attenuated hPitx1-induced apoptosis of MCF-7 cells, indicating that p53 is directly involved in hPitx1-induced apoptosis in MCF-7 cells. Both wild-type and mutant p53 proteins were expressed similarly (data not shown).
Depletion of hPitx1 by siRNA decreases expression of p53 and its target genes
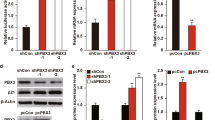
We next determined the effect of depleting endogenous hPitx1 by plasmid vector-based siRNA on p53 expression in MCF-7 cells. We designed and synthesized three pairs of oligonucleotides encoding short hairpin transcripts directed against a portion of the hPitx1 mRNA. The oligonucleotides were cloned into pSilencer 2.1 U6 vector. pSilencer-hPitx1 plasmids were stably transfected into MCF-7 cells. The effect of hPitx1 siRNA on hPitx1 expression level was determined by RT-PCR and Western blot in comparison with the negative control pSilencer-CK-transfected cells. The expression of endogenous hPitx1 mRNA was abrogated in MCF-7 cells by one of the three pSilencer-hPitx1 plasmids (Figure 3a). Western blotting demonstrated that hPitx1 protein was also effectively depleted in the pSilencer-hPitx1 stable. Moreover, depletion of hPitx1 by siRNA resulted in decreases of p53, as well as its downstream target genes p21 and PTFGβ, at both mRNA and protein levels.

Depletion of hPitx1 in MCF-7 cells by siRNA decreased expression of p53 and its target genes. (a) MCF-7 cells were stably transfected with plasmid expressing hPitx1-specific siRNA construct pSilencer-hPitx1 (Pitx1-RNAi) or the negative siRNA control plasmid pSilencer-CK (CK-RNAi) and the mRNA levels of indicated genes were determined by RT-PCR (left panel). The knockdown efficiency was quantitated with a UVP-system and normalized to β-actin relative to control siRNA. The protein levels were analyzed by Western blot (right panel). (b) The hPitx1-depleted (Pitx1-RNAi) and control (CK-RNAi) MCF-7 cells were transfected with the p53 luciferase reporter plasmid pGL3-p53pro or the promoterless empty pGL3 vector plasmid together with pCMVβ-Gal normalization control plasmid for transfection efficiency normalization. Luciferase activity was measured as in Figure 2d. ***P<0.001, relative to vector
As observed in Figure 2d, forced expression of hPitx1 was able to transactivate the p53 promoter in MCF-7 cells. We therefore tested whether the siRNA-reduced endogenous expression of hPitx1 would attenuate p53 promoter activity. The hPitx1-depleted and control MCF-7 cells were transfected with the pGL3-p53pro or the empty pGL3 vector together with the pCMVβ-Gal normalization control plasmid. As observed in Figure 3b, depletion of hPitx1 by siRNA decreased p53 promoter activity by fivefold, but was without effect on the basal level of luciferase expression of the promoterless pGL3 vector. Therefore, endogenous hPitx1 modulates p53 transcription in MCF-7 cells.
Depletion of p53 by siRNA impairs hPitx1-induced apoptosis
To determine the role of endogenous p53 in hPitx1-induced apoptosis, the endogenous level of p53 mRNA was decreased using an siRNA specifically targeting p53. Based on a highly effective p53 siRNA target sequence described previously,24 a p53 siRNA plasmid pSilencer-p53 was constructed and stably transfected into MCF-7 cells. As expected, the basal level of p53 in p53 siRNA-transfected cells was considerably reduced compared with negative control siRNA-transfected cells as determined by Western blot analysis (Figure 4a). The high efficiency of p53 depletion was documented previously.20, 24 Depletion of endogenous p53 in MCF-7 dramatically reduced hPitx1-induced apoptosis. The EGFP-tagged hPitx1-expressing vector or the empty vector pEGFP were transiently transfected into p53-depleted and control MCF-7 cells. As shown in Figure 4b, the apoptosis caused by the control EGFP was minimal (6%) and was similar in p53-depleted and control cells. However, apoptosis induced by EGFP-hPitx1 was reduced by threefold (46 versus 14%) in p53-depleted cells compared with the control cells. As observed in wild-type MCF-7 cells (Figure 1b), typical nuclear condensation of apoptotic cells was observed in EGFP-hPitx1-expressing control siRNA cells but not in the p53-depleted cells (Figure 4c).

Depletion of p53 by siRNA impairs hPitx1-induced apoptosis in MCF-7 cells. (a) Western blot of p53 with β-actin as loading control in MCF-7 cells stably transfected with a plasmid expressing p53-specific siRNA construct pSilencer-hPitx1 (p53-RNAi) or the negative siRNA control plasmid pSilencer-CK (CK-RNAi). (b, c) The p53-depleted (P53-RNAi) and control (CK-RNAi) MCF-7 cells were transfected with EGFP-tagged hPitx1 expression plasmid pEGFP-hPitx1-MycII (EGFP-hPitx1) or empty pEGFP vector plasmid (EGFP). At 24 h after transfection, apoptosis was determined as in Figure 1c using fluorescence microscopy (b) and representative microscopic pictures are shown (c). (d) The p53-depleted (p53-RNAi) and control (CK-RNAi) MCF-7 cells were stably transfected with the c-Myc-tagged hPitx1 (Myc-hPitx1) or the empty vector plasmid (Vector). Protein levels of p53 and hPitx1 were determined by Western blotting with the antibodies indicated. The p53 Western blot was overexposed to show the weak signal in the vector stably transfected p53-depleted cells. ***P<0.001, relative to pEGFP vector transfection. Scale bar=20 μm
We next reasoned that if p53 was the mediator of hPitx1-induced apoptosis in wild-type MCF-7 cells, it should be possible to stably force the expression of hPitx1 in p53-depleted cells. We therefore transfected the c-Myc-tagged hPitx1 expression plasmid (pUSE-hPitx1-MycII) or control empty pUSE vector into the p53-depleted and siRNA control MCF-7 cells. After selection, the expression of hPitx1 was determined by Western blot. As observed in Figure 4d, there was no detectable c-Myc-tagged exogenous hPitx1 in hPitx1-transfected siRNA control cells, whereas the forced expression was readily detectable in p53-depleted MCF-7 cells. The forced expression of hPitx1 was further confirmed by using a monoclonal antibody against hPitx1. It is notable that forced expression of hPitx1 in p53-depleted cells increased the basal level of p53, in agreement with our observation above (Figure 2b and c).
hPitx1-induced apoptosis in MG-63 cells is not mediated by p53
Unlike MCF-7 cells, MG-63 osteosarcoma cells are known to lack the p53 gene.25 To test whether forced expression of hPitx1 would also produce apoptosis independent of p53, MG-63 cells were transiently transfected with EGFP-tagged hPitx1 expression vector or pEGFP vector as control. Forced expression of EGFP-hPitx1 in MG-63 cells also caused apoptosis in comparison to EGFP-only-expressing cells. However, compared with MCF-7 cells (Figure 1c), the apoptosis in MG-63 cells was delayed and diminished (Figure 5a). At 1, 2 and 3 days after transfection, the percentages of apoptotic cells expressing EGFP-hPitx1 gradually increased and were 18, 23 and 36%, respectively. However, the percentage of apoptotic cells expressing EGFP did not change significantly over time and remained approximately similar at 5, 5 and 6%, respectively. These results are indicative that hPitx1-induced apoptosis, in addition to being p53-dependent in MCF-7 cells, also occurs in a p53-independent manner in MG-63 cells.

Forced expression of hPitx1 produced apoptotic cell death in p53-deficient MG-63 cells and disrupted cell division in both MCF-7 and MG-63 cells. (a) MG-63 cells were transfected with EGFP-tagged hPitx1-expressing plasmid pEGFP-hPitx1-MycII or empty pEGFP vector plasmid. On each day after transfection, cells were fixed and nuclei were stained with Hoechst 33258. Nuclei of green cells were examined with fluorescence microscopy and apoptosis resulting from the transient expression of EGFP (open bar) or EGFP-hPitx1-MycII (solid bar) was presented as the percentages of apoptotic cells with apoptotic nuclei out of the number of all green cells examined. For each point, 200–300 cells were examined. (b) MCF-7 and MG-63 cells at 20% confluence were transfected with EGFP-tagged hPitx1-expressing plasmid pEGFP-hPitx1-MycII (hatched bar) or empty pEGFP vector (open bar). At 48 h after transfection, cells were fixed and nuclei were stained with Hoechst 33258. The number of cells at metaphase and anaphase were counted with fluorescence microscopy. The percentages of green cells out of all dividing cells examined are presented as the mean±S.D. of three independent experiments and analyzed by Student's t-test. (c) MCF-7 cells were transfected with the EGFP-tagged hPitx1-expressing plasmid or empty pEGFP vector. At 18 h after transfection, cells were analyzed by flow cytometry. The percentage of transfected cells in each phase of the cell cycle (sub 2N, G0/G1, S, G2/M and >4N) are shown. (d) Disrupted anaphase of MG-63 cells. Sister chromosomes failed to separate from each other in a cell expressing EGFP-hPitx1 (upper panel) compared to an EGFP-expressing cell in which two sister chromosomes successfully migrated into two cells (lower panel). Scale bar=5 μm. ***P<0.001, relative to pEGFP vector transfection
Forced expression of hPitx1 disrupts cell division
We next examined other potential cellular effects of hPitx1. To determine if hPitx1 expression affects cell division, we plated MCF-7 and MG-63 cells at low confluence (20%) upon transfection with the EGFP-hPitx1 or EGFP-expressing plasmids. At 2 days after transfection, we determined the percentage of dividing cells within the EGFP–labeled population. As observed in Figure 5b, expression of EGFP-hPitx1 disrupted cell division in both MCF-7 and MG-63 cells. In MCF-7 cells, transient forced expression of hPitx1 almost completely inhibited cell division. Approximately 30% of dividing cells were EGFP-expressing 48 h after transfection, whereas only 1–2% of dividing cells were EGFP-hPitx1-expressing. In MG-63 cells, overexpression of hPitx1 had less effect on cell division. At 48 h after transfection, 45.5 and 30% of dividing cells were found to be EGFP- and EFGP-hPitx1-expressing cells, respectively.
Flow-activated cell sorter (FACS) analysis was utilized to examine the effect of forced expression of hPitx1 on MCF-7 cell-cycle progression. MCF-7 cells were transiently transfected with the EGFP-tagged hPitx1 expression vector or pEGFP vector as control. In view of the severe cell death induced by forced expression of hPitx1 (Figure 1c), transfected cells were incubated for 18 h before harvesting for flow cytometry. As observed in Figure 5c, there were significant differences in flow cytometric patterns between EGFP- and EGFP-hPitx1-expressing cells. The percentage of cells in G2/M phase decreased and that in sub-2N increased in EGFP-hPitx1-expressing cells compared with the control EGFP-expressing cells, indicative of induction of apoptosis and a significant inhibition of cell-cycle progression at the G1–S checkpoint.
The less severe disruption of cell division by hPitx1 in MG-63 cells made it possible to observe the effects of hPitx1 microscopically. For example, the sister chromosomes separated normally in EGFP-expressing cells and moved to opposite poles in anaphase (Figure 5d, lower panel), whereas the sister chromosomes in the EGFP-hPitx1-expressing cell failed to do so and stayed in the same hemisphere instead (Figure 5d, upper panel), which may result in aneuploidy.
Identification of hPitx1-R141P as a dominant-negative mutant
To further examine the effects of hPitx1, we decided to construct a dominant-negative molecule to inhibit Pitx1 function. Pitx2 mutations have often been detected in the homeodomain in patients with iris hypoplasia, iridogoniodysgenesis syndrome or Axenfeld-Rieger syndrome (ARS). One of the Pitx2 mutants, R91P, was observed to not only dominantly inhibit wild-type Pitx2 function, but also reverse the effects of Pitx1 in CV-1 cells.26 Pitx1 is known to share DNA-binding and transactivating characteristics with Pitx2.27 We therefore constructed and tested the effect of the hPitx1-R141P mutant, the equivalent of Pitx2-R91P mutant, on gene activation. MCF-7 cells were transfected with the LHβ luciferase reporter plasmid pGL2-bLHβ in combination with wild-type hPitx1 and R141P mutant expression plasmids in different ratios. As observed in Figure 6a, the R141P mutant inhibited wild-type hPitx1-induced transactivation of the LHβ promoter in a dose-dependent manner. Fourfold excess of the R141P mutant over wild-type hPitx1 reduced the transcriptional activity of hPitx1 by 20% and sixfold excess of the R141P mutant over wild-type hPitx1 reduced the transcriptional activity of hPitx1 almost to the basal level. The hPitx-R141P mutant reduced the basal level of LHβ transactivation by 50%, indicative that the mutant was able to inhibit the activity of endogenous hPitx1 in MCF-7 cells. Therefore, the R141P mutant of hPitx1 functioned as a dominant-negative inhibitor of hPitx1-stimulated transactivation and was able to inhibit the transcriptional activity of wild-type hPitx1. Unlike wild-type hPitx1, the expression of the hPitx1-R141P mutant could be detected in both transiently and stably transfected MCF-7 cells by the expression plasmid pUSE-hPitx1-MycII-R141P (Figure 1a), and did not result in apoptosis as did the wild-type hPitx1 (Figure 1d).

Mapping of hPitx1-responsive elements in the human p53 promoter. (a) Dominant-negative hPitx1 mutant hPitx1-R141P. The inhibitory effect of R141P mutant of hPitx1 on wild-type hPitx1 was examined with a luciferase reporter plasmid pGL2-bLHβ containing the bovine LHβ promoter in MCF-7 cells. Cells were co-transfected with 0.4 μg of the reporter plasmid along with 0.2 μg of wild-type hPitx1 and various amounts of hPitx1-R141P expression plasmids per well in six-well plates, keeping the total amount of plasmid DNA constant. (b) R141P mutant of hPitx1 aborted its transactivational ability on the p53 promoter. MCF-7 cells were co-transfected with p53 luciferase reporter plasmid pGL3-p53pro or the promoterless pGL3 vector plasmid in conjunction with wild-type hPitx1 (hatched bar) or R141P mutant (solid bar) expression plasmid as well as the control empty vector (open bar). Unlike wild-type hPitx1, R141P mutant attenuated its transcriptional activation on p53 promoter. (c) The nucleotide sequences of human p53 promoter from −302 to −1 in lower case (GenBank Acc no. J04238) and partial exon I from +1 to +131 in upper case (GenBank Acc no. NM_000546). The two hPitx1-responsive elements TAATCC (or GGATTA in reverse) and CAATCC (or GGATTG in reverse) are indicated as RE1 and RE2 in bold, respectively. The DNA probe (+44 to +71) used for gel shift assays (Figure 7b) is underlined. The positions of the two PCR primers used in chromatin immunoprecipitation assay (Figure 7c) are underlined and in bold italics. (d) Schematic diagrams of p53 promoter constructs in pGL3 luciferase reporter vector. The RE1 TAATCC (shaded circle •) or the RE2 CAATCC (shaded square ▪) were replaced by GAATTC (cross ×) in the mutant constructs. Luc, the luciferase coding sequence. (e) The effects of wild-type hPitx1 and R141P mutant on the wild-type p53 promoter and the hPitx1 RE-mutated p53 promoter were examined in MCF-7 cells. Cells were co-transfected with the promoterless pGL3 vector plasmid, wild-type p53 luciferase reporter plasmid pGL3-p53pro or hPitx1 RE-mutated p53 luciferase reporter plasmids (pGL3-p53pro-mu1, pGL3-p53pro-mu2 and pGL3-p53pro-mu12) as indicated, in conjunction with wild-type hPitx1 (hatched bar) or R141P mutant (solid bar) expression plasmid as well as the control empty vector (open bar). Where appropriate, transfections were performed in triplicate for each condition within a single experiment and luciferase activity was measured 20 h after transfection. Data are presented as the mean±S.D. ***P<0.001, relative to vector co-transfection
We next determined the effect of the hPitx1-R141P mutant on p53 transactivation. MCF-7 cells were transfected with the p53 luciferase reporter plasmid pGL3-p53pro or the promoterless empty pGL3 vector plasmid in combination with wild-type hPitx1 or R141P mutant expression plasmids as well as the control empty vector. Wild-type hPitx1 increased p53 promoter activity as previously demonstrated, whereas the hPitx1-R141P mutant reduced p53 promoter activity to 50% of the basal level (Figure 6b).
hPitx1 activates p53 transcription via DNA binding
The consensus sequence for the Pitx-1-binding element has been defined as 5′-TAA(T/G)CC-3′.2 The sequence of the 433-bp genomic fragment of the human p53 gene in MCF-7 cells cloned in the p53 luciferase reporter plasmid pGL3-p53pro was identical to that previously published.28 Analysis of the cloned sequence revealed two potential Pitx1 transcription factor regulatory elements (RE). One was ‘TAATCC’ (or GGATTA in reverse, RE1) and the other was ‘CAATCC’ (or GGATTG in reverse, RE2) (Figure 6c). Using PCR-based site-directed mutagenesis, we constructed three p53 promoter mutant reporter plasmids pGL3-p53pro-mu1, pGL3-p53pro-mu2 and pGL3-p53pro-mu12 in which either RE1 or RE2, or both RE1 and RE2, were replaced with ‘GAATTC’ (Figure 6d). To examine whether hPitx1 transactivated the p53 gene promoter via these Pitx1 consensus elements, these p53 mutant reporters were tested for responses to hPitx1 induction relative to the wild-type reporter pGL3-p53pro. As observed in Figure 6e, MCF-7 cells were transfected with the promoterless empty pGL3 vector plasmid, pGL3-p53pro, pGL3-p53pro-mu1, pGL3-p53pro-mu2 or pGL3-p53pro-mu12, in conjunction with wild-type hPitx1 or R141P mutant expression plasmids, as well as the empty vector as control. Mutation of the hPitx1 RE1 reduced p53 promoter basal activity (empty vector co-transfection) to 50% of that of the wild-type p53 promoter, and hPitx1-induced transcriptional activation (hPitx1 co-transfection) to 25% of that of the wild-type p53 promoter. However, mutation of RE2 or both RE1 and RE2 abrogated p53 promoter activity almost to the same level as that of the empty pGL3 control vector. The hPitx1-R141P mutant inhibited the basal p53 promoter activity level presumably by preventing the transcriptional activity of the endogenous hPitx1. Thus, hPitx1 directly activated p53 gene expression via the hPitx1 RE1 and RE2 in the p53 promoter with RE2 being the predominant hPitx1 RE. Alignment of the sequences of human and murine p53 genes indicates that the RE2 is conserved between species and RE1 is not (data not shown). Therefore, it is not surprising that RE2 is the major hPitx1 RE.
hPitx1 binds to the p53 promoter in vitro and in vivo
One likely mechanism for hPitx1 to transactivate the p53 gene promoter is by direct DNA binding to the promoter. Pitx1 has been shown to transactivate the POMC, LHβ and FSHβ genes by binding directly to promoter elements.2, 3 As the second Pitx1 RE is the major responsive site for the transcriptional activation of hPitx1 on the p53 promoter, we first examined in an electrophoretic mobility shift assay the in vitro binding of hPitx1 to a DNA probe containing RE2 spanning +44 to +71 of the first exon of p53 gene. Whole-cell lysates were prepared from MCF-7 cells transiently transfected with expression plasmid pUSE-hPitx1-MycII or empty vector. Myc-tagged hPitx1 formed a complex with the 32P-labeled oligonucleotide probe RE2 (Figure 7b, lane 3), which was completely abrogated by excess unlabeled probe RE2 (Figure 7b, lane 4). As demonstrated above, MCF-7 cells expressed endogenous hPitx1 and complex formation with much lower intensity was also observed with the lysate of empty vector-transfected MCF-7 cells (Figure 7b, lane 2). To verify the specificity of the protein–DNA interaction, the lysate containing the Myc-tagged hPitx1 protein was preincubated with either anti-c-Myc monoclonal antibody 9E10 or mouse control IgG before the addition of the 32P-labeled DNA probe RE2. The formation of a complex between Myc-tagged hPitx1 and the probe was attenuated by the specific anti-c-Myc monoclonal antibody 9E10 (Figure 7b, lane 6), but not by control IgG (Figure 7b, lane 5), indicating that the binding of the c-Myc antibody to the epitope-tagged hPitx1 protein prevented protein–DNA interaction. Thus hPitx1 binds directly to a hPitx1-responsive element within the p53 promoter.

hPitx1 binds to the p53 promoter in vitro and in vivo. (a) DNA probe sequences used in electrophoretic mobility shift assays. Probe RE2 consists of the hPitx1 RE2 consensus CAATCC in reverse orientation (in bold) and its flanked sequences in the human p53 promoter (GenBank Acc no. NM_000546). (b) hPitx1 binds the RE2 site within the exon I of the p53 gene. MCF-7 cells were transfected with Myc-tagged hPitx1 expression plasmid pUSE-hPitx1-MycII or control empty vector plasmid. Cells were harvested after 36 h in extraction buffer. Whole-cell lysates were prepared as described in Materials and methods. A 1 μg portion of lysate proteins from cells transfected with control vector plasmid (MCF7-vector, lane 2) or pUSE-hPitx1-MycII plasmid (MCF7-Myc-hPitx1, lanes 3-6) was incubated with 32P-labeled probe RE2 (lanes 1–6) at room temperature for 30 min in binding buffer. In some experiments, 200-fold unlabeled (‘cold’) probe RE2 was included (lane 4). For specificity binding experiments, mouse control IgG (lane 5) and monoclonal antibody 9E10 against c-Myc tag (lane 6) was pre-incubated with the cell lysates on ice for 4 h before adding the 32P-labeled probes. The mixtures were separated on 4% native polyacrylamide gel followed by exposure onto X-ray film. Free and protein-bound probes are indicated. (c) Analysis of recruitment of Pitx1 to p53 gene promoters. Chromatin immunoprecipitation (IP) analysis was performed using Pitx1 antibody or normal serum IgG (as control) from MCF-7 cells transiently transfected with the expression plasmid pUSE-hPitx1-MycII (P) or control empty vector (V) as described in Materials and methods. The presence of the p53 promoter DNA before immunoprecipitation was confirmed by PCR (input). PCR products were resolved in 1.2% agarose gels containing ethidium bromide. M, one kb plus DNA ladder (Invitrogen)
To determine whether hPitx1protein binds to the endogenous Pitx1 RE1 and RE2 regions of the p53 promoter and therefore directly regulates p53 gene expression, chromatin immunoprecipitation assays were performed using MCF-7 cells transiently transfected with the expression vector containing hPitX1 cDNA or the control empty vector (Figure 7c). A specific DNA fragment of expected size of 229 bp spanning the two Pitx1 RE sites was detected by PCR after immunoprecipitation of chromatin with a Pitx1-specific antibody, but not with the control IgG (or the human Artemin antibody, data not shown). Use of nonrelated primers (β-actin) demonstrated lack of PCR amplification in Pitx1 immunoprecipitates of chromatin (data not shown). Moreover, transient forced expression of hPitx1 significantly increased the amount of fragment precipitated by the Pitx1-specific antibody compared with the control vector transfection. Therefore, hPitx1 was indeed able to bind to the endogenous p53 promoter.
Upregulation of PTGFβ and p21 by hPitx1 is mediated by p53
Apoptosis as a result of increased p53 expression has been intensively studied.11 A multitude of p53 target genes have been implicated in this process.7 Among them, PTGFβ29 and cyclin-dependent kinase inhibitor p217, 14 are two well-known direct transcriptional targets of p53. To determine whether hPitx1 was able to activate the expression of these two p53 downstream target genes via p53 activation, a luciferase reporter plasmid containing wild-type PTGFβ promoter, which possesses two functional p53-responsive elements, was co-transfected into MCF-7 cells with plasmids expressing hPitx1 and its dominant-negative mutant hPitx1-R141P, wild-type p53 and its dominant-negative mutant p53-143A, and/or HPV-E6, the human papilloma virus type-16 E6 oncoprotein that promotes the degradation of p53 through its interaction with the cellular E6-associated protein (E6-AP), and together as the E3 ubiquitin ligase.13 As observed in Figure 8a, both p53 and hPitx1 activated PTGFβ transcription, whereas hPitx1-R141P, p53-143A and HPV-E6 inhibited PTGFβ transcription compared with control (vector co-transfection). The transcriptional activity of hPitx1 on the PTGFβ promoter was completely attenuated by the dominant-negative p53 mutant p53-143A and reduced by p53-degradation-promoting protein HPV-E6, indicating that PTGFβ transcriptional activation by hPitx1 was p53-dependent. The p53 mutant p53-143A and HPV-E6 also enhanced the inhibitory effects of dominant-negative mutant hPitx1-R141P. Moreover, neither p53 nor hPitx1 were able to transactivate the PTGFβ gene promoter with both the p53-responsive sites mutated, consistent with previous observations that mutation of both sites completely abolished p53 responsiveness.29 Similar results were obtained for p21, for which hPitx1 was shown to indirectly transactivate the p21 promoter via p53 activation (Figure 8b).

hPitx1 activates p53 downstream target genes PTGFβ and p21 in MCF-7 cells. (a) Cells were co-transfected with p53 RE containing wild-type PTGFβ promoter reporter plasmid pGL3-PTGFβ-w/p53RE or both the p53 RE mutated PTGFβ promoter reporter plasmid pGL3-PTGFβ-w/o-p53RE, in conjunction with control vector or plasmids expressing hPitx1and its mutant hPitx1-R141P, p53 and its mutant p53-143A, and the human papilloma virus type-16 E6 oncoprotein HPV-E6 separately or in combination as indicated. Transfection was carried out in serum-free RPMI 1640 media. Luciferase activity was measured 20 h post-transfection. Experiments were carried out in six-well plates in three replicates. (b) Same as in (a), except that the p53 RE-containing wild-type p21 promoter reporter plasmid pGL2-p21-w/p53 was used. (c) Forced overexpression of hPitx1 increased PTGFβ expression. Cells were transfected with Myc-tagged hPitx1expression plasmid pUSE-hPitx1-MycII (hPitx1) or the empty vector plasmid pUSE (vector). Cells were harvested 24 h after transfection. Total RNA was isolated and the mRNA levels of hPitx1, PTGFβ and β-actin were detected by RT-PCR using a one-step RT-PCR kit from Qiagen. Soluble whole-cell extracts were run on a 12% SDS-PAGE and transferred to nitrocellulose for Western blot analyses for protein expression. The same blot was first immunoblotted by the monoclonal antibody against PTGFβ, followed by the monoclonal antibody against Pitx1 and then by the monoclonal antibodies against β-actin to ensure equal loading of the cell lysate proteins after stripping. Data are presented as the mean±S.D. and analyzed by Student's t-test. *P<0.05 and ***P<0.001, relative to vector co-transfection. ###P<0.001, relative to hPitx1 co-transfection. &P<0.05 and &&&P<0.001, relative to hPitx1-R141P co-transfection
To further confirm the biological relevance of hPitx1 with p53 target genes, we analyzed the effects of hPitx1 on the expression of PTGFβ at both the mRNA and protein level. As observed in Figure 8c, forced transient expression of hPitx1 in MCF-7 cells dramatically increased not only PTGFβ mRNA as determined by RT-PCR, but also its protein expression as revealed by Western blot analysis.
Discussion
We have demonstrated that hPitx1 directly binds to consensus elements in the p53 promoter and stimulates p53 gene transcription and subsequent apoptosis in human mammary carcinoma cells. This is supported by the following lines of evidence: (1) transient forced expression of hPitx1 increased p53 expression at both mRNA and protein levels; (2) hPitx1 bound directly to the p53 promoter and transactivated p53 transcription, a process that was inhibited by the dominant-negative mutant hPitx1-141P; (3) reduction of hPitx1 expression by siRNA resulted in decreased expression of p53 and of its target genes p21 and PTGFβ; (4) apoptosis induced by forced expression of hPitx1 in p53-replete MCF-7 cells was enhanced by cotransfection of p53 but abrogated by cotransfection of the dominant-negative p53 mutant p53-143A; (5) depletion of p53 in MCF-7 cells by siRNA largely prevented apoptosis induced by forced expression of hPitx1. These observations, although different in mechanism, are in agreement with a recently published report6 where Pitx1 was identified to suppress tumorigenicity by downregulation of the Ras pathway through Rasal1, a Ras GTPase-activating protein. The potential tumor suppressive capacity of Pitx1 is correlated with reduced expression of Pitx1 in prostate and bladder cancer compared to normal tissues.6 Pitx1, as a negative regulator of oncogenicity, therefore constitutes a potential regulatory target for therapeutic intervention.
Pitx1 is known to share DNA-binding and transactivating characteristics with Pitx2.27 In this regard, it is interesting that Pitx2 has been shown to bind to HPV type 18 E6 protein and inhibit p53 degradation in vitro, leading to the accumulation of functional p53 protein and the induction of cell-cycle arrest in HeLa cells.30 Whether Pitx2 also transcriptionally activates p53 remains to be determined, but the two family members at least share functional equivalence in terms of p53 function. Here, we have demonstrated that hPitx1 binds directly to the p53 promoter in vitro and in vivo resulting in the transcriptional activation of the p53 gene. The p53 promoter contains two consensus sequences for hPitx1 protein binding, one located in the promoter itself and the other in the first exon of the p53 gene. By mutagenesis of the consensus elements, we demonstrated that both binding sites are essential for the transcriptional activation of the p53 gene by hPitx1, but that the consensus element within the first exon is the predominant functional site. This is not surprising, because it is well known that the first exon of p53 gene is an important regulatory region for p53 gene transcriptional regulation. Depletion of hPitx1 by hPitx1-specific siRNA or forced expression of the dominant-negative mutant hPitx1-141P both resulted in a decrease in p53 promoter activity. Thus, we have identified p53 as a direct transcriptional target of hPitx1.
MCF-7 cells harbor a wild-type p53 gene.23 In contrast, the human osteogenic sarcoma cell line MG-63 contains rearrangements in the p53 gene with no evidence of p53 expression in vitro either at the level of mRNA or protein.25 In this report, we observed that forced expression of hPitx1 in MG-63 also caused apoptosis. However, the apoptosis in MG-63 cells was delayed compared with that observed in MCF-7 cells. Induction of p53 often leads to an irreversible inhibition of cell growth, most decisively by activating apoptosis.14 The potential irreversibility of p53-dependent apoptosis in MCF-7 cells may explain the increased apoptotic potency of hPitx1 in MCF-7 cells compared with MG-63 cells. The dramatic reduction in the percentage of apoptotic cells induced by hPitx1 in p53-depleted MCF-7 cells strongly suggests p53 mediation of apoptosis. Interestingly, whereas it was not possible to establish MCF-7 cells with stable forced expression of hPitx1, we successfully forced the expression of hPitx1 in: (1) cells in which p53 expression was depleted by siRNA; and (2) cells with forced expression of the dominant negative mutant hPitx1-141P. As hPitx1 still induced apoptosis in p53-depleted MCF-7 cells, it is apparent that hPitx1-induced apoptosis also occurs independently of the presence of p53 and such an observation is consistent with the recent report of hPitx1 suppressing Ras activity to produce apoptosis.6
p53 mediates apoptosis by regulating transcription of target genes that contain a p53-binding regulatory sequence. hPitx1, by increasing p53 expression, would therefore potentially also regulate at least a subset of p53-regulated genes. One key downstream target of p53, the cyclin-dependent kinase inhibitor p21, mediates both G1 and G2/M phase arrest.7 An increase in p21 above threshold level results in G1 arrest due to inhibition of cdks and presumably p21–PCNA interaction. We demonstrated here that hPitx1 stimulated p21 gene transcription in mammary carcinoma cells and that this transactivation was mediated by p53. We have also demonstrated that another p53-regulated gene, PTGF-β, is regulated by hPitx1 in a p53-dependent manner. The TGFβ superfamily of cytokines contains a number of hormone-like polypeptides that play key roles in a variety of biological processes including cell-cycle progression, differentiation and apoptosis.31 PTGFβ was recently identified as a novel member of this family with high expression in placenta and prostate tissues. It is directly transactivated by p53 and is another key downstream mediator of p53.32, 33 The expression of PTGFβ was undetectable by Northern blot, but increased expression of this protein has previously been demonstrated to lead to G1 cell-cycle arrest and apoptosis in MCF-7 cells.32 In the present study, we have demonstrated that forced expression of hPitx1 dramatically increased PTGFβ expression at both the RNA and protein levels (Figure 8a and c). Although the precise contributions of p21 and PTGFβ to the overall growth arrest and apoptotic response to forced expression of hPitx1 remain to be determined, our results clearly demonstrate that hPitx1 activates p53-responsive genes. We observed that hPitx1 disrupts cell division in both MG-63 and MCF-7 cells. Transient forced expression of hPitx1 reduced cell division almost completely in MCF-7 cells, whereas in MG-63 it was reduced by only 34%. It is well known that p53 controls cell-cycle progression at the G1/S boundary by increasing transcription of the Cdk inhibitor p21.34 Therefore, it was not surprising that forced expression of hPitx1 resulted in cell-cycle arrest in G1/S phase, in addition to inducing apoptosis as (1) we demonstrated that hPitx1 was able to stimulate p21 gene transcription and translation and (2) depletion of hPitx1 decreased both p53 and p21 expression at the mRNA and protein levels.
Differential Pitx1 expression patterns in normal versus tumor tissue of different organs can be observed in the gene-expression database Oncomine.6 The expression of Pitx1 is significantly reduced in prostate and bladder cancers relative to their normal tissue. In breast cancer, estrogen receptor-positive breast carcinomas express significantly lower level of Pitx1 than do estrogen receptor-negative breast carcinomas, (P=0.007).35 Pitx1 mRNA expression was also shown to decrease in Barrett's esophagus compared with matching normal squamous esophagus specimens with a further decrease in Barrett's-associated cancer.36
In conclusion, we have demonstrated that hPitx1 stimulates mammary carcinoma apoptosis in a p53-dependent manner. This is achieved by direct hPitx1-mediated transcriptional activation of the p53 gene which subsequently activates its downstream effector molecules. Given that hPitx1 also inhibits oncogenicity in a non-p53-dependent manner,6 it may constitute a powerful therapeutic target for inhibition of neoplastic growth.
Materials and Methods
Cell lines
Human mammary carcinoma MCF-7 and osteosarcoma MG-63 cell lines were obtained from the ATCC. MCF-7 cells were grown in RPMI 1640 and MG-63 cells in DMEM media, supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine in a 37°C, humidified incubator with 5% CO2.
Preparation of total RNA
Total RNA was isolated from MCF-7 cells with Trizol reagent (Invitrogen) at 1 ml/10 cm2 according to the manufacturer's instructions and resuspended in diethyl pyrocarbonate-treated nuclease-free water. RNA samples were further treated with DNase I for 30 min at 37°C. The reaction was stopped by addition of 25 mM EDTA and incubation at 65°C for 15 min. RNA samples were then purified by extraction in phenol/chloroform (pH 5.2, phenol : chloroform : isoamyl alcohol 25 : 24 : 1), followed by an additional chloroform extraction and ethanol precipitation. Quantification and purity of the RNA was assessed by A260/A280 absorption, and RNA quality was assessed by agarose gel electrophoresis. RNA also samples with ratios of A260/A280 greater than 1.6 were stored at −80°C for further analysis.
Generation of expression and luciferase reporter plasmids
The coding region of hPitx1 was first amplified by RT-PCR from total RNA isolated from MCF-7 cells with a pair of primers: 5′-CATGGACGCCTTCAAGGGGGGC-3′ (sense) and 5′-CTGTTGTACTGGCACGCGTTGAG-3′ (antisense). The amplified DNA fragment was then cloned into a modified pBlueScript vector (Stratagene) between BamHI and BstBI sites using standard molecular cloning technologies so that the coding region is in frame with 3′ sequence encoding two copies of c-Myc-epitope (EQKLISEEDL) at C terminus and followed by the 3′-untranslated region sequence of mouse growth hormone. The resultant plasmid was termed pBS-hPitx1-MycII. Finally, the BamHI–EcoRI fragment of the pBS-hPitx1-MycII was subcloned into the pUSE amp (+) (BD Biosciences) vector between BamHI and EcoRI sites to generate the mammalian expression plasmid pUSE-hPitx1-MycII. The dominant-negative hPitx1 expression plasmid pUSE-hPitx1-MycII-R141P was generated from pUSE-hPitx1-MycII by PCR-based site-directed mutagenesis. EGFP-tagged hPitx1 expression plasmid pEGFP-hPitx1-MycII was obtained as follows. To facilitate cloning, the multiple cloning sites of pEGFP-C1 vector (BD Biosciences) were modified such that the original EcoRI site was shifted to immediately after the BspEI site and the XhoI site to just before the PstI site. Then the NcoI (5′-fill-in by Klenow) and KpnI fragment containing c-Myc-tagged full-length of hPitx1 coding sequence was released from the pBS-hPitx1-MycII and inserted into the modified pEGFP-C1 vector between EcoRI (5′-fill-in by Klenow) and KpnI sites, in frame with 5′ sequence encoding the N-terminal EGFP, to obtain the pEGFP-hPitx1-MycII plasmid.
p53 reporter plasmid, pGL3-p53pro, was constructed as follows. The p53 promoter region (−302/+131, GenBank accession numbers J04238 and NM_000546 for genomic and exon I sequences, respectively) was amplified by PCR with Vent DNA polymerase using human genomic DNA purified from MCF-7 cells as the template. The following oligonucleotides were used as the primers: 5′-AACGTTAGGGTGTGGATATTACG-3′ (sense) and 5′-ACGGTGGCTCTAGACTTTTGAG-3′ (antisense). The resulting DNA fragment was introduced into SmaI-digested promoterless luciferase reporter pGL3 basic vector (Promega).
p53 promoter mutant reporter plasmids were generated by PCR. Two potential hPitx1 transcription factor REs ‘TAATCC’ (reverse GGATTA) or ‘CAATCC’ (reverse GGATTG) were found in the p53 promoter (Figure 6c). One is at position −15 to −10 (GGATTA, RE1) and the other at position +54 to +59 (GGATTG, RE2). Either (pGL3-p53pro-mu1 or pGL3-p53pro-mu2) or both (pGL3-p53pro-mu12) REs were replaced with ‘GAATTC’ in the p53 promoter mutant reporter plasmids using PCR-based mutagenesis on the pGL3-p53pro plasmid (Figure 6d).
Bovine luteinizing hormone β (bLHβ) promoter reporter plasmid pGL2-bLH β (-776) was kindly provided by Dr. J Nilson, Washington State University, USA. The PTGFβ promoter (PTGFβ-w/-p53RE) or the PTGFβ promoter in which the two p53-binding sites were deleted (PTGFβ-w/o-p53RE) was subcloned into the pGL3 basic vector as described previously.37 The luciferase reporter plasmid pGL2-p21pro-w/-p53RE contains the 2.4 kilobase pairs HindIII fragment from the p21 promoter cloned into the pGL2 basic vector.38
RNA interference constructs
For design of siRNA oligonucleotides targeting hPitx1, a DNA sequence of type AA(N19) was selected to correspond to the nucleotides 1144–1164 of hPitx1 cDNA (GenBank accession number NM_002653). BLAST search against the human genome sequence showed that only the hPitx1 gene was targeted. The target sequence of p53 siRNA corresponded to nucleotides 1024–1044 of human p53 cDNA (GenBank accession number NM_000546) was described previously.24 Pairs of oligonucleotides, 5′-GATCCGCAACATCAACAACCTCACCTTCAAGAGAGGTGAGGTTGTTGATGTTGTTTTTTGGAAA-3′ and 5′-AGCTTTTCCAAAAAACAACATCAACAACCTCACCTCTCTTGAAGGTGAGGTTGTTGATGTTGCG-3′ for hPitx1 siRNA (pSilencer-hPitx1), and 5′-GATCCGACTCCAGTGGTAATCTACTTCAAGAGAGTAGATTACCACTGGAGTCTTTTTTGGAAA-3′ and 5′-AGCTTTTCCAAAAAAGACTCCAGTGGTAATCTACTCTCTTGAAGTAGATTACCACTGGAGTCG-3′ for p53 siRNA (pSilencer-p53), were cloned into pSilencer 2.1-U6 hygro vector (Ambion) according to the manufacturer's protocol. The negative control siRNA plasmid (pSilencer-CK) encodes siRNA having no significant sequence similarity to human gene sequences (Ambion).
Generation of knockdown/overexpression stable cell lines
The human breast cancer cell line MCF-7 was stably transfected with plasmids encoding siRNA targeted to hPitx1 or p53 transcript or with the negative control siRNA plasmid using Saint-Mix (Synvolux Therapeutics B.V., The Netherlands) transfection reagent. Cell clones were selected by addition of hygromycin to a concentration of 100 μg/ml in the medium. Transfected cell lines were generated as pools of positive cell clones. The expression of hPitx1 or p53 in the stable clones was determined by RT-PCR and Western blotting. Because we were unable to establish MCF-7 cells stably overexpressing hPitx1, p53-knocked down stable cells were further transfected with the hPitx1 expression plasmid pUSE-hPitx1-MycII or control vector, after we found out that the failure was due to hPitx1 induced p53-dependent apoptosis. These clones were selected under double selections of both hygromycin and neomycin (G-418 at a concentration of 800 μg/ml).
Cell-cycle analysis by flow cytometry
MCF-7 cells were transfected with EGFP-tagged hPitx1 expression plasmid pEGFP-hPitx1-MycII or empty pEGFP vector plasmid. After 18 h, the transfected cells and supernatants were collected together. The flow cytometry assay was performed by propidium iodide staining as described previously,39 after fixing in 2% paraformaldehyde (PFA) containing 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA and 10 mM PIPES, pH 6.8. A FACS analysis was carried out using a FACScan (Becton Dickinson) with the CELLQUEST software. Data were evaluated using WinMDI 2.8 research software (The Scripps Research Institute, La Jolla, CA, USA). A total of 20 000 events was measured per sample. The experiment was performed in triplicate.
Reverse transcriptase-PCR
One-step RT-PCR kit (Qiagen) was used to determine the presence and relative quantities of hPitx1, p53, p21 and PTGFβ transcripts. Primer sequences are as follows: (i) Pitx1 sense 5′-CTCCACCAAGAGCTTCACCTTC-3′; (ii) Pitx1 antisense 5′-CAGAGTGGGGTCCGGAAAAGC-3′; (iii) p53 sense 5′-AAGACTCCAGTGGTAATCTACTG-3′; (iv) p53 antisense 5′-ATCTAAGCTGGTATGTCCTACTC-3′; (v) p21 sense 5′-CGACTGTGATGCGCTAATGGC-3′; (vi) p21 antisense AACCTCTCATTCAACCGCCTAG-3′; (vii) PTGFβ sense 5′-CCCTGTCTCTGGCCGAGGCGAGC-3′; and (viii) PTGFβ antisense 5′- TGGGGTCTTGCAAGGCTGAGCTGAC -3′. In brief, we used the following procedure for RT-PCR: 1–1.5 μg of total RNA (diluted to 0.1 μg/μl to minimize variation of sample handling) was mixed with a master cocktail containing RT-PCR buffer, sense and antisense primers, dNTPs, RNase inhibitor and an enzyme mixture containing reverse transcriptase (Omniscript and Sensiscript) and HotStart Taq DNA polymerase at the concentrations recommended by the manufacturer (Qiagen) to a final volume of 50 μl. The temperature-cycle protocol was as follows: 30 min at 50°C for RT reaction, followed by denaturation and activation of HotStart DNA polymerase for 15 min at 95°C, and PCR amplification (10 s at 95°C, 30 s at 60°C and 1 min at 72°C for 26–30 cycles). A final extension for 10 min at 72°C was performed at the end of the cycles. β-actin was similarly RT-PCR amplified using 0.2 μg of total RNA as an internal control. Ten microliters of each of the RT-PCR products were fractionated on 1% agarose gels. The identity of RT-PCR product was confirmed by size, restriction enzyme digestion and DNA sequencing.
Immunoblotting
MCF-7 cells were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed at 4°C in lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1% Nonidet P-40, 1 μg/ml protease inhibitor cocktail (GE Healthcare) and 0.1 mM PMSF). The lysates were next sonicated and then cleared by centrifuging at 15 000 × g for 15 min at 4°C. SDS-PAGE sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 2% β-mercaptoethanol and bromophenol blue) was added to each sample and the samples were boiled for 5 min. Samples were subjected to discontinuous SDS-PAGE with a 12% resolving gel and transferred to nitrocellulose membranes (Hybond C-extra) using standard electroblotting procedures. Membranes were blocked with 5% non-fat dry milk in PBS with 0.1% Tween 20 (PBST) for 1 h at room temperature. The blots were then immunolabeled with the following primary antibodies: mouse monoclonal antibodies 9E10 against c-Myc, M01 against Pitx1 (Abnova) or DO-1 against p53, rabbit polyclonal antibody against p21 (C-19, sc-397) and goat polyclonal antibody against PTGFβ (H-20, sc-10603) (Santa Cruz biotechnology, CA), in PBST containing 1% non-fat dry milk at 4°C overnight. After incubation with appropriate secondary antibodies at room temperature, immunolabeling was detected by ECL plus™ chemiluminescence as described by the manufacturer (GE Healthcare). Blots were stripped and reprobed with monoclonal antibody against β-actin to ensure equal loading of the cell lysate proteins. Blots were stripped by incubation for 30 min at 50°C in a solution containing 62.5 mM Tris-HCl, pH 6.7, 2% SDS and 0.7% β-mercaptoethanol. Blots were then washed for 30 min with several changes of PBST at room temperature. Efficacy of stripping was determined by reexposure of the membranes to ECL plus. Thereafter, blots were reblocked and immunolabeled as described above.
Apoptosis assay
Apoptotic cell death was measured by fluorescent microscopic analysis of cell DNA staining patterns with Hoechst 33258. Cells were seeded on glass cover slips in six-well plates and transfected with pEGFP-hPitx1-MycII or pEGFP empty vector. At the end of transfection, cells were fixed with 4% PFA, permeabilized with 0.6% Tween 20 and stained with the karyophilic dye Hoechst 33258 (1 : 10 000; Molecular Probes, Eugene, OR) for 5 min at room temperature. Following washing with PBS, nuclear morphology was examined under a UV-visible fluorescence microscope (Zeiss Axioplan) with a × 40 objective. Green cells with apoptotic nuclear characteristics such as nuclear condensation and fragmentation were scored as apoptotic. Green cells (200–300) were examined on each treatment. In some experiments, the apoptotic cells were confirmed by TUNEL assay using a fluorometric in situ cell death detection kit (Roche) in accordance with the manufacturer's instructions.
Luciferase reporter assay
MCF-7 cells were plated in six-well plates the day before transfection so that they reached 60–80% confluence upon transfection. Reporter plasmids (1.0–1.5 μg/well) were transfected with Effectene (20 μl for each microgram of DNA) as per the manufacturer's instructions (Qiagen). DNA–lipid complex was diluted to a final volume of 2 ml/well with serum-free RPMI 1640 medium, added to the cells and the cells were left in the 37°C incubator for 5 h. The media were then changed to fresh serum-free RPMI 1640 for an additional incubation of 12–24 h. The cells were washed in cold PBS three times and lysed with 200 μl of the 1 × lysis buffer by a freeze-thaw cycle, and lysate was collected by centrifugation at 14 000 rpm for 2 min in a bench top centrifuge. Twenty microliters of supernatant was used for the assay of luciferase activity using a kit (Promega). The luciferase activities were normalized for the amount of total proteins in the cell lysates.
Electrophoretic mobility shift assays
The ability of hPitx1 to bind to the p53 promoter was characterized by electrophoretic mobility shift assays (EMSA). MCF-7 whole-cell lysates were prepared and EMSA performed as described previously.40 Briefly, one T75 flask of MCF-7 cells was transfected with 10 μg of expression plasmid pUSE-hPitx1-MycII or control empty vector using Effectine reagents. Cells were harvested after 36 h and resuspended in 600 μl of extraction buffer (20 mM HEPES, 150 mM NaCl, 25% glycerol, 0.5 mM dithiothreitol (DTT) and 1 μg/ml protease inhibitor cocktail). Cell suspensions were sonicated, the resulting mixture centrifuged and the supernatant frozen and stored at −80°C. The p53 promoter sequence region +44 to +71 (5′-GGGGTTGATGGGATTGGGGTTTTCCCCT-3′) containing the second putative hPitx1 RE (in bold) was used as the probe RE2. To generate probes, sense (5′-GGGGTTGATGGGATTGGGGTTTTCCC-3′) and antisense (5′-AGGGGAAAACCCCAATCCCATCAACC-3′) oligonucleotides corresponding to the RE2 probe were annealed, then the ends were filled in and labeled with α-[32P]dCTP using Klenow. EMSA was performed with 1 μg protein of the MCF-7 cell lysates in a 20 μl volume of gel shift binding buffer containing 15 mM Tris-HCl (pH 7.5), 6.5% glycerol, 0.7 mM EDTA, 0.2 mM DTT, 90 mM KCl, 1 mM bovine serum albumin, 500 ng of poly(dI-dC), 100 ng of sonicated salmon sperm DNA and 15 fmol of labeled oligonucleotides. Competition studies were performed using 200-fold unlabeled cold probe RE2. Cold competitor oligonucleotides were added 5 min before the addition of the probe. For specificity-binding experiments, monoclonal antibody 9E10 recognizing c-Myc-tagged Pitx1 or mouse control IgG was incubated with protein on ice for 4 h, after which the probe was added and the reaction mixtures were incubated at room temperature for an additional 30 min. Free DNA or protein–DNA complexes were resolved on 5% polyacrylamide gel in 0.5% TBE at 250 V at 4°C for 2 h. Gel were dried and exposed to Kodak X-OMAT film at −80°C.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation assay was performed with the EpiQuik™ chromatin immunoprecipitation kit (Epigentek, NY, USA) according to the manufacturer's instructions. Briefly, MCF-7 cells were transiently transfected with the expression plasmid pUSE-hPitx1-MycII or control empty vector. Cells were trypsinized and washed with PBS and then crosslinked in medium containing 1% of formaldehyde for 10 min at room temperature. Cells were washed in ice-cold PBS and lysed in a lysis buffer containing protease inhibitor cocktail. Chromatin was sheared to an average size of 500 bp by sonication (3 pulses of 10 s at continuous maximum power setting; Bandelin Sonopuls, Berlin, Germany). Goat anti-Pitx1 antibody Pitx1 (N-15) (sc-18922 ×) or control normal goat IgG (sc-2028) (both from Santa Cruz biotechnology, CA, USA), as well as the goat anti-human Artemin polyclonal antibody (AF2589, R&D Systems), were coated onto 96-well plates for 90 min at room temperature. A sample of chromatin was set aside before immunoprecipitation and used to represent the input DNA. Lysates corresponding to 5 × 105 cells were added and incubated for 90 min at room temperature on a rocking platform. The crosslinked-DNA fragments bound to the plates were washed and reversed by proteinase K treatment. Precipitated DNA fragments were recovered by column purification and quantified by PCR using the expand long template PCR system (Roche) with primer pairs flanking the two Pitx1 RE1 and RE2 sites in p53 promoter (sense 5′-AGTATCTACGGCACCAGGTCG-3′ and antisene 5′-ACGGTGGCTCTAGACTTTTGAG-3′; see Figure 6c). Thermal cycles were 3 min at 95°C followed by 40 cycles of 94°C, 30 s; 56°C, 30 s and 70°C, 2 min, plus a final extension at 70°C for 5 min. PCR products were resolved in 1.2% agarose gels containing ethidium bromide.
Statistics
All experiments were repeated at least three to five times. All numerical data are expressed as mean±S.D. Statistical analyses were carried out by the Student's t-test using Microsoft Excel XP.
Abbreviations
- EGFP:
-
enhanced green fluorescent protein
- hPitx1:
-
human pituitary homeobox 1
- PTGFβ:
-
placental transforming growth factor β
- siRNA:
-
small interfering RNA
- TUNEL:
-
TdT-mediated dUTP nick end labeling
References
Quentien MH, Barlier A, Franc JL, Pellegrini I, Brue T, Enjalbert A . Pituitary transcription factors: from congenital deficiencies to gene therapy. J Neuroendocrinol 2006; 18: 633–642.
Tremblay JJ, Lanctot C, Drouin J . The pan-pituitary activator of transcription, Ptx1 (pituitary homeobox 1), acts in synergy with SF-1 and Pit1 and is an upstream regulator of the Lim-homeodomain gene Lim3/Lhx3. Mol Endocrinol 1998; 12: 428–441.
Tremblay JJ, Drouin J . Egr-1 is a downstream effector of GnRH and synergizes by direct interaction with Ptx1 and SF-1 to enhance luteinizing hormone β gene transcription. Mol Cell Biol 1999; 19: 2567–2576.
Szeto DP, Rodriguez-Esteban C, Ryan AK, O'Connell SM, Liu F, Kioussi C et al. Role of the Bicoid-related homeodomain factor Pitx1 in specifying hindlimb morphogenesis and pituitary development. Genes Dev 1999; 13: 484–494.
DeLaurier A, Schweitzer R, Logan M . Pitx1 determines the morphology of muscle, tendon, and bones of the hindlimb. Dev Biol 2006; 299: 22–34.
Kolfschoten IG, van LB, Berns K, Mullenders J, Beijersbergen RL, Bernards R et al. A genetic screen identifies PITX1 as a suppressor of RAS activity and tumorigenicity. Cell 2005; 121: 849–858.
Vogelstein B, Lane D, Levine AJ . Surfing the p53 network. Nature 2000; 408: 307–310.
Vousden KH . p53: death star. Cell 2000; 103: 691–694.
Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004; 119: 861–872.
Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004; 119: 847–860.
Bates S, Vousden KH . p53 in signaling checkpoint arrest or apoptosis. Curr Opin Genet Dev 1996; 6: 12–18.
Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM et al. ‘Super p53’ mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J 2002; 21: 6225–6235.
Brooks CL, Gu W . p53 ubiquitination: Mdm2 and beyond. Mol Cell 2006; 21: 307–315.
Vousden KH, Lu X . Live or let die: the cell's response to p53. Nat Rev Cancer 2002; 2: 594–604.
Lakin ND, Jackson SP . Regulation of p53 in response to DNA damage. Oncogene 1999; 18: 7644–7655.
Takagi M, Absalon MJ, McLure KG, Kastan MB . Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005; 123: 49–63.
Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E et al. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature 2000; 405: 974–978.
Phan RT, la-Favera R . The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 2004; 432: 635–639.
Huang Q, Raya A, DeJesus P, Chao SH, Quon KC, Caldwell JS et al. Identification of p53 regulators by genome-wide functional analysis. Proc Natl Acad Sci USA 2004; 101: 3456–3461.
Wang S, el-Deiry WS . p73 or p53 directly regulates human p53 transcription to maintain cell cycle checkpoints. Cancer Res 2006; 66: 6982–6989.
Llanos S, Efeyan A, Monsech J, Dominguez O, Serrano M . A high-throughput loss-of-function screening identifies novel p53 regulators. Cell Cycle 2006; 5: 1880–1885.
Olivier M, Hainaut P . TP53 mutation patterns in breast cancers: searching for clues of environmental carcinogenesis. Semin Cancer Biol 2001; 11: 353–360.
Bohnke A, Westphal F, Schmidt A, El-Awady RA, hm-Daphi J . Role of p53 mutations, protein function and DNA damage for the radiosensitivity of human tumour cells. Int J Radiat Biol 2004; 80: 53–63.
Brummelkamp TR, Bernards R, Agami R . A system for stable expression of short interfering RNAs in mammalian cells. Science 2002; 296: 550–553.
Masuda H, Miller C, Koeffler HP, Battifora H, Cline MJ . Rearrangement of the p53 gene in human osteogenic sarcomas. Proc Natl Acad Sci USA 1987; 84: 7716–7719.
Quentien MH, Pitoia F, Gunz G, Guillet MP, Enjalbert A, Pellegrini I . Regulation of prolactin, GH, and Pit-1 gene expression in anterior pituitary by Pitx2: an approach using Pitx2 mutants. Endocrinology 2002; 143: 2839–2851.
Tremblay JJ, Goodyer CG, Drouin J . Transcriptional properties of Ptx1 and Ptx2 isoforms. Neuroendocrinology 2000; 71: 277–286.
Tuck SP, Crawford L . Characterization of the human p53 gene promoter. Mol Cell Biol 1989; 9: 2163–2172.
Wong J, Li PX, Klamut HJ . A novel p53 transcriptional repressor element (p53TRE) and the asymmetrical contribution of two p53 binding sites modulate the response of the placental transforming growth factor-β promoter to p53. J Biol Chem 2002; 277: 26699–26707.
Wei Q . Pitx2a binds to human papillomavirus type 18 E6 protein and inhibits E6-mediated P53 degradation in HeLa cells. J Biol Chem 2005; 280: 37790–37797.
Massague J . TGFβ signaling: receptors, transducers, and Mad proteins. Cell 1996; 85: 947–950.
Li PX, Wong J, Ayed A, Ngo D, Brade AM, Arrowsmith C et al. Placental transforming growth factor-β is a downstream mediator of the growth arrest and apoptotic response of tumor cells to DNA damage and p53 overexpression. J Biol Chem 2000; 275: 20127–20135.
Tan M, Wang Y, Guan K, Sun Y . PTGF-β, a type β transforming growth factor (TGF-β) superfamily member, is a p53 target gene that inhibits tumor cell growth via TGF-β signaling pathway. Proc Natl Acad Sci USA 2000; 97: 109–114.
Wang S, El-Deiry WS . p53, Cell cycle arrest and apoptosis. In: Hainaut P and Wiman KG (eds). 25 Years of p53 Research. Dordrecht: Springer, 2005 pp 141–163.
Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005; 365: 671–679.
Lord RV, Brabender J, Wickramasinghe K, DeMeester SR, Holscher A, Schneider PM et al. Increased CDX2 and decreased PITX1 homeobox gene expression in Barrett's esophagus and Barrett's-associated adenocarcinoma. Surgery 2005; 138: 924–931.
Graichen R, Liu D, Sun Y, Lee KO, Lobie PE . Autocrine human growth hormone inhibits placental transforming growth factor-β gene transcription to prevent apoptosis and allow cell cycle progression of human mammary carcinoma cells. J Biol Chem 2002; 277: 26662–26672.
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75: 817–825.
Riccardi C, Nicoletti I . Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc 2006; 1: 1458–1461.
Chalepakis G, Fritsch R, Fickenscher H, Deutsch U, Goulding M, Gruss P . The molecular basis of the undulated/Pax-1 mutation. Cell 1991; 66: 873–884.
Acknowledgements
This work was funded by the Marsden Fund, Royal Society of New Zealand, The Breast Cancer Research Trust (NZ), Foundation for Research, Science and Technology and The National Research Centre for Growth and Development (NRCGD, New Zealand).
We thank Dr John Nilson, Washington State University, USA for the bovine luteinizing hormone β promoter reporter plasmid pGL2-bLH β (−776).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by K Vousden
Rights and permissions
About this article
Cite this article
Liu, D., Lobie, P. Transcriptional activation of p53 by Pitx1. Cell Death Differ 14, 1893–1907 (2007). https://doi.org/10.1038/sj.cdd.4402209
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402209
Keywords
This article is cited by
-
Well-TEMP-seq as a microwell-based strategy for massively parallel profiling of single-cell temporal RNA dynamics
Nature Communications (2023)
-
Loss of resilience contributes to detrusor underactivity in advanced age
Biogerontology (2023)
-
PITX1 inhibits the growth and proliferation of melanoma cells through regulation of SOX family genes
Scientific Reports (2021)
-
Overexpression of the double homeodomain protein DUX4c interferes with myofibrillogenesis and induces clustering of myonuclei
Skeletal Muscle (2018)
-
Profilin potentiates chemotherapeutic agents mediated cell death via suppression of NF-κB and upregulation of p53
Apoptosis (2016)
