
Abstract
The role of Toll-like receptors (TLRs) in pathogen recognition has been expeditiously advanced in recent years. However, investigations into the function of TLRs in non-infectious tissue injury have just begun. Previously, we and others have demonstrated that fragmented hyaluronan (HA) accumulates during tissue injury. CD44 is required to clear HA during tissue injury, and impaired clearance of HA results in unremitting inflammation. Additionally, fragmented HA stimulates the expression of inflammatory genes by inflammatory cells at the injury site. Recently, we identified that HA fragments require both TLR2 and TLR4 to stimulate mouse macrophages to produce inflammatory chemokines and cytokines. In a non-infectious lung injury model, mice deficient in both TLR2 and TLR4 show an impaired transepithelial migration of inflammatory cells, increased tissue injury, elevated lung epithelial cell apoptosis, and decreased survival. Lung epithelial cell overexpression of high molecular mass HA protected mice against acute lung injury and apoptosis, in part through TLR-dependent basal activation of NF-κB. The exaggerated injury in TLR2 and TLR4 deficient mice appears to be due to impaired HA-TLR interactions on epithelial cells. These studies identify that host matrix component HA and TLR interactions provide signals that initiate inflammatory responses, maintain epithelial cell integrity, and promote recovery from acute lung injury.
Similar content being viewed by others


Introduction
A major advancement in our understanding of the mechanisms of host defense against pathogen invasion is the identification that the innate immune system uses Toll family receptors to signal for the presence of microbes and initiate host defense (see reviews in 1, 2). Toll-like receptors (TLRs) act as the pattern recognition receptors to detect pathogen attack and trigger host defense responses, as envisioned by Dr Janeway 3. Since Janeway and Medzhitov cloned and identified the first human homolog of the Toll protein 4, more than 10 mammalian TLRs have been reported (see reviews in 5, 6, 7). The merging work on genes in the Drosophila that control dorsal-ventral patterning and the long sought after gene that controls lipopolysaccharide (LPS) responsiveness led to the discovery of the Toll family proteins that mediate pathogen recognition by macrophages 4, 8. The spectra of the ligands for each TLR have been reported extensively (see reviews in 5, 6, 7, 9). Understanding their complex role in host defense offers tremendous potential for developing novel therapeutic drugs for more effective treatment of infectious and immune diseases.
While the importance of Toll family receptors in sensing pathogens is well recognized, it is also plausible that they may function in non-infectious diseases since TLR expression is also regulated in conditions other than infection 10. Studies into the function of TLRs in non-infectious tissue injury have just begun. This review will focus on our recent understanding on TLR-matrix interactions in non-infectious diseases 11.
TLRs in innate immunity
TLR2 was initially suggested to be involved in LPS recognition and signaling 12, 13. However, later studies demonstrated that TLR2 is a major receptor to recognize Gram-positive cell-wall components, including peptidoglycan and lipoteichoic acid, as well as mycobacterial cell-wall components such as lipoarabinomannan and mycolylarabinogalactan, and yeast cell-wall zymosan 8, 14. In vitro, TLR2-deficient macrophages produced reduced tumor necrosis factor and interleukin-6 in response to Staphylococcus aureus when compared to wild-type macrophages 8, 14.
Tlr4 gene is the long-sought-after causative gene for the LPS hyporesponsiveness in C3H/HeJ and 10ScCr mice 15. C3H/HeJ mice have a defective response to bacterial endotoxin because of a homozygosity for a codominant allele, Lpsd 15. Akira's group generated TLR4-deficient mice and demonstrated that TLR4 on macrophages recognizes the Gram-negative bacterial wall component LPS 8. Destructive mutations of TLR4 predispose to the development of Gram-negative sepsis, leaving most aspects of immune function intact 8. LPS interacts with LPS-binding protein and CD14 to present LPS to TLR4, which activates inflammatory gene expression through NF-κB and MAPK signaling 16.
There are four TLRs that are intracellularly situated and can be stimulated by nucleic acids during viral infections to initiate antiviral immunity, including interferon production 17. TLR3 recognize double-stranded RNA 18 and West Nile virus 19, TLR7 and TLR8 can be stimulated by antiviral derivatives such as imidazoquinoline and ixoribine, and guanosine- and uridine-rich single-stranded RNA oligonucleotides derived from human immunodeficiency virus-1 20. DNA from bacteria has stimulatory effects on mammalian immune cells, which depend on the presence of unmethylated CpG dinucleotides in the bacterial DNA. In contrast, mammalian DNA has a very low frequency of CpG dinucleotides, and these are mostly methylated. CpG DNA induces a strong Th1-like inflammatory response, while mammalian DNA does not have immunostimulatory activity. The cellular response to CpG DNA is mediated by TLR9 21.
TLR signaling pathway
TLR family receptors share structural similarity as well as a similar signaling pathway with interleukin-1 receptors. Upon ligand binding to TLR, the adaptor molecule MyD88 is recruited to TLR complex as a dimer 22, 23. Binding of MyD88 promotes association with interleukin-1 receptor-associated kinase 4 (IRAK4) and IRAK-1 24, 25, 26. Tumor necrosis factor-associated factor 6 (TRAF6) is recruited to IRAK-1 27. The complex IRAK-4/IRAK-1/TRAF6 dissociates from the receptor and then interacts with another complex consisting of transforming growth factor-β-activated kinase (TAK1), TAK1-binding protein 1 (TAB1), and TAB2 28, 29. TAK1 is subsequently activated in the cytoplasm, leading to the activation of IkB kinase kinases (IKKs) 28, 30. IKK activation leads to phosphorylation and degradation of IκB, and consequent release of NF-κB. Once translocated into the nucleus, NF-κB activates the expression of inflammatory chemokines and cytokines.
In addition to MyD88, other Toll/interleukin-1-receptor (TIR)-domain-containing adaptor proteins were identified and characterized. For example, TIRAP (TIR-domain-containing adaptor protein) 31, 32, TRIF (TIR-domain-containing adaptor protein inducing interferon-β) 33, and TRAM (TRIF-related adaptor molecule) 34 mediate MyD88-independent induction of interferon-β, which in turn activates the expression of interferon-inducible genes such as CXCL10.
Endogenous ligands for TLRs
In addition to the array of ligands that represent various pathogens, one cannot ignore the increasing amount of publications reporting endogenous ligands for TLRs. There are about four groups of putative endogenous ligands for TLR reported. First, several inflammatory proteins or peptides were reported to signal through TLRs. For example, heat-shock protein (Hsp)60 family chaperones were suggested as ligands for TLR2 and/or TLR4 in macrophages and B cells 35, 36, 37. High mobility group box 1 (HMGB1) is released extracellularly during acute inflammatory responses. Park and colleagues reported that stimulation of neutrophils, monocytes, or macrophages by HMGB1 required both TLR2 and TLR4 resulting in increased nuclear translocation of NF-κB and enhanced expression of proinflammatory cytokines 38. Murine β-defensin 2 acted directly on immature dendritic cells as an endogenous ligand for TLR-4, inducing up-regulation of costimulatory molecules and dendritic cell maturation 39. Second, some collectin molecules may signal through TLRs. For instance, the collectin surfactant protein-A is involved in innate host defense and the regulation of inflammatory processes in the lung. Surfactant protein-A-induced activation of the NF-κB signaling pathway and up-regulation of cytokine synthesis were reported to be critically dependent on the TLR4 functional complex 40. Third, nucleic acids of mammalian origin may act as endogenous ligands for TLRs and may promote systemic lupus erythematosus 41. In this study, Barrat and associates reported that mammalian DNA and RNA, when complexed with autoantibodies, are potent self-antigens for TLR9 and TLR7, respectively, and induce IFN-α production by plasmacytoid predendritic cells 41. The last group was the breakdown species of glycosaminoglycans. Oligosaccharides of hyaluronan (HA) activated dendritic cells and endothelial cells via TLR4 42, 43. We showed that HA fragments signal through TLR2 and TLR4 in macrophages 11. Maturation of dendritic cells by soluble heparan sulfate was reported to be mediated through TLR4 44.
However, the view of endogenous TLR ligands faces some skepticism due largely to the agonist contamination issue since most “ligands” used in these studies were prepared in microbial systems 45, 46, 47, 48. Many candidate endogenous ligands were shown to activate through more than one TLR 37, 38, 49, 50. For example, Hsps were reported as endogenous ligands for TLRs 35, 49. However, subsequent studies identified that LPS 48 and non-LPS contaminants 47 in Hsp preparations were responsible for cytokine induction. Secondly, the lack of biochemical studies on endogenous ligands and their respective TLRs thus far impedes the acceptance of the concept of endogenous ligands for TLR. Although many of the studies showed that the candidate endogenous ligands stimulated cells to produce cytokines via TLRs in vitro, biochemical analysis demonstrating the direct binding between a TLR and its endogenous ligand would be the key to coin the concept. Recently, Schaefer and colleagues 51 demonstrated direct binding between TLR4 and biglycan by using gel filtration, coimmunoprecipitation and electrospray ionization tandem mass spectrometry. Paradoxically, even genuine TLR agonists can be contaminated with agonists of other TLRs. For example, the initial mistaken assignment of TLR2 as the endotoxin receptor was likely because the agonist used in the studies was contaminated 8, 12, 52. Care has to be taken to ensure that TLR activation by endogenous ligands is genuine and not attributable to the contamination of the agonist preparation by the presence of LPS 11, 51. More importantly, increasing amount of evidence from in vivo studies 11, 51, where ligand contamination is less likely to contribute to TLR signaling, supports the concept of the endogenous ligands for TLRs. For example, Schaefer and associates 51 demonstrated that biglycan signals through TLR4 and TLR2 in a set of elegant in vivo experiments, and a critical question would be how these ligands function in various disease states.
TLRs and lung injury
It is becoming clear that TLRs not only play a role in the recognition of pathogens and in the initiation of immune responses but also have a fundamental role in non-infectious disease pathogenesis. Recent studies suggested that TLR may regulate and contribute to the pathogenesis of diseases with non-infectious tissue injury, since elevated expression of TLRs has been described in such diseases or pathological states 10, 53, and TLR polymorphisms have been associated with the susceptibility to certain diseases 54, 55, 56, 57. For example, inactivation of the MyD88 pathway led to a reduction in atherosclerosis through a decrease in macrophage recruitment to the artery wall that was associated with reduced chemokine levels 58. TLR4 (Asp299Gly) polymorphism, which attenuates receptor signaling and diminishes the inflammatory response to Gram-negative pathogens, was associated with a decreased risk of atherosclerosis 59. These studies suggested that the activation of TLR pathway promotes the development of atherosclerosis. The biological roles of TLRs in non-infectious lung pathogenesis have been explored and can be classified into three areas (Table 1): (i) In host defense, TLR4 recognizes diesel exhaust particles 60 and pollutant ozone 61, supporting that TLR4 may be one of the recognition receptors to sense pollutants in the airways. (ii) In allergic diseases, TLR9 ligand-induced pulmonary indoleamine 2,3-dioxygenase activity by resident lung cells rather than by pulmonary dendritic cells inhibits T-helper cell (Th)2-driven lung inflammation and airway hyper-reactivity in the experimental asthma, providing evidence for the inverse relationship between microbial exposure and the prevalence of allergic asthma and autoimmune diseases in Westernized countries 62. This study also highlights the notion that activation of innate immunity can impede adaptive Th responses 62. (iii) In tissue injury and repair, several recent studies examined the expression patterns of TLR2 and/or TLR4 in smoke-induced lung injury and in patients with chronic obstructive pulmonary disease 63, 64, 65. Hemorrhage-induced lung TNF-α production, neutrophil accumulation, and protein permeability, but not NF-κB activation, were dependent on a functional TLR4 66. TLR4-TLR2 cross-talk activated a positive-feedback signal leading to alveolar macrophage priming and exaggerated lung inflammation in response to invading pathogens during hemorrhage-induced acute lung injury 67. On the other hand, a recent study demonstrated that TLR4 has a protective role in oxidant-mediated injury, by maintaining appropriate levels of antiapoptotic responses in the face of oxidant stress 68. This study is in accord with our study in a bleomycin-induced lung injury model 11.
HA as a signaling molecule
During tissue injury, there is increased turnover of ECM. A variety of lung diseases such as asthma, emphysema and pulmonary fibrosis are associated with abnormal ECM turnover. In chronic lung diseases, the ECM is modified by the inflammatory milieu, and degradation products generated by oxidants and other mechanisms take on unique properties not attributable to the precursor molecules. HA is a major ECM component, a non-sulfated glycosaminoglycan composed of repeating polymeric disaccharides D-glucuronic acid and N-acetyl glucosamine 69. It is distributed widely in nature, such as the Streptococcus coat. In mammals, HA is abundant in heart valves, skin, skeletal tissues, the vitreous of the eye, umbilical cord, and synovial fluid. HA has the property of a biological lubricant, reducing friction during movement and providing resiliency under static conditions. Under physiologic conditions, HA exists as a high molecular weight polymer (>106 Da) and undergoes dynamic regulation resulting in the accumulation of lower molecular weight species following tissue injury 70, 71. One often compares its structure with LPS. Although they both contain repeating di- or oligosaccharides, HA does not contain lipids. Saccharide units in HA are simple D-glucuronic acid and N-acetyl glucosamine. Core polysaccharide in LPS consists of a short chain of sugars with variations, for example, 2-keto-3-deoxyoctonoic acid-heptose-heptose-glucose-galactose-glucose-N-acetyl glucosamine. The O antigen chain of LPS consists of repeating oligosaccharide subunits made up of 3-5 sugars. The individual chains vary in length ranging up to 40 repeat units 72.
A historical study by West and colleagues 73, 74 showed that oligosaccharides derived from high molecular weight HA stimulate angiogenesis in vivo and endothelial proliferation in vitro. We and others have demonstrated that HA degradation products generated in vitro in the 200 000 Da range induce the expression of a variety of genes, including chemokines, cytokines, growth factors, signal transduction molecules, and adhesion molecules in macrophages, eosinophils, dendritic cells, kidney epithelial cells, and fibroblasts, suggesting that endogenously generated HA fragments may be a dynamic molecule that influences cell behavior by regulating inflammatory processes 43, 75, 76, 77, 78, 79, 80, 81. These studies support a concept in which the modified matrix regulates inflammation and repair processes during tissue injury.
CD44 and lung injury
HA binds to numerous cell surface proteins. Among them, CD44 is a major cell surface receptor for HA 82. CD44 is a type 1 transmembrane glycoprotein that is expressed on most cell types. The interaction of HA and CD44 has been implicated in the regulation of a variety of biological processes, including tumor growth and metastasis, wound healing, T-cell recruitment to sites of inflammation, macrophage activation, neutrophil migration, and endothelial cell activation (see reviews in 83, 84). For example, HA promotes leukocyte trafficking to the inflammatory sites during tissue injury 85. A recent study demonstrated that gdT cells are required for HA deposition in the ECM and subsequent macrophage infiltration into wound sites 86. HA fragments can deliver maturational signals to antigen-presenting dendritic cells and can provide costimulatory signals to specific T cells in regulating dendritic cell maturation and trafficking 43. However, CD44 null mice were found to develop normally 87. In order to investigate the role of HA in lung injury and repair, we examined the inflammatory response in CD44-deficient mice 71. CD44-deficient mice and wild-type C57Bl/6 mice were instilled intratracheally with bleomycin, a widely used model for pulmonary injury and fibrosis. CD44-deficient mice displayed an increased mortality, and increased numbers of neutrophils, macrophages, and lymphocytes in bronchoalveolar lavage fluid 71. In addition, there was overwhelming HA accumulation in the injury lungs in the CD44-deficient mice relative to the wild-type control mice 71. Thus, the CD44-deficient state was associated with both an inability to resolve lung inflammation and a failure to properly remove HA degradation products from the lung tissue. These data suggest that the production of HA following acute tissue injury may serve the very important function of initiating the host innate immune response by providing an essential signal to macrophages, which then produce chemokines that recruit other leukocyte subsets required to debride the tissue injury and begin restoring tissue integrity 71.
TLRs and HA signaling
CD44 appears to be involved in HA binding in vitro since the blockade with anti-CD44 antibodies partially inhibits HA fragment signaling in macrophages 88. However, it is not sufficient to mediate HA signaling 11, 89. Thus, there must be another receptor system required for HA signaling. Since HA is a repeating disaccharide structure with features of “pathogen-associated molecular patterns”, and since recent reports suggest that HA oligomers can signal through TLR4 in dendritic cells 43 and endothelial cells 42, we investigated the potential role of TLRs in non-infectious lung injury 11 where matrix turnover is an important regulator of the inflammatory response 43, 86. Both TLR2 and TLR4 signaling pathways appear to require MyD88. MyD88-deficient mice (and macrophages) are unresponsive to LPS in vitro and in vivo 22, 23, 90. To investigate the potential role for TLRs in mediating HA signaling, we utilized elicited peritoneal macrophages from MyD88-deficient, TLR1-, TLR2-, TLR3-, TLR4-, TLR5-, and TLR9-deficient mice. Stimulation of chemokine gene expression by HA fragments is abolished in the MyD88-deficient macrophages 11. Chemokine MIP-2 expression is reduced, but remains present in both TLR2- and TLR4-deficient macrophages, respectively. TLR4- and TLR2-deficient mice were then crossed to generate TLR2- and TLR4-double-deficient (TLR2−/−TLR4−/−) mice. HA fragment-induced chemokine and cytokine expression was completely abolished in TLR2−/−TLR4−/− peritoneal macrophages 11. These data suggest that the polymeric properties of HA may mimic cell-wall components from both Gram-positive and Gram-negative organisms. In contrast, targeted deletion of TLR1, TLR3, TLR5, and TLR9 had no effect on HA fragment-induced chemokine expression. As our data demonstrated HA induces chemokine expression through TLR2 and TLR4, the next question would be: do HA fragments directly bind to TLR4 and TLR2? Biochemical studies such as photoaffinity labeling or direct binding should be performed to elucidate whether and how HA fragments bind to TLRs and to examine whether any other binding partners are involved.
To determine if these findings were relevant to acute lung injury in patients, we then examined circulating HA fragments produced in vivo and purified from the serum of patients with acute lung injury. The human HA degradation products were of similar molecular mass (peak 200 kDa) to the in vitro generated HA degradation products and stimulated chemokine production in wild-type, but neither TLR2−/−TLR4−/− nor MyD88−/− peritoneal macrophages 11.
TLR deficiency and lung injury
The observation that CD44 deficiency led to unremitting inflammation following lung injury clearly implicated an alternative signaling pathway by which macrophages responded to HA fragments. Our discovery that both TLR2 and TLR4 are required for macrophages to express inflammatory genes in response to HA fragments suggested that TLR2/TLR4 deficiency should lead to an impaired inflammatory response to non-infectious lung injury. To test this hypothesis, we challenged TLR2−/−TLR4−/− mice with bleomycin-induced lung injury. TLR2−/−TLR4−/− mice developed a reduced inflammatory response to lung injury with a decrease in transepithelial neutrophil migration and reduced expression of MIP-2 11. However, these mice displayed an increase in mortality. This surprising finding led us to inspect the degree of tissue injury and epithelial cell integrity. We found evidence to suggest increased tissue injury and epithelial cell apoptosis 11. Thus, TLR2/TLR4 deficiencies appear to protect the host from acute inflammation, but may impede epithelial cell repair processes important in tissue injury recovery.
HA-cell interaction in lung injury
Next, we tested whether the blockade of HA action with an HA-blocking peptide could produce a similar phenotype as in TLR2−/−TLR4−/−. An HA-blocking peptide has been previously shown to be effective in vivo in inhibiting HA-cell interactions 42, 85, 91. Inhibition of HA binding with the peptide in vivo recapitulated the phenotype observed in the TLR2−/−TLR4−/− mice after lung injury, enhanced lung injury and increased epithelial cell apoptosis, and decreased transmigration of neutrophils 11. Increasing production of high molecular weight HA by overexpression of HA synthase-2 under the control of the lung-specific CC10 promoter is protective against mortality, lung injury, epithelial cell apoptosis following high-dose bleomycin 11. Therefore, HA-cell interaction regulates the responses to lung injury.
HA-TLR interaction on epithelial cells
Previous studies in kidney injury models have demonstrated that fragmented HA markedly stimulates chemokine MCP-1 production by renal tubular cells 92, triggers cell adhesion molecule expression through a mechanism involving activation of NF-κB and activating protein-1 93, and promotes proximal tubule cell migration and re-epithelialization, through MAPK activation 94. Quinn's group has shown that low molecular weight HA stimulated cytokine IL-8 production by lung epithelial cells, via JAK2 kinase, not MEK1/2, pathway 95. It is plausible that injured epithelial cells respond to HA fragments by producing chemokines and cytokines to recruit inflammatory cells to the injury sites. To investigate the hypothesis that HA and TLR interactions are important in lung injury and repair processes, we asked if HA on the epithelial cell surface plays a role in lung injury. Isolated lung alveolar epithelial cells have increased apoptosis at baseline and exhibit greater total apoptosis in response to bleomycin. The exogenous addition of high molecular weight HA is protective against apoptosis 11. We found that bleomycin induces both NF-κB activation and apoptosis in primary lung epithelial cells 11. It is likely that HA regulates basal NF-κB activation in epithelial cells. NF-κB is known to regulate apoptosis 96, 97 and we have previously shown that HA fragments can activate NF-κB in macrophages 77. Primary epithelial cells from TLR2−/−TLR4−/− mice were found to have a significant increase in spontaneous apoptosis relative to wild-type cells 11. Intriguingly, we also found that cell surface HA is severely abrogated in TLR2−/−TLR4−/− epithelial cells 11. The mechanism underlying the abolishment of cell surface HA in TLR2−/−TLR4−/− epithelial cells is unknown. Nevertheless, these data suggest that epithelial cell-surface HA promotes basal NF-κB activation in a TLR-dependent manner, and that this activation has a protective effect against injury 98.
Conclusion
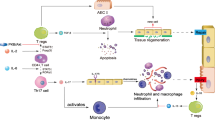
As part of inflammatory responses, HA fragments are generated following tissue injury in both animal models and patients. The removal of HA fragments requires the HA receptor CD44. The failure to remove HA from the lung following injury results in unremitting inflammation. The persistent HA fragment presence leads to chronic inflammation and tissue damage. Here we propose that the interactions of HA and TLRs serve two critical functions in host defense against non-infectious lung injury (Figure 1). Soluble HA fragments can stimulate macrophages to produce chemokines and cytokines that recruit inflammatory cells to the site of injury. On the other hand, native high molecular weight HA limits the extent of lung epithelial cell injury by providing a basal NF-κB activation and inhibiting apoptosis and promoting the repair of parenchymal cell injury through a TLR-dependent mechanism. Shaping the balance of HA in favor of high molecular weight form could favor the host recovery from acute lung injury.

HA signals through TLR2 and TLR4 in both epithelial cells and macrophages. (A) High molecular weight hyaluronan (HMW-HA) on the cell surface or surrounding epithelial cells signals through to TLR2 and TLR4, providing cells with basal NF-κB activation. In turn, the tonic NF-κB activity prevents epithelial cells from undergoing apoptosis upon injury. Thus, HMW-HA on epithelial cells provides cells with a survival signal. On other hand, low molecular weight hyaluronan (LMW-HA) fragments generated during tissue injury stimulate chemokine/cytokine expression in macrophages, leading to inflammatory responses. (B) The loss of TLR2 and TLR4, or MyD88 on epithelial cells causes the loss of basal NF-κB activation and leads to an increased apoptosis of epithelial cells upon injury. In macrophages, LMW-HA cannot transduce signals due to TLR deficiency. This leads to a decreased NF-κB activation and subsequently decreases inflammatory responses.
References
Aderem A, Ulevitch RJ . Toll-like receptors in the induction of the innate immune response. Nature 2000; 406:782–787.
Kopp E, Medzhitov R . Recognition of microbial infection by Toll-like receptors. Curr Opin Immunol 2003; 15:396–401.
Janeway CA Jr . Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 1989; 54(Part 1):1–13.
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr . A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997; 388:394–397.
Lauw FN, Caffrey DR, Golenbock DT . Of mice and man: TLR11 (finally) finds profilin. Trends Immunol 2005; 26:509–511.
Iwasaki A, Medzhitov R . Toll-like receptor control of the adaptive immune responses. Nat Immunol 2004; 5:987–995.
Akira S, Takeda K . Toll-like receptor signalling. Nat Rev Immunol 2004; 4:499–511.
Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity 1999; 11:443–451.
Takeda K, Kaisho T, Akira S . Toll-like receptors. Annu Rev Immunol 2003; 21:335–376.
Frantz S, Kobzik L, Kim YD, et al. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest 1999; 104:271–280.
Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 2005; 11:1173–1179.
Kirschning CJ, Wesche H, Merrill Ayres T, Rothe M . Human toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J Exp Med 1998; 188:2091–2097.
Yang RB, Mark MR, Gray A, et al. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature 1998; 395:284–288.
Underhill DM, Ozinsky A, Hajjar AM, et al. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 1999; 401:811–815.
Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998; 282:2085–2088.
Bochkov VN, Kadl A, Huber J, et al. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature 2002; 419:77–81.
Doyle S, Vaidya S, O'Connell R, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 2002; 17:251–263.
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA . Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001; 413:732–738.
Wang T, Town T, Alexopoulou L, et al. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 2004; 10:1366–1373.
Diebold SS, Kaisho T, Hemmi H, et al. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004; 303:1529–1531.
Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000; 408:740–745.
Medzhitov R, Preston-Hurlburt P, Kopp E, et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Molecular Cell 1998; 2:253–258.
Adachi O, Kawai T, Takeda K, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998; 9:143–150.
Muzio M, Ni J, Feng P, Dixit VM . IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 1997; 278:1612–1615.
Huang J, Gao X, Li S, Cao Z . Recruitment of IRAK to the interleukin 1 receptor complex requires interleukin 1 receptor accessory protein. Proc Natl Acad Sci USA 1997; 94:12829–12832.
Cao Z, Henzel WJ, Gao X . IRAK: a kinase associated with the interleukin-1 receptor. Science 1996; 271:1128–1131.
Ye H, Arron JR, Lamothe B, et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature 2002; 418:443–447.
Shibuya H, Yamaguchi K, Shirakabe K, et al. TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science 1996; 272:1179–1182.
Takaesu G, Kishida S, Hiyama A, et al. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell 2000; 5:649–658.
Ninomiya-Tsuji J, Kishimoto K, Hiyama A, et al. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 1999; 398:252–256.
Horng T, Barton GM, Flavell RA, Medzhitov R . The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 2002; 420:329–333.
Fitzgerald KA, Palsson-McDermott EM, Bowie AG, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 2001; 413:78–83.
Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003; 301:640–643.
Yamamoto M, Sato S, Hemmi H, et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol 2003; 4:1144–1150.
Ohashi K, Burkart V, Flohe S, Kolb H . Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol 2000; 164:558–561.
Cohen-Sfady M, Nussbaum G, Pevsner-Fischer M, et al. Heat shock protein 60 activates B cells via the TLR4-MyD88 pathway. J Immunol 2005; 175:3594–3602.
Vabulas RM, Ahmad-Nejad P, da Costa C, et al. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem 2001; 276:31332–31339.
Park JS, Svetkauskaite D, He Q, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 2004; 279:7370–7377.
Biragyn A, Ruffini PA, Leifer CA, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 2002; 298:1025–1029.
Guillot L, Balloy V, McCormack FX, et al. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J Immunol 2002; 168:5989–5992.
Barrat FJ, Meeker T, Gregorio J, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med 2005; 202:1131–1139.
Taylor KR, Trowbridge JM, Rudisill JA, et al. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem 2004; 279:17079–17084.
Termeer C, Benedix F, Sleeman J, et al. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med 2002; 195:99–111.
Johnson GB, Brunn GJ, Kodaira Y, Platt JL . Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol 2002; 168:5233–5239.
Tsan MF, Gao B . Endogenous ligands of Toll-like receptors. J Leukocyte Biol 2004.
Hirschfeld M, Ma Y, Weis JH, et al. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol 2000; 165:618–622.
Gao B, Tsan MF . Recombinant human heat shock protein 60 does not induce the release of tumor necrosis factor alpha from murine macrophages. J Biol Chem 2003; 278:22523–22529.
Gao B, Tsan MF . Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor alpha release by murine macrophages. J Biol Chem 2003; 278:174–179.
Vabulas RM, Ahmad-Nejad P, Ghose S, et al. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem 2002; 277:15107–15112.
Asea A, Rehli M, Kabingu E, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem 2002; 277:15028–15034.
Schaefer L, Babelova A, Kiss E, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest 2005; 115:2223–2233.
Hoshino K, Takeuchi O, Kawai T, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 1999; 162:3749–3752.
Frantz S, Kelly RA, Bourcier T . Role of TLR-2 in the activation of nuclear factor kappaB by oxidative stress in cardiac myocytes. J Biol Chem 2001; 276:5197–5203.
Berdeli A, Celik HA, Ozyurek R, et al. TLR-2 gene Arg753Gln polymorphism is strongly associated with acute rheumatic fever in children. J Mol Med 2005; 83:535–541.
Hamann L, Gomma A, Schroder NW, et al. A frequent toll-like receptor (TLR)-2 polymorphism is a risk factor for coronary restenosis. J Mol Med 2005; 83:478–485.
Cooke GS, Segal S, Hill AV . Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med 2002; 347:1978–1980; author reply 1978–1980.
Sabroe I, Whyte MK, Wilson AG, et al. Toll-like receptor (TLR) 4 polymorphisms and COPD. Thorax 2004; 59:81.
Bjorkbacka H, Kunjathoor VV, Moore KJ, et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med 2004; 10:416–421.
Kiechl S, Lorenz E, Reindl M, et al. Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med 2002; 347:185–192.
Inoue K, Takano H, Yanagisawa R, et al. The role of toll-like receptor 4 in airway inflammation induced by diesel exhaust particles. Arch Toxicol 2006; 80:275–279.
Kleeberger SR, Reddy S, Zhang LY, Jedlicka AE . Genetic susceptibility to ozone-induced lung hyperpermeability: role of toll-like receptor 4. Am J Respir Cell Mol Biol 2000; 22:620–627.
Hayashi T, Beck L, Rossetto C, et al. Inhibition of experimental asthma by indoleamine 2,3-dioxygenase. J Clin Invest 2004; 114:270–279.
Karimi K, Sarir H, Mortaz E, et al. Toll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophages. Respir Res 2006; 7:66.
Pons J, Sauleda J, Regueiro V, et al. Expression of Toll-like receptor 2 is up-regulated in monocytes from patients with chronic obstructive pulmonary disease. Respir Res 2006; 7:64.
Droemann D, Goldmann T, Tiedje T, et al. Toll-like receptor 2 expression is decreased on alveolar macrophages in cigarette smokers and COPD patients. Respir Res 2005; 6:68.
Barsness KA, Arcaroli J, Harken AH, et al. Hemorrhage-induced acute lung injury is TLR-4 dependent. Am J Physiol Regul Integr Comp Physiol 2004; 287:R592–599.
Fan J, Li Y, Vodovotz Y, et al. Hemorrhagic shock-activated neutrophils augment TLR4 signaling-induced TLR2 upregulation in alveolar macrophages: role in hemorrhage-primed lung inflammation. Am J Physiol Lung Cell Mol Physiol 2006; 290:L738–L746.
Zhang X, Shan P, Qureshi S, et al. Cutting edge: TLR4 deficiency confers susceptibility to lethal oxidant lung injury. J Immunol 2005; 175:4834–4838.
Laurent TC 1988. The Chemistry, Biology and Medical Applications of Hyaluronan and its Derivatives. Portland Press, London.
Fraser JR, Laurent TC, Laurent UB . Hyaluronan: its nature, distribution, functions and turnover. J Intern Med 1997; 242:27–33.
Teder P, Vandivier RW, Jiang D, et al. Resolution of lung inflammation by CD44. Science 2002; 296:155–158.
Heine H, Rietschel ET, Ulmer AJ . The biology of endotoxin. Mol Biotechnol 2001; 19:279–296.
West DC, Hampson IN, Arnold F, Kumar S . Angiogenesis induced by degradation products of hyaluronic acid. Science 1985; 228:1324–1326.
West DC, Kumar S . Hyaluronan and angiogenesis. Ciba Found Symp 1989; 143:187–201; discussion 201–187, 281–185.
Horton MR, Burdick MD, Strieter RM, et al. Regulation of hyaluronan-induced chemokine gene expression by IL-10 and IFN-gamma in mouse macrophages. J Immunol 1998; 160:3023–3030.
McKee CM, Penno MB, Cowman M, et al. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages. The role of HA size and CD44. J Clin Invest 1996; 98:2403–2413.
Noble PW, McKee CM, Cowman M, Shin HS . Hyaluronan fragments activate an NF-kappa B/I-kappa B alpha autoregulatory loop in murine macrophages. J Exp Med 1996; 183:2373–2378.
Ohkawara Y, Tamura G, Iwasaki T, et al. Activation and transforming growth factor-beta production in eosinophils by hyaluronan. Am J Respir Cell Mol Biol 2000; 23:444–451.
Fitzgerald KA, Bowie AG, Skeffington BS, O'Neill LA . Ras, protein kinase C zeta, and I kappa B kinases 1 and 2 are downstream effectors of CD44 during the activation of NF-kappa B by hyaluronic acid fragments in T-24 carcinoma cells. Journal of Immunology 2000; 164:2053–2063.
Oliferenko S, Kaverina I, Small JV, Huber LA . Hyaluronic acid (HA) binding to CD44 activates Rac1 and induces lamellipodia outgrowth [published erratum appears in J Cell Biol 2000 Apr 3;149(1):following 236]. Journal of Cell Biology 2000; 148:1159–1164.
Termeer CC, Hennies J, Voith U, et al. Oligosaccharides of hyaluronan are potent activators of dendritic cells. Journal of Immunology 2000; 165:1863–1870.
Aruffo A, Stamenkovic I, Melnick M, et al. CD44 is the principal cell surface receptor for hyaluronate. Cell 1990; 61:1303–1313.
Ponta H, Sherman L, Herrlich PA . CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol 2003; 4:33–45.
Cichy J, Pure E . The liberation of CD44. J Cell Biol 2003; 161:839–843.
Mummert ME, Mohamadzadeh M, Mummert DI, et al. Development of a peptide inhibitor of hyaluronan-mediated leukocyte trafficking. J Exp Med 2000; 192:769–779.
Jameson JM, Cauvi G, Sharp LL, et al. gammadelta T cell-induced hyaluronan production by epithelial cells regulates inflammation. J Exp Med 2005; 201:1269–1279.
Schmits R, Filmus J, Gerwin N, et al. CD44 regulates hematopoietic progenitor distribution, granuloma formation, and tumorigenicity. Blood 1997; 90:2217–2233.
Hodge-Dufour J, Noble PW, Horton MR, et al. Induction of IL-12 and chemokines by hyaluronan requires adhesion-dependent priming of resident but not elicited macrophages. Journal of Immunology 1997; 159:2492–2500.
Khaldoyanidi S, Moll J, Karakhanova S, et al. Hyaluronate-enhanced hematopoiesis: two different receptors trigger the release of interleukin-1beta and interleukin-6 from bone marrow macrophages. Blood 1999; 94:940–949.
Yang RB, Mark MR, Gurney AL, Godowski PJ . Signaling events induced by lipopolysaccharide-activated toll-like receptor 2. Journal of Immunology 1999; 163:639–643.
Mummert ME, Mummert D, Edelbaum D, et al. Synthesis and surface expression of hyaluronan by dendritic cells and its potential role in antigen presentation. J Immunol 2002; 169:4322–4331.
Beck-Schimmer B, Oertli B, Pasch T, Wuthrich RP . Hyaluronan induces monocyte chemoattractant protein-1 expression in renal tubular epithelial cells. J Am Soc Nephrol 1998; 9:2283–2290.
Oertli B, Beck-Schimmer B, Fan X, Wuthrich RP . Mechanisms of hyaluronan-induced up-regulation of ICAM-1 and VCAM-1 expression by murine kidney tubular epithelial cells: hyaluronan triggers cell adhesion molecule expression through a mechanism involving activation of nuclear factor-kappa B and activating protein-1. J Immunol 1998; 161:3431–3437.
Ito T, Williams JD, Al-Assaf S, et al. Hyaluronan and proximal tubular cell migration. Kidney Int 2004; 65:823–833.
Mascarenhas MM, Day RM, Ochoa CD, et al. Low molecular weight hyaluronan from stretched lung enhances interleukin-8 expression. Am J Respir Cell Mol Biol 2004; 30:51–60.
Lawrence T, Bebien M, Liu GY, et al. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature 2005; 434:1138–1143.
Chen LW, Egan L, Li ZW, et al. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med 2003; 9:575–581.
O'Neill LA . TLRs play good cop, bad cop in the lung. Nat Med 2005; 11:1161–1162.
Sha Q, Truong-Tran AQ, Plitt JR, et al. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol 2004; 31:358–364.
Acknowledgements
This work was supported by US National Institutes of Health Grants HL57486 and AI52487.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jiang, D., Liang, J., Li, Y. et al. The role of Toll-like receptors in non-infectious lung injury. Cell Res 16, 693–701 (2006). https://doi.org/10.1038/sj.cr.7310085
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7310085
Keywords
This article is cited by
-
Pulmonary toxicity assessment of polypropylene, polystyrene, and polyethylene microplastic fragments in mice
Toxicological Research (2024)
-
Immunoregulatory nanomedicine for respiratory infections
Nature Reviews Bioengineering (2023)
-
Ruscogenin attenuates particulate matter-induced acute lung injury in mice via protecting pulmonary endothelial barrier and inhibiting TLR4 signaling pathway
Acta Pharmacologica Sinica (2021)
-
Sterile inflammation in thoracic transplantation
Cellular and Molecular Life Sciences (2021)
-
Beneficial effect of low-level laser therapy in acute lung injury after i-I/R is dependent on the secretion of IL-10 and independent of the TLR/MyD88 signaling
Lasers in Medical Science (2017)
