
Abstract
PAX6, a paired box transcriptional factor, is considered as the master control gene for morphogenesis of the eye. Human PAX6 mutations have been associated with a range of eye abnormalities, including aniridia, various anterior segment defects and foveal hypoplasia. We carried out a mutational analysis of the PAX6 gene in 54 unrelated patients with aniridia or related syndromes. A deleterious variation was evidenced in 17 sporadic cases (50%) and in 13 (72%) familial cases. Twenty-four different mutations, 17 of which are novel, were found. The spectrum of PAX6 mutations was highly homogeneous: 23 mutations (96%) leading to premature stop codons (eight nonsense and four splice site mutations, 11 insertions and deletions) and only one (4%) missense mutation. Twenty-two mutations were associated with aniridia phenotypes whereas two were associated with atypical phenotypes. These latter encompassed a missense mutation (R19P) in an individual with a microphthalmia-sclerocornea and a splice site mutation (IVS4+5G>C) in a family presenting with a congenital nystagmus. Both represented the most probably hypomorphic alleles. Aniridia cases were associated with nonsense or frameshifting mutations. A careful examination of the phenotypes did not make it possible to recognise significant differences whenever the predicted protein was deprived of one or another of its functional domains. This strongly suggested that most of the truncating mutations generated null alleles by nonsense mediated mRNA decay. Our observations support the concept of dosage effects of the PAX6 mutations as well as presenting evidence for variable expressivity.
Similar content being viewed by others

Introduction
The PAX6 gene (paired box homeotic gene 6) has a key role as a master regulator in the development of the eye and central nervous tissues. It was identified as the candidate gene for aniridia (MIM 106210) which is a relatively rare panocular disease, characterised by complete or partial hypoplasia of the iris. Aniridia occurs in the general population at a frequency of around 1 in 60 000–100 000. The eye and visual defects associated with aniridia include some or all of the following: corneal opacification, cataract, glaucoma, lens dislocation, ciliary body hypoplasia, foveal hypoplasia, strabismus, and nystagmus.1,2,3
Two-thirds of aniridia cases are familial. The trait is autosomal dominant with a high degree of penetrance, but variable expressivity. In the remaining third no previous family history is found. A small number of the patients have WAGR syndrome (Wilms tumour, aniridia, genitourinary anomalies, mental retardation), a contiguous gene syndrome arising from deletion of chromosome 11p13, which encompasses both the PAX6 and WT1 genes.3,4
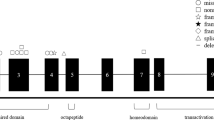
The aniridia gene (AN) has been localised on chromosome 11p135,6,7,8,9 and isolated by positional cloning.10 PAX6 gene spans 22 kilobases, is divided into 14 exons and encodes by alternative splicing two proteins of 422 and 436 amino acids.11 The predicted protein harbours functional domains: a 128 amino acid paired domain (PD) and a 61 amino acid homeodomain (HD) separated by a 78 amino acid linker segment (LNK) and followed by a 153 amino acid C terminal region rich in Proline, Serine, and Threonine (PST).10,11
Heterozygous PAX6 mutations are responsible for aniridia and have also been found casually in patients with Peters' anomaly, congenital cataracts, autosomal dominant keratitis, and isolated foveal hypoplasia.2,3,12,13,14 To date about 172 distinct mutations quite uniformly distributed across the gene have been described (for a review see Prosser 1998,3 the PAX6 mutation database: http://www.hgu.mrc.ac.uk/Softdata/PAX6). Nearly all reported mutations lead to premature protein termination and the paucity of missense mutations remains unclear. The molecular basis of these various ocular defects is loss of function of one allele, resulting in a 50% reduction in overall activity (haploinsufficiency) rather than the accumulation of dominant or dominant-negative forms of the protein.3 Compound heterozygosity is thought to be lethal15,16 as leading to severe malformations of the foetuses.
Here, we report a mutational analysis of PAX6 gene in 54 unrelated patients with aniridia or one of various closely related syndromes. Mutations were found in 30 of them and 17 of these mutations have not been previously described. A review of clinical features in our series, supported the concept of dosage effect of the PAX6 mutations as well as presenting evidence for variable expressivity and showed few if any phenotype/genotype correlation.
Patients, materials and methods
Patients
Fifty-four unrelated patients with aniridia or a closely related syndrome were screened for mutations of the PAX6 gene after their informed consent was obtained according to French law.
In aniridia patients, 16 cases were familial and 34 were sporadic. As for variant phenotypes, two sporadic cases of isolated foveal hypoplasia, one familial microphthalmia sclerocornea and one congenital nystagmus associated with iris and foveal hypoplasia were included. We also performed similar analyses in two familial and two sporadic cases of Peters' anomaly.
Mutation screening
Genomic DNA was prepared from venous blood according to standard protocols. The coding exons 4–13 of the PAX6 gene with intronic flanking sequences were amplified by PCR using previously published primers.11,17 All PCR products were submitted to single-strand conformation analysis (SSCA) to detect electrophoretic mobility variants in exons 4 to 13. In addition Denaturing Gradient Gel Electrophoresis (DGGE) was performed for exons 9 and 10.
For SSCA, denatured PCR products were separated on a non denaturant MDE gel (TEBU, FMC Bioproducts-Europe, Denmark), run at 4°C overnight at 3 W and 4–5 h at 12 W (constant power) and then visualised by silver staining.18 DGGE analysis of exons 9 and 10, used GC-rich specific primers (GC clamp incorporated at the 5′ end of direct primer of exon 9 and at the 3′ end of the reverse primer of exon 10). Each amplified DNA sample was subjected to electrophoresis at 160 V in a 6.5% polyacrylamide gel (37.5 : 1) containing a linear denaturant gradient (10 to 40% and 0 to 50% urea-formamide for exons 9 and 10 respectively). Following electrophoresis, the gels were stained with ethidium bromide and photographed under UV transillumination. DNA from amplified exons displaying variant migration patterns were sequenced. PCR fragments were purified with QIAquick Gel Extraction (QIAGEN SA, France) and sequenced using the Perkin-Elmer dye-terminator cycle sequencing kit (PE Biosystem, UK). Reactions were run in ABI373Strech or ABI310 sequencers (PE Biosystem, UK) and sequences analyses were carried out using Mac Vector program (version 6.5, Oxford Molecular Genetic, UK).
Whenever possible, some mutations were screened by restriction enzyme digestion of PCR-amplified exon DNAs, using specific enzymes as the R240X and the R317X mutations create or abolish respectively a DdeI and HaeIII restriction site.
Microsatellite genotyping
Polymorphic markers D11S995 (591A/B), D11S935 (AFM254zb9) and AFM205yg5 closely flanking the PAX6 gene, and two intragenic polymorphisms: a dinucleotide repeat in the promoter,19 (Genbank access #NT 009237) and IVS8CA/GT (Genome Data Base #215669) were used in order to screen deletions by familial segregation analysis.
RT-PCR
Total RNAs were extracted from frozen pellets of lymphoblastoid cells with RNeasy Midi kits (QIAGEN SA, France). RT-PCRs were performed using Omniscript RT kit according to the manufacturer's instructions (QIAGEN SA, France). The products of the RT were then amplified by PCR using previously published methods.20 As for the IVS4+5G>C, a semi-nested PCR used the C139 primer in exon 320 in order to by-pass a skipping of exon 4.
Electronic analysis of the splice site variants was conducted using the online human splice site prediction programs at http://www.fruitfly.org/seq_tools/splice.html and http://www.itba.mi.cnr.it/webgene.
Results
Mutational spectrum
Screening of the entire coding sequence of the PAX6 gene was performed in the 54 unrelated patients allowing identification of 30 heterozygous mutations, including 24 distinct variants of which 17 are novel (indicated in bold in Table 1).
The majority (23/34) of mutations was predicted to lead to an absent or truncated protein (11 insertions and deletions, including a complete deletion of the PAX6 gene, 8 nonsense mutations and four splice site mutations). Only one missense mutation (R19P) was detected. It affected an highly conserved residue in PAX6 paired-domains of all species so far analysed. The substitution of the arginine by a proline in codon 19 probably led to a spatial conformational change of the PAX6 protein.
We observed four point mutations in splice sites of which two are novel. One was located in the donor splice site of exon 4 (IVS4+5G>C) and the other in the acceptor splice site of exon 7 (IVS6−1G>C). The splice site scores were electronically predicted to be dramatically reduced suggesting the impairment of splicing. In both cases, the routine RT-PCR using published primers20 detected only normal mRNA isoforms. Concerning the IVS4+5G>C mutation effect, as the direct nested primer C127 (located in exon 4) lay within a possible skipped area, an extensive search of variant mRNA has been conducted using new sets of primers in nested PCR reactions. This made it possible to detect a mutant mRNA with a skipping of exon 4 (manuscript in preparation). In the case of the IVS6-1G>C, the sensitivity of RT–PCR was questioned. As four normal isoforms11,23,24,25 of the PAX6 mRNA have been systematically recovered we excluded a lack of detection and concluded that the mutant mRNA was unstable.
Only three of the eight nonsense mutations were novel: C40X, Q179X and Q301X. Seven mutations affected CpG sites. While R240X and R317X were found three times each in our population, mutations Q179X, R203X, R261X, W265X and Q301X were found once each in patients from distinct families. Five of these have been previously reported in the database and the most representative ones are R240X (17/212 cases), R317X (12/212 cases) and R203X (7/212 cases).
All deletions/insertions mutations identified in our study were novel. Notably an in frame 15 bp deletion, 427–441del, was found in exon 5. It deprived the predicted protein of the five (PDSTR) amino acids encoded by codons 22 to 26. This mutation arose in a highly conserved region among PDs corresponding to the amino terminus of the first α-helix and it is predicted to alter the ability of the PAX6 protein to bind its target DNA sequences.
In two families microsatellite segregation analyses detected large deletions in patients 98/216 and 99/423 (Table 1). FISH analysis confirmed that these deletions respected the WT1 gene (data not shown).
The mutations reported in our study were scattered all over the gene, including the PD (46%), LNK (9%), HD (27%) and PST region (18%). They, at least, deprived the protein of one or another of its functional domains.
Phenotype/genotype correlation
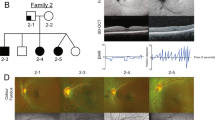
All but two mutations were associated with the aniridia phenotype. A missense mutation (R19P) occurred in a familial form of an unusual microphthalmia-sclerocornea phenotype and a splice site mutation (IVS4+5G>C) in a family presenting with congenital nystagmus, partial hypoplasia or atypical coloboma of iris and foveal hypoplasia.
Review of clinical features showed few if any phenotype/genotype correlation (Table 2). The nature of mutation did not appear to predict the severity of the disease. Notably, the complete deletion of the PAX6 gene encountered in patient 99/423 did not induce distinctive or more severe clinical manifestations than those associated with nonsense mutations, even the most distal ones (Q301X, patient 99/866; R317X, patient 00/523). Moreover examination of the phenotype did not make it possible to recognise significant differences whenever the protein was deprived of one or another of its functional domains. Although an inter- familial difference was found in expressivity in individuals bearing the same mutation (ie R240X, C40X), similar phenotypes were observed in distal (R317X patient 309) and in proximal (C40X patient 97/228) truncative mutations. However we noticed that the R261X (patient 97/083) and W265X (patient 167) mutations that deprived the protein of the PST domain only led in our patients to more severe ocular anomalies than those depriving the protein of both the HD and PST domains as the R203X (patient 98/218).
It is worthwhile to note that missense mutations appeared to be both rare events and associated with uncommon phenotypes.
No mutation was found in patients with Peters' anomaly.
Discussion
Mutation spectrum and diagnostic implications
We screened 54 unrelated families or sporadic cases, over all the coding sequence of the PAX6 gene and described 17 novel mutations.
Despite an association of several methods (direct analysis by SSCA, DGGE or indirect familial study with microsatellites) a deleterious variation was evidenced in only 50–72% of cases. This detection rate is lower than expected considering previous reports.25,26 We cannot exclude that it could be due to technical pitfalls as well as to the relatively high percentage of sporadic cases. Indeed our detection rate was 72% in familial cases corresponding to the detection rates previously published. In other respects, as we focused our study on protein coding regions, mutations might lie in regulatory regions of the PAX6 gene27 or correspond to gene rearrangements (inversions or duplications) that would have been missed by our approach. Alternatively, other gene(s) involved in eye development may be implicated.
It is difficult to comment meaningfully on the PAX6 mutation pattern as most categories are represented, from large scale rearrangements to single base substitutions and small insertions and deletions.3,4 Not surprisingly hot spots of mutations are constituted by CpG sites. The Arginine of codon 240 and 317 are to date the most recurrent point mutations found in the PAX6 gene according to the classical deamination of methylated Cytosine mechanism, as 29 families carry one of these two nonsense mutations28 (PAX6 mutations database).
The PD and HD contain a disproportionate number of mutations from that expected on the basis of their size alone. In the case of the HD, the hypermutable nucleotide 1080 is responsible for the increase, but in the PD the number of different mutations is much higher than expected and it is difficult to find a sequence-based explanation for this increase.3
Phenotype–genotype correlations
Mutation detection has already proven to be useful for genetic counselling and prenatal diagnosis in aniridia families. Because sporadic congenital aniridia or anterior segment dysgenesis may be associated with a deletion of both PAX6 and WT1 and may predispose to Wilms tumour (WAGR syndrome), the detection of an intragenic PAX6 mutation prevents the need for routine kidney ultrasound screening. In all cases, detection of a PAX6 mutation facilitates full genetic counselling.3,29,30 Regardless WAGR syndromes, a mild mental retardation has been reported in a few cases of aniridia patients bearing PAX6 point mutations (namely R44X, I87R and Q135X). The frameshifting de novo insertion of 31 nucleotides in position 473 of the cDNA, leading to a premature STOP, 28 codons downstream was found in a young boy with intellectual impairment. Owing to the paucity of mental retardation in patients bearing similar PAX6 mutations, such an association might be fortuitous.
Nearly all pathogenic alterations so far reported appear to be loss-of-function mutations that predispose to aniridia. Most of the nonsense mutations are assumed to mediate mutant mRNA which are underexpressed.26 A nonsense-mediated mRNA decay is currently observed as a major control mechanism of the mRNA quality in cells (for a review see31,32). This would explain that premature stop codons bearing mRNA could have a similar effect to a deleted allele. In that way, alleles that were presumed to encode truncated proteins most probably do not lead to protein synthesis. So it enlightens the fact that all attempts made to correlate the phenotype according to the site of the nonsense mutations were unsuccessful. Nevertheless, some hypomorphic alleles may arise from reduced, rather than eradicated activity and result in less severe or phenotypically variable aniridia, or related phenotypes.3 On the other hand, the deprivation of the transactivation domain induced by R261X and W265X while DNA binding through PD and HD remains functional, was found to lead to particularly severe phenotypes in our patients. They raise the hypothesis of a dominant negative effect rather than haploinsufficiency and several functional data questioned that mechanism.33,34 This suggests that some mRNA harbouring premature stop codons escape the degradation pathway and make truncated proteins synthesised.35
The rare missense mutations of the PAX6 gene tend to provoke atypical aniridia or milder phenotypes. It is possible that they provide a variety of functional errors of the PAX6 protein, and hence result in several variants of aniridia.16,25,36,37 According to these observations, the R19P mutation (patient 98/217) is associated with an uncommon familial phenotype. By analogy, the non aniridia phenotype encountered in the patient bearing the IVS4+5G>C mutation is not in favour of haploinsufficiency. This mutation mimics the previously described IVS4+3AA>GC mutation which leads to exon 4 skipping. As this exon contains the translation start site, the absence of protein is expected. Nevertheless, the mutant mRNA was recovered in cultured cells. So an in frame putative alternative ATG in exon 3 could direct the synthesis of a slightly modified protein.
Taken together all these data reinforce the concept of dosage effects16 of the majority of ‘truncating’ PAX6 mutations as they result in null alleles as well as presenting evidence for an inter- and intra-familial variability of clinical expression.38
References
Nelson, LB, Spaeth, GL, Nowinski, TS, Margo, CE & Jackson, LA : A review. Surv Ophthalmol, (1984). 28, 621–642.
Hanson, IM, Fletcher, JM & Jordan, T et al : Mutations at the Pax6 locus are found in heterogeneous anterior segment malformations including Peters' anomaly. Nature Genet, (1994). 6, 168–173.
Prosser, J & van Heyningen, V : Pax6 mutations reviewed. Hum Mutation, (1998). 11, 93–108.
Chao, LY, Huff, V, Strong, LC & Saunders, GF : Mutation in the PAX6 gene in twenty patients with aniridia. Hum Mutation, (2000). 15, 332–339.
Gessler, M, Simola, KO & Bruns, GA : Cloning of breakpoints of a chromosome translocation identifies the AN2 locus. Science, (1989). 244, 1575–1578.
Davis, LM, Everest, AM, Simola, KO & Shows, TB : Long-range restriction map around 11p13 aniridia locus. Somat Cell Mol Genet, (1989). 15, 605–615.
Rose, EA, Glaser, T & Jones, C et al : Complete physical map of the WAGR region of 11p13 localizes a candidate Wilms' tumor gene. Cell, (1990). 60, 495–508.
Lyons, LA, Martha, A, Mintz-Hittner, HA, Saunders, GF & Ferrell, RE : Resolution of the two loci for autosomal dominant aniridia, AN1 and AN2, to a single locus on chromosome 11p13. Genomics, (1992). 13, 925–930.
Compton, DA, Weil, MM, Jones, C, Riccardi, VM, Strong, LC & Saunders, GF : Long range physical map of the Wilms' tumor-aniridia region on human chromosome 11. Cell, (1988). 55, 827–836.
Ton, CCT, Hirvonen, H & Miwa, H et al : Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell, (1991). 67, 1059–1074.
Glaser, T, Walton, DS & Maas, RL : Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nature Genet, (1992). 2, 232–239.
Jordan, T, Hanson, I & Zaletayev, D et al : The human PAX6 gene is mutated in two patients with aniridia. Nature Genet, (1992). 1, 328–332.
Azuma, N, Nishina, S, Yanagisawa, H, Okuyama, T & Yamada, M : PAX6 missenses mutation in isolated foveal hypoplasia. Nature Genet, (1996). 13, 141–142.
Mirzayans, F, Pearce, WG, MacDonald, IM & Walter, MA : Mutations of the Pax6 gene in patients with autosomal dominant keratitis. Am J Hum Genet, (1995). 57, 539–548.
Hodgson, SV & Saunders, KE : A probable case of the homozygous condition of the aniridia gene. J Med Genet, (1980). 6, 478–480.
Glaser, T, Jepeal, L, Edwards, JG, Young, SR, Favor, J & Maas, RL : Pax6 gene dosage effect in a family with congenital cataracts, aniridia, anophtalmia and central nervous system defects. Nature Genet, (1994). 7, 463–471.
Love, J, Axton, R, Churchill, A, van Heyningen, V & Hanson, I : A new set of primers for mutation analysis of the human Pax6 gene. Hum Mutation, (1998). 12, 128–134.
Budowle, B, Chakraborty, R, Guisti, A, Eisenberg, A & Allen, C : Analysis of the VNTR locus D1S80 by the PCR followed by high-resolution PAGE. Am J Hum Genet, (1991). 48, 137–144.
Sander, T, Syagailo, Y, Samochowiec, J, Okladnova, O, Lesh, KP & Janz, D : Association analysis of a regulatory promoter polymorphism of the PAX6 gene with idiopathic generalized epilepsy. Epilepsy Res, (1999). 36, 61–67.
Hanson, IM, Seawright, A & Hardman, K : Pax6 mutations in aniridia. Hum Mol Genet, (1993). 7, 915–920.
Crolla, J, Cross, I, Atkey, N, Wright, M & Oley, C : FISH studies in a patients with sporadic aniridia and t(7;11)(q31.2;p13). J Med Genet, (1996). 33, 66–68.
Fantes, JA, Bickmore, WA, Fletcher, JM, Ballesta, F, Hanson, IM & van Heyningen, V : Submicroscopic deletions at the WAGR locus, revealed by nonradioactive in situ hybridization. Am J Hum Genet, (1992). 51, 1286–1294.
Calvas, P, Rozet, JM, Gerber, S, Munnich, A & Kaplan, J : Novel PAX6 paired-domain mutations in congenital aniridia and identification of a new alternative splicing of PAX6 mRNA. Am J Hum Genet, (1996). Suppl.59 A394, 2297
Jaworski, C, Sperbeck, S, Graham, C & Wistow, G : Alternative splicing of Pax6 in bovine eye and evolutionary conservation of intron sequences. BBR communications, (1997). 240, 196–202.
Grønskov, K, Rosenberg, T, Sand, A & Brondum Nielsen, K : Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype. Eur J Hum Genet, (1999). 7, 274–286.
Axton, R, Hanson, I, Danes, S, Sellar, G, van Heyningen, V & Prosser, J : The incidence of PAX6 mutation in patients with simple aniridia: an evaluation of mutation detection in 12 cases. J Med Genet, (1997). 34, 279–286.
Lauderdale, J, Wilensky, J, Oliver, E, Walton, E, Walton, D & Glaser, T : 3′deletions cause aniridia by preventing PAX6 gene expression. PNAS, (2000). 97, 13755–13759.
Kondo-Saitoh, A, Matsumoto, N & Sasaki, T et al : Two nonsense mutations of Pax6 in two japanese aniridia families: cases report and review of the literature. Eur J Ophthalmol, (2000). 10, 167–172.
Gupta, S, De Becker, I, Guernsey, D & Neuman, P : Polymerase chain reaction-based risk assessment for Wilms tumor in sporadic aniridia. Am J Ophthalmol, (1998). 125, 687–692.
Churchill, A, Hanson, I & Markham, A : Prenatal diagnosis of aniridia. Ophthalmology, (2000). 107, 1153–1156.
Marquat, LE & Carmichael, GG : Quality control of mRNA function. Cell, (2001). 104, 173–176.
Byers, PH : Killing the messengers: new insights into nonsense-mediated mRNA decay. J Clin Invest, (2002). 109, 3–6.
Singh, S, Tang, HK, Lee, JY & Saunders, G : Truncation mutations in the transactivation region of the PAX6 result in dominant negative mutants. J Biol Chem, (1998). 273, 21531–21541.
Singh, S, Chao, L, Mishra, R, Davies, J & Saunders, G : Missense mutation at the C-terminus of PAX6 negatively modulates homeodomain function. Hum Mol Genet, (2001). 10, 911–918.
Deckwardt, S, Neu-Yilik, G, Thermann, R, Frede, U, Hentze, MW & Kulozik, AE : Abnormally spliced β-globin mRNAs: a single point mutation generates transcripts sensitive and insensitive to nonsense-mediated mRNA decay. Blood, (2002). 99, 1811–1816.
Hanson, IM, Churchill, A & Love, J et al : Missense mutations in the most ancient residues of the Pax6 paired domain underlie a spectrum of human congenital eye malformation. Hum Mol Genet, (1999). 8, 165–172.
Sonoda, S, Isashiki, Y, Tabata, Y, Kimura, K, Kakiuchi, T & Ohba, N : A novel Pax6 gene mutation (P118R) in a family with congenital nystagmus associated with a variant form of aniridia. Graefe's Arch Clin Exp Ophthalmol, (2000). 238, 552–558.
Syagailo, Y, Wilke, K & Okladnova, O et al : Different ocular abnormalities in individuals of a three generation family caused by a new nonsense mutation in the PST domain of the Pax6 gene. Hum. Mutation, (1998). on line mutation in brief 189
Acknowledgements
This work was supported by grants of the DRRC, from the CHU of Toulouse. We wish to thank all clinicians who sent us careful clinical examination and DNA samples especially Dr J Kaplan, Pr N Philipp, Dr P Malzac and Dr H Dollfus. We are grateful to Dr V van Heyningen of the MRC Human Genetics Unit, Edinburgh for having kindly provided the FAT5 and B2.1 cosmids. We would also like to thank Nicole Janisse, Anne-Marie Mazarguil and Sandra Ibos for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vincent, MC., Pujo, AL., Olivier, D. et al. Screening for PAX6 gene mutations is consistent with haploinsufficiency as the main mechanism leading to various ocular defects. Eur J Hum Genet 11, 163–169 (2003). https://doi.org/10.1038/sj.ejhg.5200940
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200940
Keywords
This article is cited by
-
Detection of a novel PAX6 variant in a Chinese family with multiple ocular abnormalities
BMC Ophthalmology (2022)
-
A novel variant in PAX6 as the cause of aniridia in a Chinese family
BMC Ophthalmology (2021)
-
Phenotype–genotype correlations and emerging pathways in ocular anterior segment dysgenesis
Human Genetics (2019)
-
Genetics of anophthalmia and microphthalmia. Part 1: Non-syndromic anophthalmia/microphthalmia
Human Genetics (2019)
-
Epistasis between Pax6Sey and genetic background reinforces the value of defined hybrid mouse models for therapeutic trials
Gene Therapy (2018)
