
Abstract
Familial adenomatous polyposis (FAP), an autosomal dominantly inherited colorectal cancer predisposition syndrome, displays considerable inter- and intrafamilial phenotypic heterogeneity, which represents a major problem in genetic counselling of APC mutation carriers. The Min mouse model indicated a putative disease modifier locus on chromosome 4, which is syntenic to human chromosome 1p35–36. This finding was subsequently supported by parametric and nonparametric linkage analyses in FAP families, however, without identifying functional variants in candidate genes. Recently, germline mutations in the base-excision repair gene MYH (1p33–34) have been described in patients with multiple adenomas, pointing to a possible role as disease modifier in FAP. Here, we present critical reassessment of one of the largest FAP kindreds published, which was previously used in linkage mapping of 1p35–36. In this family, all affected members harbour the same APC germline mutation (5945delA), but display marked phenotypic variability, in particular regarding the occurrence of extracolonic disease that segregates in several branches of the family tree. Using updated clinical information, additional mutation carriers and polymorphic markers, fine mapping of the critical region as well as mutation analysis of the MYH gene were performed. These investigations allowed us to significantly exclude (i) the 1p33–36 region as a modifier locus and (ii) MYH as a modifier gene for extracolonic disease in this FAP kindred. Our results do not eliminate 1p33–36 from suspicion in other families, but clearly indicate that in our family linkage analysis of further putative candidate regions is necessary to identify a disease modifier locus in FAP.
Similar content being viewed by others
Introduction
Familial adenomatous polyposis (FAP) is an autosomal dominantly inherited predisposition to colorectal cancer caused by germline mutations in the adenomatous polyposis coli (APC) gene.1 It is a clinically heterogenous disease in which patients develop hundreds to thousands of adenomas throughout the large intestine. Extracolonic disease may also occur, for example upper gastrointestinal (GI) polyposis, soft tissue tumours (in particular desmoid tumours), osteomas and congenital hypertrophy of the retinal pigment epithelium (CHRPE).2,3,4 Despite established genotype–phenotype correlations, considerable phenotypic variation with respect to colonic and extracolonic disease has been reported in patients harbouring identical APC mutations,5,6,7,8 indicating that other genetic factors (modifier genes) are likely to play important roles in disease development.3,5,9,10
In support of this, loci that modify polyp multiplicity have been identified in different mouse models of FAP (Min and Apc1638N).11,12,13 The Mom1 and Mom2 loci were found to affect tumour number in Min mice, and their synteny regions on the human chromosomes 1p35–36,11 18q21 and 18q2314,15 are known to be frequently deleted/lost in a variety of human cancers, including colon tumours.16,17,18,19 In Apc1638N mice, additional (X-ray responsive) modifier loci have been suggested.20,21
In view of the significant differences between the mouse and human FAP phenotype, the observations in the mouse do not necessarily apply to the human situation and mouse candidate modifiers should be evaluated with respect to the occurrence of both colonic and extracolonic disease manifestations.11,12,22,23 For the Mom1 locus, the secretory phospholipase A2 (Pla2g2a) gene has been suggested to be a strong candidate for suppression of the Min phenotype.24,25 So far, no functional variants of Pla2G2A have been detected in humans,26,27,28 and the possibility that other genes in the region can function as modifiers lead our group and others to further investigations of 1p35–36. However, these studies neither significantly excluded nor confirmed a human FAP modifier locus for extracolonic disease at 1p35–36.22,29
Recently, germline mutations in the base-excision repair gene MYH (region 1p33–34) have been described in patients with multiple adenomas, some of which had extracolonic disease (duodenal polyps).30,31 This may implicate MYH as a possible FAP modifier, as MYH mutations/variants in combination with germline APC mutations could be expected to enhance the FAP disease phenotype.
To assess the role of the 1p32–36 region as a candidate modifier locus for extracolonic disease, we reinvestigated a large Swiss FAP kindred (No. 1460), part of which was previously used in linkage analysis, and where a lod score of 2.08 was obtained for an autosomal recessive model.29 In the 7 years since this analysis has been performed, 13 additional members were identified and updated clinical information on the known mutation carriers was gathered, which enabled us to perform an extended linkage analysis of the 1p32–36 region as well as mutation analysis of the new candidate modifier gene MYH.
Methods
Patient data
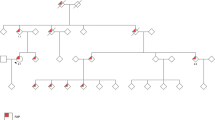
The study was performed on a large, genetically isolated Swiss FAP kindred (n>200), whereof all affected members (n=63) share the same APC germline mutation, at codon 1982 in exon 15n, 5945delA (confirmed by sequencing and protein truncation test).29,32 In 50 members (Figure 1; Table 1) belonging to the pedigree branches with extracolonic manifestations, histopathological data and reports from colonoscopies, gastro-duodenal endoscopies, computer tomographies, surgery, autopsies, as well as information from regular dental examinations, were collected and re-evaluated for the present study. Only patients with verified data from clinical and histopathological reports were used for linkage analysis. Written informed consent was obtained from all individuals.

Extract from FAP kindred no. 1460 displaying branches with extracolonic disease manifestations. Only APC mutation carriers and their married-in members are included. Symbol description: (a) upper right quadrant, presence of desmoids and fibromas; (b) lower right quadrant, upper gastrointestinal polyps; (c) lower left quadrant, osteomas; (d) upper left quadrant, other extracolonic manifestations. Patients included in MYH gene mutation analysis are marked by asterisk.
Genotyping of polymorphic markers
Genotyping was performed using fluorescently labelled primers from the ABI Prism Linkage mapping Set-MD10 (PE Applied Biosystems;33,34,35) and primers of additional microsatellite markers from the 1p32–36, selected according to their map location and their heterozygosity status (http://www.ucsc.genome.org,36 ftp://bioinformatics.weizmann.ac.il/pub/databases/genethon/Gmap/Nature-1995/data/35). PCR reactions were performed according to the manufacture's protocol and analysed on an ABI PRISM® 3700 DNA Analyzer (Applied Biosystems). Genotyping was exclusively performed in APC mutation carriers and their married-in members.
Linkage analysis
Microsatellite data was checked for genotyping errors using the PEDCHECK program,37 two-point parametric lod score linkage analysis was performed using the MLINK program from the LINKAGE package38 for both autosomal dominant (disease allele frequency 0.0781) and autosomal recessive (disease allele frequency 0.1000) models. Marker allele frequencies were set to be equal. Penetrance of 1.00 and 0.95 was used for the recessive model, and of 0.90 and 0.85, and age-dependent penetrance for the dominant model. Liability classes for age-dependent penetrance were estimated from our pedigree data and defined as a function of age at diagnosis.39 Following liability classes were used: 0.157 (⩽20 years), 0.368 (⩽30 years), 0.684 (⩽40 years), 0.895 (⩽50 years), 0.947 (⩽60 years) and, 0.999 (⩽70 years). Penetrance of phenocopies was set to be 0.001. Only verified APC mutation carriers (and their married-in members) were included in the analysis, as only in these members extracolonic manifestations are expected to result from both APC and modifier gene mutations. Polyposis patients with colonic disease only were classified as having an ‘unaffected’ affection status, married-in members as having an ‘unknown’ phenotype. Patients presenting with extracolonic disease manifestation(s) were evaluated applying two different sets of criteria: (a) stringent criteria: only patients displaying at least adenomatous polyps in the upper GI tract and/or desmoids were classified as ‘affected’ with the others being classified as ‘unknown’; (b) loose criteria: all patients with confirmed extracolonic manifestation(s) were scored as ‘affected’ (Table 1). One patient with bronchial carcinoma was classified as ‘unknown’ in all analyses.
MYH mutational analysis
Coding regions and exon–intron boundaries of MYH (GenBank accession number NM_012222) were screened by dHPLC using the 3500HT WAVE nucleic acid fragment analysis system (Transgenomic). Samples with different elution profiles, in comparison to control samples run in parallel, were then directly sequenced in forward and reverse orientations. The sequencing reactions were perfomed using the Big Dye Teminator Cycle Sequencing kit (Applied Biosystems) and analysed on an ABI PRISM 310 Genetic Analyser (Applied Biosystems).
Results
Clinical data
In previous studies,29,32 we reported a large FAP kindred (no. 1460) whose affected members present with a highly variable phenotype, on the level of both colonic as well as extracolonic disease manifestations. A total of 50 family members of this kindred, belonging to subbranches displaying extracolonic disease were clinically re-evaluated for this study (Table 1). In general, the polyposis phenotype among APC mutation carriers was found to be relatively mild, as could be expected by the site of the germline mutation, with 26 (65%) patients displaying attenuated polyposis (less than 100 polyps). The polyposis phenotype was very variable, ranging from severe forms with more than 1000 polyps (two patients) to the very mild form, where no polyps (three patients at age 22, 29, and 47) or less then 10 polyps (three patients at age 32, 33, and 47) were present.
In 26 patients, extracolonic tumours developed, the majority of these being desmoids (16/26; 61.5%) and upper gastrointestinal polyps (16/26; 61.5%). Adenomatous origin of the polyps was confirmed in nine patients, other were fundus gland polyps, which developed to a great extent in three patients.
Linkage analysis
Simulation linkage analysis, previously performed in family no. 1460, using the same diagnostic criteria and parameters as employed for linkage analysis (see Methods) revealed a maximum expected lod score of 3.8–5.3 for autosomal dominant models, and 1.9–2.7 for autosomal recessive models. In all, 28 polymorphic markers spanning 58.7 cM (50.2 Mb) 36 of the 1p32–36 region (Figure 2) were used for two-point linkage analysis under an autosomal dominant model with age-dependent penetrance. No evidence for the existence of a dominant modifier locus for extracolonic FAP disease was found. The lod scores throughout the region 1p33–36 were below −2 (except for three markers, D1S3669, D1S255 and D1S2733, with lod scores of −1.6, −1.9 and −0.6, respectively), thus excluding this region that includes both Pla2G2A and MYH, as a possible modifier locus (Table 2). To exclude the possibility that our negative results were due to choosing the wrong mode of inheritance, the analysis was also performed using an autosomal recessive model at penetrance 0.95 and 1.00, as well as an autosomal dominant model at reduced penetrance of 0.90 and 0.85, respectively (data not shown). All analyses, under both stringent and loose diagnostic criteria, resulted in negative lod scores below −2, except for the markers D1S3669, D1S417 and D1S231 (lod scores below −1.5, 0.1, and 0.8, respectively), hence excluding the 1p32–36 region as a modifier locus of extracolonic disease in our FAP kindred.

Physical map of the 1p32–36 region. Marker order, physical distance (Mb) and position of the secretory phospholipase A2 gene (Pla2g2a) and base-excision repair gene (MYH) were determined according to the UCSC genome bioinformatics site.36
MYH mutation analysis
For MYH gene mutation analysis, six patients (i) coming from different pedigree branches and (ii) displaying different extracolonic disease manifestations were selected (Figure 1; Table 1). No DNA variants could be detected in all but one of them. Patient 1501-1 with multiple desmoids, osteomas, stomach and duodenal adenomas, but only one colonic polyp was found to harbour a heterozygous V22M variant of the MYH gene (exon 2). Subsequent mutation analysis on all family members identified only one more patient (1460-16) and his nonaffected father as V22M carriers. The variant represents an already described polymorphism, which was previously reported at a population frequency of 9–10%.30,31
Discussion
The present study excludes the 1p33–36 region as a modifier gene locus for extracolonic disease in our large Swiss FAP kindred no. 1460. The analysis was performed on updated family information, and used both more affected family members and more microsatellite markers. Since our initial investigation, restricted to the 1p35–36 region in 1996, 13 additional patients either developed extracolonic tumours or were newly referred to our department. Out of these, four were classified as having an ‘affected’ or ‘unknown’ diagnosis, depending on the stringency of the affection criteria used. Four patients previously classified as ‘affected’ were for the present analysis scored as ‘unknown’, because original data provided by the patient's record could not be confirmed from histopathological records. Furthermore, unlike the previous analysis, only APC mutation carriers and their spouses were used for linkage analysis. These differences may explain why the lod score for the autosomal recessive model dropped from a previously observed 2.08 (D1S211) to below −2 (instead of marker D1S211, markers D1S2645 and D1S2733 were used), and for the autosomal dominant model from 1.77 (D1S197) to below −0.7 (Table 2).
The rationale for considering upper GI polyps and desmoids together in linkage analysis was three-fold: (i) in all parent–child pairs, where both presented with extracolonic disease, upper GI polyps and desmoids were observed (Figure 1); (ii) overall, 50% of patients with desmoid disease also displayed upper GI polyps (and vice versa); (iii) the occurrence of upper GI polyps together with desmoids in FAP has been well documented in the literature.40,41,42,43
Although our linkage results for an autosomal recessive mode of inheritance resulted in significant exclusion of the 1p32–36 region, we put our emphasis on autosomal dominant models which seem to be more appropriate in our FAP kindred for several reasons. Firstly, the ratio of APC mutation carriers with, compared to those without, extracolonic disease varied between 0.42 and 0.52, depending on the affection criteria applied. Secondly, in some of the subbranches of family no. 1460, extracolonic manifestations are clearly transmitted through generations (Figure 1). When comparing 12 parent–child pairs with extracolonic disease present, transmission of extracolonic disease through the generations could be observed in 100% of informative pairs, suggesting an autosomal dominant mode of inheritance. Using the stringent phenotype criteria, that is, only including patients with at least upper GI adenomatous polyps or desmoids, transmission was seen in eight out of nine pairs (88%).
Phenotype analysis revealed the same clinical heterogeneity as previously reported.32 Furthermore, when comparing the group of patients with <100 and those with >100 colorectal polyps, no statistically significant relationship could be found between polyp number and the occurrence of extracolonic disease in general (χ2=0.44, P=0.50). The same was true if only desmoids (χ2=1.20, P=0.27) or only upper GI polyps were taken into account (χ2=0.10, P=0.75). This indicates that the severity of colonic polyposis does not correlate with the presence of extracolonic disease manifestations; hence, polyp number and extracolonic disease may represent two genetically related but distinct entities. We cannot, however, exclude an underestimation of the actual frequency of extracolonic disease which, in particular, may be true for patients screened before the event of cross-sectional imaging (for the detection of desmoid tumours) and side-viewing endoscopy (duodenal polyps).
Our linkage analysis data are in agreement with the results from the mutation screening in PLA2G2A27 and MYH, where, except for the heterozygous V22M variant present in two patients belonging to different branches of the family tree (and observed in 2% of Swiss control population, unpublished results), no other DNA alterations could be identified in the coding region of the MYH gene.
In conclusion, our data on this large Swiss FAP kindred significantly exclude the 1p33–36 region as a modifier locus and MYH as a modifier gene for extracolonic disease. Since simulation linkage analysis revealed a maximum expected lod score of 3.8–5.3 for autosomal dominant and 1.9–2.7 for autosomal recessive models, future work will concentrate on performing a genome-wide linkage analysis in this FAP kindred, which should help in the identification of a modifier locus in FAP.
References
Lynch HT, de la Chapelle A : Hereditary colorectal cancer. N Engl J Med 2003; 348: 919–932.
Gardner EJ, Richards RC : Multiple cutaneous and sub-cutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet 1953; 5: 139–147.
Houlston R, Crabtree M, Phillips R, Tomlinson I : Explaining differences in the severity of familial adenomatous polyposis and the search for modifier genes. Gut 2001; 48: 1–5.
Griffioen G, Bus PJ, Vasen HF, Verspaget HW, Lamers CB : Extracolonic manifestations of familial adenomatous polyposis: desmoid tumours, and upper gastrointestinal adenomas and carcinomas. Scand J Gastroenterol Suppl 1998; 225: 85–91.
Friedl W, Caspari R, Sengteller M et al: Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut 2001; 48: 515–521.
Giardiello FM, Krush AJ, Petersen GM et al: Phenotypic variability of familial adenomatous polyposis in 11 unrelated families with identical APC gene mutation. Gastroenterology 1994; 106: 1542–1547.
Brensinger JD, Laken SJ, Luce MC et al: Variable phenotype of familial adenomatous polyposis in pedigrees with 3′ mutation in the APC gene. Gut 1998; 43: 548–552.
Rozen P, Samuel Z, Shomrat R, Legum C : Notable intrafamilial phenotypic variability in a kindred with familial adenomatous polyposis and an APC mutation in exon 9. Gut 1999; 45: 829–833.
Crabtree MD, Tomlinson IPM, Talbot IC, Phillips RK : Variability in the severity of colonic disease in familial adenomatous polyposis results from differences in tumour initiation rather than progression and depends relatively little on patient age. Gut 2001; 49: 540–543.
Crabtree MD, Tomlinson IP, Hodgson SV, Neale K, Phillips RK, Houlston RS : Explaining variation in familial adenomatous polyposis: relationship between genotype and phenotype and evidence for modifier genes. Gut 2002; 51: 420–423.
Dietrich WF, Lander ES, Smith JS et al: Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell 1993; 75: 631–639.
Smits R, van der Houven van Oordt W, Luz A et al: Apc1638N: a mouse model for familial adenomatous polyposis-associated desmoid tumors and cutaneous cysts. Gastroenterology 1998; 114: 275–283.
Fodde R, Smits R : Disease model: familial adenomatous polyposis. Trends Mol Med 2001; 7: 369–373.
Silverman KA, Koratkar R, Siracusa LD, Buchberg AM : Identification of the modifier of Min 2 (Mom2) locus, a new mutation that influences Apc-induced intestinal neoplasia. Genome Res 2002; 12: 88–97.
Silverman KA, Koratkar RA, Siracusa LD, Buchberg AM : Exclusion of Madh2, Madh4, and Madh7 as candidates for the modifier of Min 2 (Mom2) locus. Mamm Genome 2003; 14: 119–129.
Schwab M, Praml C, Amler LC : Genomic instability in 1p and human malignancies. Genes Chromosomes Cancer 1996; 16: 211–229.
Thiagalingam S, Lengauer C, Leach FS et al: Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet 1996; 13: 343–346.
Shih IM, Zhou W, Goodman SN, Lengauer C, Kinzler KW, Vogelstein B : Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res 2001; 61: 818–822.
Candusso ME, Luinetti O, Villani L et al: Loss of heterozygosity at 18q21 region in gastric cancer involves a number of cancer-related genes and correlates with stage and histology, but lacks independent prognostic value. J Pathol 2002; 197: 44–50.
van der Houven van Oordt CW, Smits R, Williamson SL et al: Intestinal and extra-intestinal tumor multiplicities in the Apc1638N mouse model after exposure to X-rays. Carcinogenesis 1997; 18: 2197–2203.
van der Houven van Oordt CW, Smits R, Schouten TG et al: The genetic background modifies the spontaneous and X-ray-induced tumor spectrum in the Apc1638N mouse model. Genes Chromosomes Cancer 1999; 24: 191–198.
Tomlinson IP, Neale K, Talbot IC et al: A modifying locus for familial adenomatous polyposis may be present on chromosome 1p35–p36. J Med Genet 1996; 33: 268–273.
Fodde R, Smits R, Hofland N, Kielman M, Meera Khan P : Mechanisms of APC-driven tumorigenesis: lessons from mouse models. Cytogenet Cell Genet 1999; 86: 105–111.
MacPhee M, Chepenik KP, Liddell RA, Nelson KK, Siracusa LD, Buchberg AM : The secretory phospholipase A2 gene is a candidate for the Mom1 locus, a major modifier of ApcMin-induced intestinal neoplasia. Cell 1995; 81: 957–966.
Cormier RT, Hong KH, Halberg RB et al: Secretory phospholipase Pla2g2a confers resistance to intestinal tumorigenesis. Nat Genet 1997; 17: 88–91.
Riggins GJ, Markowitz S, Wilson JK, Vogelstein B, Kinzler KW : Absence of secretory phospholipase A2 gene alterations in human colorectal cancer. Cancer Res 1995; 55: 5184–5186.
Dobbie Z, Muller H, Scott RJ : Secretory phospholipase A2 does not appear to be associated with phenotypic variation in familial adenomatous polyposis. Hum Genet 1996; 98: 386–390.
Tomlinson IP, Beck NE, Neale K, Bodmer WF : Variants at the secretory phospholipase A2 (PLA2G2A) locus: analysis of associations with familial adenomatous polyposis and sporadic colorectal tumours. Ann Hum Genet 1996; 60: 369–376.
Dobbie Z, Heinimann K, Bishop DT, Muller H, Scott RJ : Identification of a modifier gene locus on chromosome 1p35–36 in familial adenomatous polyposis. Hum Genet 1997; 99: 653–657.
Al-Tassan N, Chmiel NH, Maynard J et al: Inherited variants of MYH associated with somatic G:C–>T:A mutations in colorectal tumors. Nat Genet 2002; 30: 227–232.
Sieber OM, Lipton L, Crabtree M et al: Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med 2003; 348: 791–799.
Scott RJ, van der Luijt R, Spycher M et al: Novel germline APC gene mutation in a large familial adenomatous polyposis kindred displaying variable phenotypes. Gut 1995; 36: 731–736.
Weissenbach J, Gyapay G, Dib C et al: A second-generation linkage map of the human genome. Nature 1992; 359: 794–801.
Gyapay G, Morissette J, Vignal A et al: The 1993-94 Genethon human genetic linkage map. Nat Genet 1994; 7: 246–339.
Dib C, Faure S, Fizames C et al: A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 1996; 380: 152–154.
Kent WJ, Sugnet CW, Furey TS et al: The human genome browser at UCSC. Genome Res 2002; 12: 996–1006.
O'Connell JR, Weeks DE : PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 1998; 63: 259–266.
Lathrop GM, Lalouel JM, Julier C, Ott J : Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 1984; 81: 3443–3446.
Terwilliger JD, Ott J : Handbook of human genetic linkage. Baltimore and London: The Hohns Hopkins University Press; 1994.
Kashiwagi H, Konishi F, Kanazawa K, Miyaki M : Sisters with familial adenomatous polyposis affected with thyroid carcinoma, desmoid tumour and duodenal polyposis. Br J Surg 1996; 83: 228.
Enomoto M, Konishi M, Iwama T, Utsunomiya J, Sugihara KI, Miyaki M : The relationship between frequencies of extracolonic manifestations and the position of APC germline mutation in patients with familial adenomatous polyposis. Jpn J Clin Oncol 2000; 30: 82–88.
Bertario L, Russo A, Sala P et al: Multiple approach to the exploration of genotype–phenotype correlations in familial adenomatous polyposis. J Clin Oncol 2003; 21: 1698–1707.
Truta B, Allen BA, Conrad PG et al: Genotype and phenotype of patients with both familial adenomatous polyposis and thyroid carcinoma. Fam Cancer 2003; 2: 95–99.
Acknowledgements
We thank all of the patients and families who participated in this study, their respective doctors and pathologists for contributing clinical information. We are very grateful to Michèle Attenhofer and Marianne Haeusler for excellent technical assistance and to Prof Jurg Ott and Dr Ian Tomlinson for critical reading of the manuscript. This work was supported by grants from the Swiss National Science Foundation and the Swiss Cancer League.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Plasilova, M., Russell, A., Wanner, A. et al. Exclusion of an extracolonic disease modifier locus on chromosome 1p33–36 in a large Swiss familial adenomatous polyposis kindred. Eur J Hum Genet 12, 365–371 (2004). https://doi.org/10.1038/sj.ejhg.5201157
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201157
Keywords
This article is cited by
-
CD36 polymorphisms and the age of disease onset in patients with pathogenic variants within the mutation cluster region of APC
Hereditary Cancer in Clinical Practice (2021)
-
Recurrent desmoids determine outcome in patients with Gardner syndrome: a cohort study of three generations of an APC mutation-positive family across 30 years
International Journal of Colorectal Disease (2013)
-
Pathogenese des sporadischen und hereditären Kolonkarzinoms
Der Onkologe (2009)
-
Modifiers of risk in familial adenomatous polyposis
Current Colorectal Cancer Reports (2006)
-
Familial Adenomatous Polyposis: Experience from a Study of 1164 Unrelated German Polyposis Patients
Hereditary Cancer in Clinical Practice (2005)
