
Abstract
The phenotype of Bardet–Biedl syndrome (BBS) is defined by the association of retinitis pigmentosa, obesity, polydactyly, hypogenitalism, renal disease and cognitive impairement. The significant genetic heterogeneity of this condition is supported by the identification, to date, of eight genes (BBS1–8) implied with cilia assembly or function. Triallelic inheritance has recently been suggested on the basis of the identification of three mutated alleles in two different genes for the same patient. In a cohort of 27 families, six BBS genes (namely BBS1, BBS2, BBS4, BBS6, BBS7 and BBS8) have been studied. Mutations were identified in 14 families. Two mutations within the same gene have been identified in seven families. BBS1 is most frequently implied with the common M390R substitution at the homozygous state (n=2), or associated with another mutation at BBS1 (n=3). Compound heterozygous mutations have been found in BBS2 (one family) and BBS6 (one family). In seven other families, only one heterozygous mutation has been identified (once in BBS1, twice for BBS2 and three times in BBS6). Although our study did not reveal any families with bona fide mutations in two BBS genes, consistent with a triallelic hypothesis, we have found an excess of heterozygous single mutations. This study underlines the genetic heterogeneity of the BBS and the involvement of possibly unidentified genes.
Similar content being viewed by others

Introduction
Bardet–Biedl syndrome (BBS) is one of the most frequent syndromes associated with retinitis pigmentosa and a classical cause of inherited syndromic obesity in childhood.1, 2 BBS is characterized by the following associations: early-onset retinitis pigmentosa, obesity, polydactyly, hypogonadism, learning disabilities and kidney malformations.3, 4 A tremendous amount of knowledge about BBS has recently accumulated, demonstrating the extensive genetic heterogeneity and giving clues for the ciliary pathogenesis of this complex condition. It has also been suggested that its inheritance may depart from classic autosomal recessive inheritance and may involve, in some families, three mutated alleles in two genes (triallelic inheritance).5, 6, 7, 8, 9
To date, eight BBS genes have been identified: BBS1 (11q13);10 BBS2 (16q21);11 BBS3 (3p12–13);12, 13 BBS4 (15q22.3–q23);14 BBS5 (2q31);15 BBS6 (20p12);16, 17, 18 BBS7 (4q27);19 BBS8 (14q32.11).20 The BBS6 gene has also been found mutated in the clinically closely related McKusick Kaufman syndrome.21, 22 Point mutations in the eight known genes account only for about 50% of BBS patients, suggesting that there are several more genes to be found.
The identification of BBS8, as a protein involved at the level of the basal body of ciliated cells with either a role in ciliogenesis or mediating communication between the cilium and the interior of the cell, has opened the road to a general understanding of the disorder especially as functional overlap between some BBS genes has emerged.20 BBS4 has recently been shown to target proteins to the pericentriolar region, and to be involved with microtubule anchoring and cell cyle progression.23 The Bbs4-null mice discloses common features with the human phenotype and confirms the connection of BBS4 with ciliated cells.24 In this study, we have analyzed 27 families for six BBS genes (BBS1, BBS2, BBS4, BBS6, BBS7 and BBS8). We confirm that, apart from BBS1 and to a lesser extent BBS6, the other genes (including the recently identified BBS7 and BBS8) contribute to a small proportion of BBS patients. We also address the question of possible digenic triallelic inheritance in this cohort.
Patients and methods
BBS patients
The patients were selected with diagnostic criteria defined by the presence of at least three of the major features: retinitis pigmentosa, obesity, polydactyly, hypogonadism, learning disabilities and kidney malformation or failure.4
The 27 families (33 patients) analyzed in this study were recruited from Medical Genetics Departments and Ophthalmology Departments located all over France. Among the 27 families, 19 were sporadic cases and the remaining had at least two affected individuals.
The ethnic origin of the families is classified as three families were from North-Africa, three were Gypsies and the remaining (21 families) were of Caucasian origin.
PCR amplification of the BBS genes
DNA extraction from blood samples was performed by standard saline procedure. PCR amplification of the exons and exon–intron boundaries were realized with 50 ng of genomic DNA template. Primer sequences were modified according to the literature.5, 7, 14, 18, 20, 22 PCR protocols and primer sequences are available on request.
Screening for the recurrent M390R recurrent BBS1 mutation
We developed a restriction test to screen rapidly the patients for the recurrent mutation M390R of BBS1, as the T → G transversion in exon 12 disrupts the cleavage site for NlaIII restriction enzyme (CATG/) (Figure 1). PCR analysis of exon 12 of BBS1 was performed with the forward primer 5′-CCATCCCCTGTCTTGCTTTC-3′ and the reverse primer 5′-CATGCTTCATTTCCACCTCC-3′ using 50 ng of DNA and 10 pmol of each primer in a standard 25 μl reaction. PCR amplification was performed in a Whatman Biometra thermocycler under the following conditions: 3 min at 94°C, 35 cycles at 94°C for 30 s, 59°C for 30 s, 72°C for 30 s and a final extension step at 72°C for 10 min; 10 μl of the PCR product was digested with 2 U of NlaIII in the appropriate buffer at 37°C during 3 h.

Detection of the recurrent BBS1 mutation M390R by NlaIII digest. BBS1 exon 12 was amplified as described and PCR was digested by the NlaIII restriction enzyme (CATG/). The T → G mutation results in the loss of normal cleavage of the 229-bp PCR fragment to a 121- and a 104-bp fragment (and a residual 4 bp nonvisualized). Digest of DNAs from the heterozygous parents gives rise to the normal 121 and 104 bands and an undigested 229-bp band; DNA from the affected children was not digested. C is a normal control.
Mutation screening of the BBS1, BBS2, BBS4, BBS6, BBS7 and BBS8 genes
Mutation screening of the six BBS genes was performed by DHPLC analysis using at least three melting temperatures for each amplicon, followed by direct sequencing of the variant PCR fragments. To detect homozygous mutations, subsequent analysis was performed by SSCP and/or by mixing PCR products for DHPLC. For patients found to be mutated in a BBS gene, its entire coding sequence including splice sites was analyzed by direct sequencing.
DHPLC analysis was performed on WAVE Nucleic Acid Fragment Analysis System (Transgenomic, Inc.). The optimal melting temperatures (Tm) were determined for each PCR fragment by individualization of each denaturation domain using the WaveMaker Software 3.3.3 (Trangenomics Inc.). For BBS7 and BBS8, PCR samples from two unrelated patients were mixed to form heteroduplexes in approximately 1:1 ratio (following checking of size and quantity of PCR products on a 2% agarose gel) by heating mixed samples at 95°C for 8 min, and then cooling to 25°C by decreasing 0.5°C every 19 s.
SSCP (single-strand conformation polymorphism) analysis (Pharmacia Biotech Genephor and Pharmacia Biotech automated gel stainer) of BBS1, BBS2, BBS4 and BBS6 was performed at 15°C using standard procedure.
Bidirectional sequencing of the purified PCR products was performed using the ABI Big Dye Terminator Sequencing kit on an ABI3100 automated capillary sequencer (Applied Biosystems). Detailed protocols are available on request.
Splice sites scoring programs (http://l25.itba.mi.cnr.it/~webgene/www.spliceview.html, http://www.fruitfly.org/seq_tools/splice.html) were used to evaluate the effect of mutations affecting splice sites and to detect potential consequences of silent, or missense changes and intronic variations on splicing. A predictive program, rescue ESE web server (http://genes.mit.edu/burgelab/rescue-ese), was used to search for potential exonic splicing enhancers in polymorphic exonic variants (silent or missense variants).
DNA analysis with microsatellite markers
Familial segregation of fluorescent microsatellite STRPs markers across BBS1, BBS2, BBS4 and BBS6 loci was performed. Experimental conditions are available on request. Haplotypes were constructed for each relevant patient or relatives.
Microsatellite sequences were obtained from the UCSC Genome Browser Bioinformatics site (http://genome.ucsc.edu/cgi-bin/hgGateway): for the BBS1 locus (D11S4076, D11S1883, D11S1889), for the BBS2 locus (D16S3140, D16S408, D16S3071, D16S673), for the BBS4 locus (D15S1050, D15S204, D15S980, D15S1026) and for the BBS6 locus (D20S162, D20S901, D20S160, D20S894).
Two additional microsatellites, D11S0887 and D11S0406 localized respectively 60 kb upstream and 75 kb downstream of the BBS1 gene were used (S Bahram and H Inoko personal communication). A BBS4 intragenic (intron 4) microsatellite STRP, MIG4, was used (Tania Attié, personal communication). We characterized a novel BBS6 intragenic microsatellite STRP (MIG6) localized in intron 1 and performed fluorescent analysis with the following primers 5′-CTCCAACCTGACAGCTAGG-3′ and 5′-CCTGCACTTCCTGATAGCC-3′.
cDNA analysis
Sequencing of cDNA was performed following extraction of RNA with RNeasy Mini kit (Qiagen). The RNA of the proband of family I.3 was sequenced with three sets of primers covering the entire BBS1 coding sequence (details available on request).
Southern blot
Southern blot analysis of DNA was performed according to a classical protocol available on request. The DNA of the proband of family I.5 was digested with the following restriction enzymes: EcoRI, HindIII and XbaI. Two radioactive cDNA probes covering the entire coding sequence of BBS4 were used for hybridization (details available on request).
Results
Mutations were detected in 14 families and no mutations could be detected for the remaining 13 families (Table 1, Figure 2). Seven families (50% of families with mutations detected) were found to carry two mutations in the same gene: five for BBS1 (two homozygotes and three compound heterozygotes), one for BBS2 (compound heterozygote) and one for BBS6 (compound heterozygote). For seven families, only one obvious or putative mutation was found in a heterozygous state: one for BBS1, two for BBS4, three for BBS6 and one for BBS7.

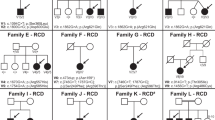
Pedigrees of the families involved in the study with corresponding mutations.
BBS1 mutations
BBS1 mutations were present in six of the 27 families. Five out of the six corresponding probant mutations carried the M390R mutation (allele frequency in BBS probands=0.13). Only two probands (I.4 and I.6) were homozygous for the M390R allele (7.4 versus 10.4% of a previous study).27 In the present series, four additional BBS1 mutations were identified, of which three have not been previously recorded: E384X, R160Q and R429X. Affected dizygotic twins of family I.15 carry both the M390R and the E549X mutations. The unaffected sibling is heterozygous for the M390R mutation. The E549X mutation was previously reported in the homozygous state in Puerto-Rican families and also in the compound heterozygous state with the M390R mutation.10, 25 The M390R mutation is associated with E384X in exon 12 in family I.22. The proband of family I.23 carries the M390R mutation and a c.479G>A change that predicts a missense mutation (R160Q) affecting a highly conserved residue, but which also most likely alters the donor splice site of exon 5, as it affects the last G of the exon (CG/gtgaga to CA/gtgaga) (Figure 3). Two affected members of family I.3 carry an R429X mutation in exon 13, which is predicted to result in an N-terminal truncated protein, but no second mutation in BBS1 could be identified, to date, for this family. This mutation was inherited from the father. Microsatellite markers analysis at the BBS1 locus showed that the two affected boys both share the same maternal haplotype. This result could suggest the presence of an undetected BBS1 mutation on the maternal allele despite RT-PCR analysis and complete DNA sequencing of the coding sequence of BBS1.

Amino-acid sequence conservation around residues affected by novel missense changes at BBS1, BBS4, BBS6 and BBS7, identified in this study. The sequences of BBS proteins/or predicted translation products from several species have been compared and aligned. The relevant amino acids are in bold, and underlined when different from Homo s. reference; other identities are represented by a gray background. Evolutionary substitutions in BBS1 (R → K) and BBS7 (I → V) are conservative. Note the variability of sequences in distant species around position 237 in BBS6. The transition 480G>A in the last nucleotide of exon5 in BBS1 is shown on the right; it is predicted to alter the donor splice site of exon5.
BBS2 mutations
Two newly described BBS2 mutations were identified in the same patient (compound heterozygote) from family I.8: 627delTT (L209fsX229) and 402delT (P134fsX200). No other BBS2 mutations were detected for other patients in this series.
BBS4 mutations
An L351R change in exon 13 of BBS4, which was not previously reported, was found in a heterozygous state in family I.5 of gypsy sinti origin. This mutation affects a residue highly conserved during evolution (Figure 3), and is predicted to affect the structure and/or function of the protein because of the replacement of a nonpolar amino acid by a positively charged amino acid. In the vicinity of codon 351, a frequent polymorphism I354T has been described that affects a less conserved position (Figure 3). The L351R mutation was inherited from the father, and an unaffected sister was heterozygous for this mutation. Microsatellite analysis at the BBS4 locus showed that the two sibs inherited a different maternal haplotype, in agreement with a possible undetected BBS4 mutation in the maternal allele. Southern blot analysis of the patient's DNA was normal excluding an important rearrangement at the level of the genomic sequence of BBS4.
For the proband of the I.21 family, a novel BBS4 mutation, 1450+2delTAGG, was identified at the heterozygous state. This mutation is predicted to abolish the splicing of exon 15 of BBS4.
BBS6 mutations
Compound heterozygous mutations in BBS6 were observed for the proband (sporadic case) of family I.27 combining a previously described frameshift mutation namely 429delCT433delAG (D143fsX157) and a novel mutation S479X. Interestingly, this 429delCT433delAG mutation has only been previously reported in Newfoundland patients in a heterozygous or homozygous state.18, 21
Three other BBS6 missense mutations or rare variants were found in a heterozygous state for three additional families.
The I339V missense was found in a sporadic French patient (family I.25). This mutation that leads to a conservative amino-acid change has been previously reported, but its pathogenecity appears to be questionable.21 A single mutated allele was found in exon 3 at position 237 (T237P) in a fetus (family I.20) with prenatal ultrasound presentation of the disease with renal abnormalities and postaxial polydactyly. Even though the T237 residue is not highly conserved during evolution, the proline substitution is likely to have a deleterious effect on the tridimensional structure of the BBS6 protein (Figure 3). Interestingly, this variation is adjacent to the previously reported S236P mutation.26, 27 The T237P mutation was not found in 48 ethnically matched controls (96 chromosomes) or in 163 BBS probands analyzed by sequencing of the open reading frame of BBS6.26
The proband of family I.17 has a T237A mutation inherited from the father and not identified in the second affected sib. Microsatellite analysis showed that the two affected sibs carry different maternal haplotypes, excluding an identical second mutation in the BBS6 gene. Microsatellite analysis at the BBS1, BBS2 and BBS4 loci revealed that the two affected sibs did not share the same haplotypes at each of these loci, excluding them as candidate genes for biallelic mutations. The T237A mutation was not found in 48 ethnically matched controls (96 chromosomes) or in 163 BBS probands previously analyzed.26
BBS7 mutations
For BBS7, a single I66F mutation was found for the sporadic case of family I.12, which was not observed in 48 ethnically matched controls or in the 84 patients analyzed by others.19 This change is likely to alter the structure of the protein and affects a highly conserved residue, such as an isoleucine or a valine, and very similar hydrophobic amino acids are found at this position from mammals to the nematode C. elegans (Figure 3). The effect of this variation proximal to the predicted beta-propeller region remains, however, to be proven as segregation could not be tested in this sporadic case.
BBS8 mutations
No mutation was detected for BBS8 for any of the 27 families studied herein.
Presumably silent variants or polymorphisms in the BBS genes
Several presumed silent variants were detected in exons or introns and are summarized in Table 2. These variants or polymorphisms are not predicted to create new splice sites, as tested by using splice sites scoring programs.
Silent exonic mutations and missense mutations were tested by enhancer splice site predictive programs. One of them, corresponding to a known polymorphism in exon 3 of BBS2 (I123V), was predicted to abolish a potential enhancer splice site. However, functional RNA studies would be necessary to test the effective consequences of this variation in RNA splicing. Identification of heterozygous polymorphisms in patients carrying heterozygous mutations allowed us to exclude deletions at the corresponding genomic region.
Discussion
Patients with BBS present with a unique association of clinical features and disclose a remarkable genetic heterogeneity. In this study, we performed, on a series of 27 families, the systematic mutation screening of six BBS genes, namely: BBS1, BBS2, BBS4, BBS6, BBS7 and BBS8. Our results are in accordance with the literature noticeably for the mutation frequency of BBS1 and excessive detection of patients where a single mutated allele was detected. Although no patient in this series demonstrated segregation of three mutant alleles, we present compelling evidence of a digenic (diallelic or triallelic) pattern of inheritance.
BBS1 and the M390R BBS1 mutation
BBS1 has been reported to be the most frequently mutated gene in BBS. The contribution of at least one BBS1 mutant allele was previously reported in 23.2% of the patients.27 This is in apparent contradiction with linkage studies estimating the contribution of BBS1 locus to be of 32–56% of BBS families.25 In this series, the contribution of BBS1 mutations was established for 26% of the families.
The major mutation M390R (located on exon 12 of BBS1) is the most common BBS1 mutated allele observed previously in 78–80% of families with BBS1 mutations.25, 27 Herein, in agreement with the previous reports, 5/6 (80%) families, with BBS1 mutations, were found to carry the M390R mutation (two as homozygous alleles and three as heterozygous alleles) representing the most common mutation found in this series of BBS patients. The M390R mutation is considered to be an ancient mutation as suggested by haplotype analysis and was described exclusively in patients of European descent.27 The origin of the families with the M390R mutation was indeed European as all of them were of French descent. In this paper, we describe three new BBS1 mutations in accordance with the low inter familial recurrence of ‘non-M390R BBS1’ mutations. One BBS1 mutation (E549X) found here in a French family has been previously described in patients of Puerto-Rican origin and may correspond to a recurring hotspot event.25, 27
No mutation in BBS8
The identification of BBS8 has been a major contribution to the understanding of the pathogenesis of the syndrome. However, no mutation could be detected in our series. Homozygous mutations in BBS8 have been identified to date in two families from Saudi Arabia and in one Pakistani family.20
Paucity of other bi-allelic families
Only two other families were clearly identified with two mutated alleles in the same gene (respectively for BBS2 and BBS6). One family disclosed compound heterozygosity for two nonsense mutations in BBS2 (Figure 1). Interestingly, the proband is a male patient aged 20, who presented with typical BBS features (obesity, early onset retinitis pigmentosa, mental retardation, hypogonadism and polydactyly). However, his eldest sister died, more then 25 years ago, at the age of 3 weeks because of a massive hydrometrocolpos, which suggested the diagnosis of Mc Kusick-Kaufman syndrome (MKKS: OMIM 236700) (no molecular investigations could be performed because DNA is not available). MKKS is a rare condition (defined as the association of postaxial polydactyly, hydrometrocolpos and congenital heart malformation), reported up to now to be associated only with BBS6 mutations.16, 22, 28
Another family showed compound heterozygosity for two truncating BBS6 mutations. The 429delCT433delAG mutation, found herein in a French family, has only been previously reported in Newfoundland patients, which suggested a founder effect in this population. The involved region is palindromic ACTttAGT with the possibility of a short base pairing that could lead to an intragenic recombination and the deletion of the AT and AG dinucleotides without affecting the TT nucleotide.18 This mutation could be a recurrent mutation, explaining the possible de novo mutation recurrence in a French family. Further analysis using linked BBS6 polymorphic markers could establish or exclude a potential common founder between the Newfoundland families and our family.
Heterozygote detection excess and oligogenic inheritance
Overall, seven of the 14 families with mutations were found to carry a single mutated allele: three families for BBS6, two families for BBS4, one family for BBS1 and one family for BBS7. For each of them, no other mutation was detected by sequencing the entire coding sequence including splice sites of the corresponding gene, and no other mutation was detected by screening the other BBS genes. However, a mutation in a regulatory and/or a noncoding region, or a large deletion or other large rearrangement cannot always be excluded by the methods used in this study although we performed, when applicable, RT-PCR analysis, Southern blot analysis and microsatellite haplotyping. These results raised the question of the pathogenicity of the single mutants: two are evident (one truncating mutation in BBS1, one splice mutation in BBS4), three are highly probable (three missense mutations that affect a highly conserved residue and/or result in a nonconservative amino-acid change: one in BBS4, one in BBS6 and one in BBS7) and two are doubtful (I339V and T237A in BBS6).
For this last mutation, the proband of family I.17 disclosed a T237A mutation that was, however, not identified in the second affected sib, ruling out the pathogenicity of this mutation if we consider classical monogenic inheritance. The putative first and second alleles would remain to be detected for family I.17. Fully informative microsatellite analysis showed that the two affected sibs have not received the same set of parental chromosomes at the BBS6 locus. Mutation analysis did not reveal any mutation in the coding and splice junction sequence of the other BBS genes tested herein. Moreover, microsatellite analysis at the BBS1, BBS2 and BBS4 loci excluded them as candidate genes for biallelic mutations.
Interestingly, the patient carrying the T237A BBS6 mutation presented with typical BBS features plus a congenital hypoplasia of the aortic arch as opposed to the other affected sib who disclosed classical BBS features and a normal aortic arch.
Other studies have, however, shown that siblings can be discordant for a third allele under a triallelic hypothesis. For instance, Badano et al7 described two families with mutations in BBS1 in which some but not all the affected sibs carry a third mutation in either BBS2 or BBS6, which seems to modulate the phenotype. More recently, Fan et al12 described a modifier effect due to a missense mutation for BBS3 in one affected sister of two sisters carrying the M390R mutation in the homozygous state. Single heterozygous mutations in BBS5 were identified in Caucasian pedigrees, suggesting also participation of this gene in complex inheritance.15
Both autosomal recessive inheritance (one locus inheritance) and oligogenic inheritance (specific alleles at more than one locus affecting a genetic trait) have been recognized to occur in BBS. Traditional autosomal recessive inheritance has been questioned as Katsanis et al recently described that for some BBS families, a total of three mutations in two genes are necessary for clinical expression.5, 8, 9, 25, 27
BBS6, BBS2, BBS3, BBS5, BBS4, BBS1 and BBS7 have been reported to be involved in triallelism with a predilection for BBS6 and BBS2.27 The third alleles reported in the literature are, however, often missense mutations for which pathogenicity can sometimes be difficult to ascertain.
Indeed, in our series of 96 control chromosomes, we have identified a missense mutation, A242S, that was previously described in a family in association with an homozygous BBS2 mutation.5, 26 This missense was also found once in 330 control chromosomes tested.26
In our series, we could not identify unaffected siblings of affected individuals who carry two mutations at one BBS locus. None of the 27 families analyzed here for six BBS genes could be demonstrated as presenting oligogenic inheritance underlying the fact that this mode of inheritance is not the rule for BBS. However, triallelic inheritance in the seven families with a single mutated allele detected in this series cannot be excluded. Moreover, our cohort of 27 families is small in comparison with the 259 families of various ethnicities studied to date by others.26 Identification of the missing BBS genes, segregation analysis in additional families and the development of functional tests to ascertain effects of the missense changes will be necessary to clarify the question of the frequency of triallelic inheritance in BBS. Our data are paradoxal in the way that the excess of heterozygotes is suggestive of oligogenic inheritance in a noticeable number of families; but, however, in this series, no clear triallelic patient has been identified. Taking into account that half of our families do not carry mutations in the known BBS genes (except BBS5 and BBS3 recently identified) and the compelling evidence for triallelism, we suggest that additional BBS loci remain to be identified.
References
Bardet G : Sur un syndrome d'obésité congénitale avec polydactylie et rétinite pigmentaire (contribution à l'étude des formes cliniques de l'obésité hypophysaire); Thesis, Paris 1920.
Biedl A : Ein geschwisterpaar mit adiposo-genitaler dystrophie. Dtsch Med Wochenschr 1922; 48: 1630.
Green JS, Parfrey PS, Harnett JD et al: The cardinal manifestations of Bardet–Biedl syndrome, a form of Laurence–Moon–Biedl. N Engl J Med 1989; 321: 1002–1009.
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA : New criteria for improved diagnosis of Bardet–Biedl syndrome: results of a population survey. J Med Genet 1999; 36: 437–446.
Katsanis N, Ansley SJ, Badano JL et al: Triallelic inheritance in Bardet–Biedl syndrome, a mendelian recessive disorder. Science 2001; 293: 2256–2259.
Burghes AHM, Vaessin HEF, De la Chappelle A : The land between mendelian and multifactorial inheritance. Science 2001; 293: 2213–2214.
Badano JL, Kim JC, Hoskins BE et al: Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet–Biedl patients with two mutaions at a second BBS locus. Hum Mol Genet 2003; 12: 1651–1659.
Fauser S, Munz M, Besch D : Further support for digenic inheritance in Bardet–Biedl syndrome. J Med Genet 2003; 40: e104.
Katsanis N : The oligogenic properties of Bardet–Biedl syndrome. Hum Mol Genet 2004; 13: R65–R71.
Mykytyn K, Nishimura DY, Searby CC et al: Identification of the gene (BBS1) most commonly involved in Bardte–Bield syndrome, a complex human obesity syndrome. Nat Genet 2002; 31: 435–438.
Nishimura DY, Searby CC, Carmi R et al: Positional cloning of a novel gene on chromosome 16q causing Bardet–Biedl syndrome. Hum Mol Genet 2001; 10: 865–874.
Fan Y, Esmail MA, Ansley SJ et al: Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet–Biedl syndrome. Nat Genet 2004; 36: 989–993.
Chiang AP, Nishimura D, Searby C et al: Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet–Biedl syndrome (BBS3). Am J Hum Genet 2004; 75: 475–484.
Mykytyn K, Braun T, Carmi R et al: Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet 2001; 28: 188–191.
Li JB, Gerdes JM, Haycraft CJ et al: Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 2004; 117: 541–552.
Slavotinek AM, Biesecker LG : Phenotypic overlap of McKusick-Kaufman syndrome with Bardet–Biedl syndrome: a literature review. Am J Med Genet 2000; 95: 208–215.
Slavotinek AM, Stone EM, Mykytyn K et al: Mutations in MKKS cause Bardet–Biedl syndrome. Nat Genet 2000; 26: 15–16.
Katsanis N, Beales PL, Woods MO et al: Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet–Biedl syndrome. Nat Genet 2000; 26: 67–70.
Badano JL, Ansley SJ, Leitch CC, Lewis RA, Lupski JR, Katsanis N : Identification of a novel Bardet–Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2. Am J Hum Genet 2003; 72: 650–658.
Ansley SJ, Badano JL, Blacque OE et al: Basal body dysfunction is a likely cause of pleiotropic Bardet–Biedl syndrome. Nature 2003; 425: 628–633.
Slavotinek AM, Searby C, Al-Gazali L et al: Mutation analysis of the MKKS gene in McKusick-Kaufman syndrome and selected Bardet–Biedl syndrome patients. Hum Genet 2002; 110: 561–567.
Stone DL, Slavotinek A, Bouffard GG et al: Mutation of a gene encoding a putative chaperonin causes McKusick-Kaufman syndrome. Nat Genet 2000; 25: 79–82.
Kim JC, Badano JL, Sibold S et al: The Bardet–Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet 2004; 36: 462–470.
Mykytyn K, Mullins RF, Andrews M et al: Bardet–Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. PNAS 2004; 202: 8664–8669.
Mykytyn K, Nishimura DY, Searby CC et al: Evaluation of complex inheritance involving the most common Bardet–Biedl syndrome locus (BBS1). Am J Hum Genet 2003; 72: 29–37.
Beales PL, Katsanis N, Lewis RA et al: Genetic and mutational analyses of a large multiethnic Bardet–Biedl cohort reveal a minor involvement of BBS6 and delineate the critical intervals of other loci. Am J Hum Genet 2001; 68: 606–616.
Beales PL, Badano JL, Ross AJ et al: Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet–Biedl syndrome. Am J Hum Genet 2003; 72: 1187–1199.
David A, Bitoun P, Lacombe D et al: Hydrometrocolpos and polydactyly: a common neonatal presentation of Bardet–Biedl and McKusick–Kaufman syndromes. J Med Genet 1999; 36: 599–603.
Acknowledgements
We wish to acknowledge the financial support of:Ministère de la Recherche (PHRC national 2002), RETINA France, LION's club du Kochersberg and la Fédération des Maladies Orphelines. We thank the constant technical help of the IGBMC (Institut de Génétique et de Biologie Moléculaire et Cellulaire, Illkirch-Graffenstaden) especially the sequencing and oligonucleotide synthesis services. We acknowledge the INSERM, CNRS and Université Louis Pasteur. We thank Dr Nicholas Katsanis for generous communication of primer sequences and gift of primers. We also thank Dr Anne Besancon from the Direction de la Recherche Clinique des Hôpitaux Universitaires de Strasbourg for her constant support to this project . We wish to thank Dr Tania Attié (Necker-Enfants Malades Hospital in Paris for the information on the intragenic microsatelitte of BBS4) and Pr Siamak Bahram for the gracious gift of BBS1 microsatellites.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hichri, H., Stoetzel, C., Laurier, V. et al. Testing for triallelism: analysis of six BBS genes in a Bardet–Biedl syndrome family cohort. Eur J Hum Genet 13, 607–616 (2005). https://doi.org/10.1038/sj.ejhg.5201372
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201372
Keywords
This article is cited by
-
The dominant findings of a recessive man: from Mendel’s kid pea to kidney
Pediatric Nephrology (2023)
-
Clinical and genetic spectrums of 413 North African families with inherited retinal dystrophies and optic neuropathies
Orphanet Journal of Rare Diseases (2022)
-
Targeted exome sequencing resolves allelic and the genetic heterogeneity in the genetic diagnosis of nephronophthisis-related ciliopathy
Experimental & Molecular Medicine (2016)
-
Genetic and clinical characterization of Pakistani families with Bardet-Biedl syndrome extends the genetic and phenotypic spectrum
Scientific Reports (2016)
-
In search of triallelism in Bardet–Biedl syndrome
European Journal of Human Genetics (2012)
