
Abstract
The prevalence of non-insulin-dependent diabetes mellitus (type II diabetes) in Polynesia is among the highest recorded worldwide and is substantially higher than in neighboring human populations. Such large differences in the frequency of a phenotype between populations may be explained by large allele frequency differences between populations in genes associated with the phenotype. To identify genes that may explain the high between-population variation in type II diabetes prevalence in the Pacific, we determined the frequency of 10 type II diabetes-associated alleles in 23 Polynesians, 23 highland New Guineans and 19 Han Chinese, calculated population-pairwise Fst values for each allele and compared these values to the distribution of Fst values from ∼100 000 SNPs from the same populations. The susceptibility allele in the PPARGC1A gene is at a frequency of 0.717 in Polynesians, 0.368 in Chinese but is absent in the New Guineans. The striking frequency difference between Polynesians and New Guineans is highly unusual (Fst=0.703, P=0.007) and we therefore suggest that this allele may play a role in the large difference in type II diabetes prevalence between Polynesians and neighboring populations.
Similar content being viewed by others

Introduction
Type II diabetes, or non-insulin-dependent diabetes mellitus [MIM 125853], has undergone an explosive increase in prevalence during the past two decades and is now considered one of the main threats to human health worldwide.1 The diabetes epidemic is primarily owing to the spread of a sedentary lifestyle and obesity, which become pronounced under modernization, urbanization and industrialization.2 Although this epidemic is apparent worldwide, it is most pronounced in some recently modernized traditional societies such as Native Americans,3 Pacific Islanders4 and Australian Aborigines.5
The prevalence of type II diabetes in several Pacific populations ranks among the highest in the world.4 For example, on the Pacific island of Nauru type II diabetes was virtually unknown 50 years ago but is now present in ∼40% of adults.6, 7 This is the second highest prevalence recorded in the literature, after that of the Pima Indians.3 This heightened susceptibility, however, is not equally severe across all Pacific populations and it has been suggested that the extreme susceptibility genotype was introduced by the Austronesian-speaking ancestors of present-day Polynesians,4, 8 who likely originated in Taiwan sometime before 3000 bp and reached the furthest islands of Polynesia by 800 bp.9 For example, Austronesian-speakers from Fiji,10 East New Britain11 and coastal Papua New Guinea12 have moderate to high susceptibilities to type II diabetes, whereas there is a notable absence of type II diabetes in both traditional living and partly urbanized non-Austronesians from highland Papua New Guinea8, 11 and the Solomon Islands.13 These latter groups are thought to be primarily derived from earlier migrations.14, 15, 16 In the Solomon Islands and New Caledonia, where Austronesian and non-Austronesian populations share the same environment, type II diabetes prevalence is higher in Austronesians than in non-Austronesians,17, 18 suggesting that the varying genetic susceptibility to type II diabetes in this region of the world may be ascribed to the relative contribution of Austronesian genetic admixture.
The loci that contribute to within-population phenotypic variation may be different from those that are responsible for between-population phenotypic variation and thus traditional within-population association and linkage analyses may have limited power to detect the loci that contribute to phenotypes that differ greatly between populations. For example, although variation in the MC1R gene is associated with skin pigmentation variation within Europeans,19, 20, 21 allele frequencies at this locus do not differ greatly between Europeans and other populations and it is, therefore, unlikely that this locus contributes to differences in skin pigmentation between Europeans and other populations.22 The alleles that underlie large between-population phenotypic differences are expected to show large frequency differences between populations. Thus, from a set of type II diabetes-associated SNPs, those at which the susceptibility allele is at unusually high frequency in Polynesians, compared to neighboring populations, are candidates to account for the high prevalence of type II diabetes in Polynesians. To identify such candidates, we genotyped and calculated Fst, a measure of genetic differentiation, for 10 type II diabetes-associated SNPs in three human populations (Polynesians, Han Chinese and highland Papua New Guineans) and compared these values to a distribution of Fst values from ∼100 000 SNPs genotyped in these same three populations.
Materials and methods
DNA samples included 23 Polynesians (nine Cook Islanders, eight Western Samoans, four Tongans and two Nuie Islanders), 23 highland New Guineans and 19 Han Chinese; samples were collected with ethical approval from the participating institutions and DNA was extracted according to standard protocols. Samples from Polynesia and New Guinea are described elsewhere,14, 23 whereas the Han Chinese are from Beijing. Type II diabetes-associated alleles were defined as variants with replicated evidence for association with type II diabetes (in ABCC8, ADRB2, CAPN10 (2 SNPs), GYS1, IRS1, KCNJ11, PPARG, PPARGC1A, SLC2A1) as suggested by two meta-analyses.24, 25 The recently described variants in TCF7L2 were not typed because this discovery was made after genotyping had been initiated for this project.26, 27 Genomic regions surrounding the SNPs of interest were amplified by PCR and restriction enzyme analyses were used to detect alleles at each SNP site. For SNPs that did not present a natural restriction site, we employed PIRA PCR.28 For the SNP in GYS1 (rs8103451), all individuals were monomorphic for the allele that is not recognized by the restriction enzyme and this was confirmed by sequencing.
An unbiased estimator of Fst was calculated for each population pairwise comparison for each type II diabetes-associated SNP and for each SNP from the Affymetrix GeneChip® Human Mapping 100 K Set according to equation (1) in Weir and Cockerham.29 The genotyping of samples with the 100 K SNP set followed previously described methods30 and included genotype data for 116 197 SNPs. However, an allele that is monomorphic in the two populations being compared receives an undefined Fst value according to Weir and Cockerham,29 and SNPs that contained genotype information from fewer than 50% of the individuals in a population were omitted in that population. Thus the number of Fst values from the 100 K set for each population pairwise comparisons were as follows: China-New Guinea=92783, China-Polynesia=93294, New Guinea-Polynesia=89012.
To determine whether our geography-based assignment of samples to populations reflects the underlying genetic structure of the samples, we examined population structure using STRUCTURE v 2.1.31 Owing to program limitations, 10 000 SNPs were randomly selected for the analyses. Two groups were assumed under the admixture model without any prior population assignment; 10 000 burn-in cycles and 50 000 replicates were used in each run. All runs were performed under the l=1 option and repeated five times. The ln of the probability of observing the data was −335837.0 for K=2. Chinese and New Guinea populations were clearly differentiated whereas the Polynesian population appeared as intermediate between them. The 95% credible intervals of the admixture proportions overlap in all Polynesian individuals. However, we observed significantly higher Chinese ancestry (13%) in one New Guinea individual when compared to other New Guineans (∼0.2%) and this individual was therefore excluded from further analyses (data not shown).
The samples from the 100 K set were identical to the samples typed for the type II diabetes-associated SNPs except seven Cook islanders and six Han Chinese from the 100 K set were replaced by seven different Cook islanders and five different Han Chinese for the diabetes SNP typing. Results were similar and conclusions unchanged when analyses were conducted on the subset of samples typed for both the 100 K set and the diabetes-associated SNPs (data not shown). The type II diabetes-associated SNP in PPARG was not typed by RFLP because it was included in the 100 K SNP set. Empirical P-values were generated by comparing the observed value to the Fst distribution from the 100 K set. To correct for multiple comparisons, for each population pairwise comparison we randomly sampled 10 SNPs 10 000 times from the 100 K set and generated corrected P-values by counting the number of times out of 10 000 one or more of the 10 Fst values was equal to or greater than the observed Fst value for each diabetes-associated SNP.
Results and discussion
A list of the sites of interest, the primers and the restriction enzymes are provided in Table 1. None of the 10 type II diabetes-associated SNPs was significantly out of Hardy-Weinberg equilibrium (data not shown). The frequencies of the type II diabetes-susceptibility alleles in the Chinese, New Guineans and Polynesians and the Fst values for each population pairwise comparison are provided in Table 2.
The susceptibility allele at SLC2A1 is at high frequency in Polynesians when compared to the Chinese (Fst=0.254, P=0.052), but not when compared to the New Guineans (Fst=0.059, P=0.500). The prevalence of type II diabetes in China is presently at 6–7%32, 33 and with increasing economic development the prevalence is on the rise.34, 35, 36 However, it is not known whether, under similar environmental conditions, type II diabetes prevalence in the Chinese is substantially lower than the prevalence in Polynesians. Thus, it remains unclear whether the high frequency of the SLC2A1 susceptibility allele contributes to the high prevalence of diabetes in Polynesians. The similar frequency of this allele between New Guineans and Polynesians could reflect genetic contributions from the former to the latter, as has been hypothesized to have occurred during the migrations of the Polynesian ancestors along the northern coast of New Guinea.37, 38
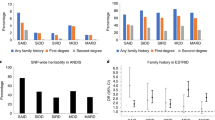
The PPARGC1A SNP exhibits the largest allele frequency difference between the populations in this study (Table 2). Especially striking is the high frequency of the susceptibility allele in Polynesians (0.72), compared to its complete absence in the New Guineans. Figure 1 shows the Fst values of the PPARGC1A SNP compared to the empirical distribution from the 100 K SNP set for all three population pairwise comparisons. In the Polynesia – New Guinea comparison, the Fst value for the PPARGC1A SNP is 0.703, which lies in the top 0.7% of the empirical Fst distribution: only 608 out of the 89012 SNPs from the 100 K set have greater Fst values. After correcting for multiple comparisons, this observation remains unusual (P=0.0655). Compared to the frequency in the Chinese (0.37), the frequency of the PPARGC1A susceptibility allele is relatively high in Polynesians: the Polynesia – China Fst is 0.194 (P=0.091). The Fst value for the China – New Guinea comparison is 0.369 (P=0.115). Although the Chinese have a frequency of the PPARGC1A susceptibility allele that is intermediate between the New Guineans and Polynesians, it is unclear how this may relate to the susceptibility of type II diabetes in the Chinese because comparisons between studies of the prevalence of type II diabetes in different populations under different environmental conditions are difficult to interpret. Nevertheless, as Polynesians have a much higher prevalence of type II diabetes than New Guineans living under similar environmental conditions, the unusually large difference in frequency between these two groups for the type II diabetes-susceptibility allele in the PPARGC1A gene supports the notion that this SNP, or SNPs in close LD to this SNP, play an important role in type II diabetes etiology in this area of the world.

Fst distributions for each population pairwise comparison generated from the 100 K SNP set. The Fst values of the PPARGC1A susceptibility allele is indicated by a dotted line and is shown in boxes along with the respective P-values.
PPARGC1A, or peroxisome proliferator-activated receptor-γ coactivator 1α, is a transcriptional co-activator that regulates the transcription of genes involved in adaptive thermogenesis, adipogenesis and oxidative metabolism39 and also regulates hepatic glucose output through the control of gluconeogenesis.40, 41 The susceptibility allele at PPARGC1A changes a glycine to serine at codon 482 and transfection assays have demonstrated that this substitution affects the protein's efficiency as a coactivator on the Tfam promoter, which may result in altered mitochondrial function and insulin resistance.42
Linkage analyses have also identified the PPARGC1A genomic region as a candidate locus for type II diabetes in the Pima Indians.43 However, as the frequency of the susceptibility allele in the Pima is only 0.18,44 which is lower than in Europeans (0.37),45 it is unlikely that this allele accounts for the observed high prevalence of type II diabetes in the Pima compared to European-Americans.3
It has previously been suggested that the Polynesians underwent strong selection pressures for energetic efficiency during their settlement of the Pacific, which required long open ocean voyages in the face of cold stress and starvation.46, 47, 48, 49 According to the thrifty gene hypothesis,50 such conditions may have led to positive selection for a thrifty metabolism in Polynesians and driven type II diabetes-susceptibility alleles to high frequency. Thus, past positive selection may at least partially explain the high prevalence of type II diabetes among contemporary Polynesian populations when compared to their neighbors in New Guinea without the heightened susceptibility. Several studies have demonstrated that a useful approach to detecting the signature of positive selection in populations under different selective pressures is to identify SNPs with high Fst values.51, 52, 53, 54 Thus, the unusually high frequency of the PPARGC1A susceptibility allele in Polynesians may reflect past positive selection.
Regardless of whether drift or selection is responsible for the high frequency of this allele in Polynesians, the fact remains that Polynesians exhibit both unusually high frequencies of type II diabetes, and unusually high frequencies of a known type II diabetes susceptibility allele. These two results indicate that the PPARGC1A susceptibility allele is a strong candidate for explaining the high frequency of type II diabetes in Polynesians and merits further investigation. Moreover, our results indicate that searching in candidate genes for alleles that exhibit large frequency differences between populations is a useful approach for identifying potential candidates for the genetic basis of phenotypic traits that vary greatly between populations.
References
Zimmet P, Alberti KGMM, Shaw J : Global and societal implications of the diabetes epidemic. Nature 2001; 414: 782.
Zimmet P : Globalization, coca-colonization and the chronic disease epidemic: can the Doomsday scenario be averted? J Internal Med 2000; 247: 301–310.
Knowler WC, Bennett P, Hamman R, Miller M : Diabetes incidence and prevalence in Pima Indians: a 19-fold greater incidence than in Rochester, Minnesota. Am J Epidemiol 1978; 108: 497–504.
Zimmet PZ, Dowse GK, Finch C, Serjeantson S, King H : The epidemiology and natural history of NIDDM--lessons from the South Pacific. Diabetes Metab Rev 1990; 6: 91–124.
O’Dea K : Westernisation, insulin resistance and diabetes in Australian Aborigines. Med J Aust 1991; 155: 258–264.
Dowse G, Zimmet P, Finch C, Collins V : Decline in incidence of epidemic glucose intolerance in Nauruans: implications for the ‘thrifty genotype’. Am J Epidemiol 1991; 133: 1093–1104.
Zimmet P, Taft P, Guinea A, Guthrie W, Thoma K : The high prevalence of diabetes mellitus on a Central Pacific Island. Diabetologia 1977; 13: 111–115.
King H, Zimmet PZ, Bennett P, Taylor R, Raper L : Glucose tolerance and ancestral genetic admixture in six semitraditional Pacific populations. Genet Epidemiol 1984; 1: 315–328.
Bellwood P, Renfrew C : Examining the farming/language dispersal hypothesis. Cambridge, United Kingdom: McDonald Institute for Archaeological Research, 2003.
Zimmet P, Taylor R, Ram P et al: Prevalence of diabetes and impaired glucose tolerance in the biracial (Melanesian and Indian) population of Fiji: a rural-urban comparison. Am J Epidemiol 1983; 118: 673–688.
King H, Finch C, Collins A et al: Glucose tolerance in Papua New Guinea: ethnic differences, association with environmental and behavioural factors and the possible emergence of glucose intolerance in a highland community. Med J Aust 1989; 151: 204–210.
Dowse GK, Spark RA, Mavo B et al: Extraordinary prevalence of non-insulin-dependent diabetes mellitus and bimodal plasma glucose distribution in the Wanigela people of Papua New Guinea. Med J Aust 1994; 160: 767–774.
Eason R, Pada J, Wallace R, Henry A, Thornton R : Changing patterns of hypertension, diabetes, obesity and diet among Melanesians and Micronesians in the Solomon Islands. Med J Aust 1987; 146: 465–469.
Stoneking M, Jorde LB, Bhatia K, Wilson AC : Geographic variation in human mitochondrial DNA from Papua New Guinea. Genetics 1990; 124: 717–733.
Dunn M, Terrill A, Reesink G, Foley RA, Levinson SC : Structural Phylogenetics and the Reconstruction of Ancient Language History. Science 2005; 309: 2072–2075.
Kayser M, Brauer S, Weiss G et al: Reduced Y-chromosome, but not mitochondrial DNA, diversity in human populations from West New Guinea. Am J Hum Genet 2003; 72: 281–302.
Beizer RA : Prevalence of Abnormal Glucose Tolerance in Six Solomon Islands Populations. Am J Phys Anthropol 1990; 81: 471–482.
Zimmet P, Canteloube D, Genelle B et al: The prevalence of diabetes mellitus and impaired glucose tolerance in Melanesians and part-Polynesians in rural New Caledonia and Ouvea (Loyalty Islands). Diabetologia 1982; 23: 393.
Rees JL : Genetics of hair and skin color. Annu Rev Genet 2003; 37: 67–90.
Makova K, Norton H : Worldwide polymorphism at the MC1R locus and normal pigmentation variation in humans. Peptides 2005; 26: 1901–1908.
Sturm RA, Teasdale RD, Box NF : Human pigmentation genes: identification, structure and consequences of polymorphic variation. Gene 2001; 277: 49–62.
Myles S, Somel M, Tang K, Kelso J, Stoneking M : Identifying genes underlying skin pigmentation differences among human populations. Hum Genet 2007; 120: 613–621.
Trent RJ, Mickleson KN, Wilkinson T et al: Globin genes in Polynesians have many rearrangements including a recently described gamma gamma gamma gamma. Am J Hum Genet 1986; 39: 350–360.
Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN : Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 2003; 33: 177.
Parikh H, Groop L : Candidate Genes for Type 2 Diabetes. Rev Endocrine Metabolic Disorders 2004; 5: 151–176.
Grant SFA, Thorleifsson G, Reynisdottir I et al: Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature 2006; 38: 320–323.
Helgason A, Palsson S, Thorleifsson G et al: Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet 2007; 39: 218–225.
Ke X, Collins A, Ye S : PIRA PCR designer for restriction analysis of single nucleotide polymorphisms. Bioinformatics 2001; 17: 838–839.
Weir BS, Cockerham CC : Estimating F-statistics for the analysis of population structure. Evolution 1984; 38: 1358–1370.
Matsuzaki H, Dong S, Loi H et al: Genotyping over 100 000 SNPs on a pair of oligonucleotide arrays. Nat Methods 2004; 1: 109–111.
Pritchard JK, Stephens M, Donnelly P : Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000; 155: 945–959.
Dong Y, Gao W, Nan H et al: Prevalence of Type 2 diabetes in urban and rural Chinese populations in Qingdao, China. Diabet Med 2005; 22: 1427–1433.
Jia WP, Pang C, Chen L et al: Epidemiological characteristics of diabetes mellitus and impaired glucose regulation in a Chinese adult population: the Shanghai Diabetes Studies, a cross-sectional 3-year follow-up study in Shanghai urban communities. Diabetologia 2007; V50: 286–292.
Wang Y, Mi J, Shan XY, Wang QJ, Ge KY : Is China facing an obesity epidemic and the consequences? The trends in obesity and chronic disease in China. Int J Obes (Lond) 2007; 31: 177–188.
Pan XR, Yang WY, Li GW, Liu J : Prevalence of diabetes and its risk factors in China, 1994. National Diabetes Prevention and Control Cooperative Group. Diabetes Care 1997; 20: 1664–1669.
Wong KC, Wang Z : Prevalence of type 2 diabetes mellitus of Chinese populations in Mainland China, Hong Kong, and Taiwan. Diabetes Res Clin Pr 2006; 73: 126–134.
Kayser M, Brauer S, Weiss G et al: Melanesian origin of Polynesian Y chromosomes. Curr Biol 2000; 10: 1237–1246.
Kayser M, Brauer S, Cordaux R et al: Melanesian and Asian origins of Polynesians: mtDNA and Y-chromosome gradients across the Pacific. Mol Biol Evol 2006; 23: 2234–2244.
Puigserver P, Spiegelman BM : Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α (PGC-1α): Transcriptional Coactivator and Metabolic Regulator. Endocr Rev 2003; 24: 78–90.
Yoon JC, Puigserver P, Chen G et al: Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001; 413: 131.
Herzig S, Long F, Jhala US et al: CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001; 413: 179.
Choi YS, Hong JM, Lim S, Ko KS, Pak YK : Impaired coactivator activity of the Gly482 variant of peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1alpha) on mitochondrial transcription factor A (Tfam) promoter. Biochem Biophys Res Commun 2006; 344: 708–712.
Pratley RE, Thompson DB, Prochazka M et al: An autosomal genomic scan for loci linked to prediabetic phenotypes in Pima Indians. J Clin Invest 1998; 101: 1757–1764.
Muller YL, Bogardus C, Pedersen O, Baier L : A Gly482Ser Missense Mutation in the Peroxisome Proliferator-Activated Receptor-γ Coactivator-1 Is Associated With Altered Lipid Oxidation and Early Insulin Secretion in Pima Indians. Diabetes 2003; 52: 895–898.
The International HapMap Consortium: A haplotype map of the human genome. Nature 2005; 437: 1299.
Baker P : Migrations, genetics, and the degenerative diseases of South Pacific Islanders; in: Boyce AJ (ed): Migration and Mobility. London: Taylor and Francis, 1984, pp 209–239.
Houghton P : The adaptive significance of Polynesian body form. Ann Hum Biol 1990; 17: 19–32.
Bindon JR, Baker PT : Bergmann's rule and the thrifty genotype. Am J Phys Anthropol 1997; 104: 201–210.
Prior I : The price of civilization. Nutr Today 1971; 6: 2–11.
Neel JV : Diabetes mellitus: a ‘thrifty’ genotype rendered detrimental by ‘progress’? Bull WHO 1962; 77: 694–703.
Akey JM, Zhang G, Zhang K, Jin L, Shriver MD : Interrogating a High-Density SNP Map for Signatures of Natural Selection. Genome Res 2002; 12: 1805–1814.
Beaumont MA, Balding DJ : Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol 2004; 13: 969–980.
Beaumont MA : Adaptation and speciation: what can Fst tell us? Trends Ecol Evol 2005; 20: 435–440.
Pollinger JP, Bustamante CD, Fledel-Alon A, Schmutz S, Gray MM, Wayne RK : Selective sweep mapping of genes with large phenotypic effects. Genome Res 2005; 15: 1809–1819.
Acknowledgements
We thank Ed Green, Dominique Quinque, Silke Brauer, Christian Becker and Mehmet Somel for technical assistance and David Hughes, Wolfgang Enard, Peter Kovacs, Svante Pääbo and Michael Stumvoll for useful discussions. This work was supported by funds from the Max Planck Society and from the German Ministry of Education and Research (Bundesministerium für Bildung und Forschung, National Genome Research Network, NGFN2) to MS as well as from the Erasmus University Medical Center Rotterdam to MK.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Myles, S., Hradetzky, E., Engelken, J. et al. Identification of a candidate genetic variant for the high prevalence of type II diabetes in Polynesians. Eur J Hum Genet 15, 584–589 (2007). https://doi.org/10.1038/sj.ejhg.5201793
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201793
Keywords
This article is cited by
-
Lack of direct evidence for natural selection at the candidate thrifty gene locus, PPARGC1A
BMC Medical Genetics (2016)
-
Positive selection of protective variants for type 2 diabetes from the Neolithic onward: a case study in Central Asia
European Journal of Human Genetics (2013)
-
Testing the thrifty gene hypothesis: the Gly482Ser variant in PPARGC1Ais associated with BMI in Tongans
BMC Medical Genetics (2011)
-
Fine-scale detection of population-specific linkage disequilibrium using haplotype entropy in the human genome
BMC Genetics (2010)
-
How culture shaped the human genome: bringing genetics and the human sciences together
Nature Reviews Genetics (2010)
