
Abstract
We carried out a GWAS meta-analysis of combined mixed-ancestry schizophrenia, schizoaffective, and bipolar cohorts that resulted in the identification of six genome-wide significant loci, including one novel locus at chr8q24.3, encompassing TSNARE1 (P = 1.28 × 10−9). The analysis included a total of 13,394 cases and 34,676 controls. While the function of TSNARE1 remains unknown, bioinformatic predictions based on phylogenetic ancestry indicate it may have a vertebrate-specific function in intracellular protein transport and synaptic vesicle exocytosis.
Similar content being viewed by others

Introduction
There is increasing epidemiological1,2,3 and genetic evidence4,5,6,7,8 of a shared genetic etiology between psychiatric disorders and, in particular, between schizophrenia (SCZ) and bipolar disorder (BP)4. Numerous genetic variants have been associated with susceptibility to schizophrenia. Many demonstrate overlapping susceptibility with bipolar disorder, including rare highly penetrant familial mutations (e.g. DISC1 gene9), numerous recurrent and de novo copy number variants (CNVs)10 and an increasing number of common single nucleotide polymorphisms (SNPs)4,5,6,11,12. To date, 12 genome-wide significant loci have been associated with SCZ and/or BP, all with odds ratios of less than 1.26,10. Taken together, evidence sugests genetic succeptibility to SCZ is highly heterogenous with multiple, common, polygenic variants contributing small effects.
The largest study to date, with genome-wide genotyping data on all samples, remains the phase I study of the PGC meta-analysis with 9,394 cases9. We carried out the largest single-phase meta-analysis of schizophrenia and bipolar cases, including data from 16 cohorts (Caucasian, African American and Asian cases and controls), to identify novel susceptibility loci.
Results
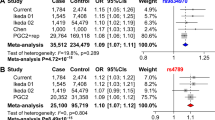
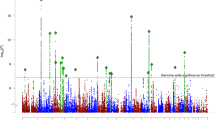
Following the meta-analysis of the combined mixed-ancestry schizophrenia, schizoaffective, and bipolar cohorts, 40 variants surpassed genome wide significance (P-values < 5 × 10−8) (Table 1). The 40 SNPs mapped to 6 loci, 5 of which had been previously associated with susceptibility to SCZ and/or BP (ITIH1, SDCCAG8, MHC, MAD1L1 and CSMD1; Figure 1). Two SNPs mapped to a novel locus containing a gene of unknown function, TSNARE1 (t-SNARE domain containing 1) on chr8q24.3 (top SNP rs10098073: fixed effects P 1.28 × 10−9 random effects P 1.28 × 10−9 OR 1.108; rs4129585 fixed effects P 2.38 × 10−8 random effects P 1.28 × 10−9 OR 1.108). In addition to the two genome-wide significant SNPs, multiple other SNPs in LD showed a trend towards association at the locus (Figure 2). Odds ratios for the most significantly associated SNP (rs10098073) across the 16 cohorts ranged from 1.003 to 1.258 (SD 0.06) with one outlier, the Dublin cohort, crossing 1 at OR 0.97. The genomic inflation factor of the meta-analysis was 1.08 indicating that population stratification had been adequately controlled. Analyzing the Caucasian cases and controls alone (n = 11,681 cases and 24,498 controls), the top TSNARE1 SNP, rs10098073, remains genome-wide significant (rs10098073 P-val 3.947 × 10−8 OR 1.12; rs4129585 P-val 1.061 × 10−7 OR 1.17).

The red dotted line indicates genome wide significance threshold of 5 × 10−8. Loci highlighted in red surpassed the genome-wide significance threshold in this study.

TSNARE1 regional association plot.
While variants at TSNARE1 have not reached genome-wide significance in previous analyses, rs10098073 showed a consistent trend towards association with schizophrenia12. For the PGC discovery and replication analyses, the P-value for rs10098073 was 2.09 × 10−5; OR 1.077; Analysis of the same SNP in the CLOZUK10 study produced a P-value of 0.0165; OR 1.101. After combining the PGC and CLOZUK studies, analysis of rs10098073 resulted in P 1.19 × 10−6; OR 1.101. The CLOZUK sample (n = 2652) has not been included in any other GWAS apart from the primary study12. There are no overlapping samples between the CLOZUK study and our sample set and therefore the CLOZUK sample can serve as an interdependent replication of the association.
Discussion
The function of the TSNARE1 gene remains unknown. A recent publication suggests it may have evolved within the vertebrate lineage from the harbinger transposon superfamily13. Bioinformatic predictions based on phylogenetic ancestry indicate it may bind SNARE (soluble N-ethylmaleimide-sensitive factor attached protein receptor) proteins and have SNAP receptor activity. TSNARE1 may therefore have a vertebrate-specific function in intracellular protein transport and synaptic vesicle exocytosis.
Taken together, our meta-analysis confirmed 5 genome-wide significant loci previously reported and identified a novel sixth locus that has not previously been shown to associate with schizophrenia at genome-wide significance. The locus spans TSNARE1, a gene of unknown function that is predicted to have a vertebrate-specific function in synaptic vesicle exocytosis.
Methods
The study included 13,394 schizophrenia and bipolar cases and 34,676 controls (detailed in Supplementary table 1). Of these, 3,182 schizophrenia (of which 377 were classified as schizoaffective) cases and 1,032 bipolar I cases were collected from 28 clinical trials conducted by Janssen Research & Development, LLC (detailed description in Supplementary methods). These samples were matched to 15,277 and 8,000 controls, respectively, from the biorepository at the Center for Applied Genomics (CAG) of the Children's Hospital of Philadelphia (CHOP). All controls were recruited at CHOP and had no diagnosis or family history of psychiatric disease based on their medical record. All Janssen cases were genotyped on the Illumina 1M-DuoV3, while CHOP controls were analyzed using either the Illumina HumanHap550 or 610 Quad arrays.
In addition, 1,157 cases meeting DSM-IV-TR criteria for schizophrenia or schizoaffective disorder from the Center for Applied Genomics (CAG) at The Children's Hospital of Philadelphia and the Department of Psychiatry at the University of Pennsylvania, School of Medicine14 and 2,107 controls from the biorepository at CAG were also included in the analysis. Samples were genotyped on the Affymetrix 6.0 array at The Children's Hospital of Philadelphia (CHOP) as previously described14.
The remaining 8,023 schizophrenia cases and 9,292 controls were part of the Schizophrenia Psychiatric Genome-Wide Association Study Consortium (PGC), as previously described11, and were obtained from the NIMH as schizophrenia distribution 9 (https://www.nimhgenetics.org/).
As different cohorts were genotyped on different platforms, we imputed genotypes based on HapMap 3 reference panel for all samples prior to meta-analysis to allow for direct comparison of variants across the entire cohort. Both fixed and random effect meta-analyses were then carried out. Detailed methods are presented in the supplementary materials.
References
Lichtenstein, P. et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 373, 234–9 (2009).
Smoller, J. W. & Finn, C. T. Family, twin, and adoption studies of bipolar disorder. Am j med genet. Part C, Seminars in medical genetics 123C, 48–58 (2003).
Maier, W. et al. Continuity and discontinuity of affective disorders and schizophrenia. Results of a controlled family study. Arch gen psych 50, 871–83 (1993).
Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet (2013).
Purcell, S. M. et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–52 (2009).
Steinberg, S. et al. Common variant at 16p11.2 conferring risk of psychosis. Mol psych (2012).
Lee, S. H. et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat genet 44, 247–50 (2012).
Lee, S. H. et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat genet (2013).
Chubb, J. E., Bradshaw, N. J., Soares, D. C., Porteous, D. J. & Millar, J. K. The DISC locus in psychiatric illness. Mol psych 13, 36–64 (2008).
Grayton, H. M., Fernandes, C., Rujescu, D. & Collier, D. A. Copy number variations in neurodevelopmental disorders. Prog neurobiol 99, 81–91 (2012).
Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat genet 43, 969–76 (2011).
Hamshere, M. L. et al. Genome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8, and extensive replication of associations reported by the Schizophrenia PGC. Mol psych (2012).
Smith, J. J., Sumiyama, K. & Amemiya, C. T. A living fossil in the genome of a living fossil: Harbinger transposons in the coelacanth genome. Mol biol evo 29, 985–93 (2012).
Glessner, J. T. et al. Strong synaptic transmission impact by copy number variations in schizophrenia. PNAS 107, 10584–9 (2010).
Acknowledgements
We are grateful to the study volunteers for participating in the research studies and to the clinicians and support staff for enabling patient recruitment and blood sample collection. Informed consent was obtained from all participants or their parents or guardians. We thank the staff in the Neuroscience Biomarkers Genomic Lab at Janssen & the Center for Applied Genomics at CHOP for sample processing and the staff at Illumina for genotyping Janssen DNA samples. We also thank Anthony Santos, Nicole Bottrel, Monique-Andree Franc, William Cafferty of Janssen Research & Development) for operational support, Stefanie Rassnick of Janssen Research & Development) for inputs in setting up the collaboration and for discussion, and Wendy P. Battisti of Janssen Research & Development, for editorial support. The work was funded in part by an Institutional Development Award to the Center for Applied Genomics from The Children's Hospital of Philadelphia; Adele and Daniel Kubert donation, University of Pennsylvania Biomedical Graduate Studies training grant; the Cotswold foundation; by 1R01MH097284-01 from the NIMH, and sponsored research to The Children's Hospital of Philadelphia by Janssen Research & Development, LLC.
Author information
Authors and Affiliations
Consortia
Contributions
H.H. and N.C. conceived and initiated the project. Q.L. managed the Janssen samples and genetic data. P.S. performed the GWAS and meta-analysis. J.G. and D.H. performed a CNV analysis of the data. D.W. performed Janssen genotype data QC. R.E.G. assisted with phenotyping of the PENN cohort. P.S. and H.H. wrote the manuscript. The Janssen-CHOP Neuropsychiatric Genomics Working Group collected the samples and performed phenotype classification. All authors contributed to the data analysis review, discussions, and contributed to the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
Dr. Cohen is a former employee of Janssen Research & Development. Drs. Wang and Li are current employees of Janssen Research & Development.
Supplementary information
Supplementary Information
Supplementary Data (DOC 6350 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Sleiman, P., Wang, D., Glessner, J. et al. GWAS meta analysis identifies TSNARE1 as a novel Schizophrenia / Bipolar susceptibility locus. Sci Rep 3, 3075 (2013). https://doi.org/10.1038/srep03075
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03075
This article is cited by
-
Methylation in MAD1L1 is associated with the severity of suicide attempt and phenotypes of depression
Clinical Epigenetics (2023)
-
Schizophrenia-associated Mitotic Arrest Deficient-1 (MAD1) regulates the polarity of migrating neurons in the developing neocortex
Molecular Psychiatry (2023)
-
Polygenic risk for schizophrenia and bipolar disorder in relation to cardiovascular biomarkers
European Archives of Psychiatry and Clinical Neuroscience (2023)
-
Exploring the mediation of DNA methylation across the epigenome between childhood adversity and First Episode of Psychosis—findings from the EU-GEI study
Molecular Psychiatry (2023)
-
MAD1L1 and TSNARE gene polymorphisms are associated with schizophrenia susceptibility in the Han Chinese population
BMC Medical Genomics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
