






Abstract
CMX001, an orally active lipid conjugate of cidofovir, is 50 times more active in vitro against herpes simplex virus (HSV) replication than acyclovir or cidofovir. These studies compared the efficacy of CMX001 to acyclovir in BALB/c mice inoculated intranasally with HSV types 1 or 2. CMX001 was effective in reducing mortality using doses of 5 to 1.25 mg/kg administered orally once daily, even when treatments were delayed 48–72 h post viral inoculation. Organ samples obtained from mice treated with CMX001 had titers 3–5 log10 plaqueforming units per gram of tissue lower than samples obtained from mice treated with acyclovir, including 5 different regions of the brain. Detectable concentrations of drug-related radioactivity were documented in the central nervous system of mice after oral administration of 14C-CMX001. These studies indicate that CMX001 penetrates the blood-brain barrier, is a potent inhibitor of HSV replication in disseminated infections and central nervous system infections, and is superior to acyclovir.
Herpes simplex virus (HSV) infects ∼1500 neonates annually, many of whom will develop central nervous system (CNS) disease; HSV encephalitis occurs in children >6 months of age and in adults at 1/150,000 annually in the United States [1–5]. Rapid diagnosis and early intervention with acyclovir have improved survival and decreased long-term neurological sequelae; however, mortality still exceeds 25%, and >50% of survivors have significant neurologic impairment [5–17]. Thus, a need exists for antiviral medications with improved distribution to the brain and greater efficacy. For the past 20 years, acyclovir has been the only FDA-approved drug and is the standard of care for treatment of disseminated infections and CNS HSV infections in neonates and herpes encephalitis in older children and adults. Experimental studies in animals infected with HSV have demonstrated that acyclovir was effective in reducing mortality and viral replication in CNS tissues, as well as in visceral organs, and that acyclovir was superior to what had been previously reported for vidarabine [18–21]. These results in animal models were highly predictive of the outcome in clinical studies. Treatment with either vidarabine or acyclovir resulted in efficacy, as evidenced by decreased mortality, but many survivors had permanent neurological sequelae, suggesting that improved therapies for these diseases were needed [5, 15, 17, 22, 23].
CMX001 (hexadecyloxypropyl cidofovir) was developed as an orally active, lipophilic form of cidofovir. It has enhanced activity in vitro and in vivo compared with cidofovir against certain herpesviruses, adenoviruses, and orthopoxviruses [24–30]. CMX001 was studied previously in animal models of cytomegaloviruses and orthopoxviruses and was shown to be effective against murine cytomegalovirus [27] and human cytomegalovirus [31], vaccinia, cowpox [32], ectromelia virus infections in mice [33–34], and in primate models using monkeypox virus [35]. It was also synergistic when coadministered in vitro or in vivo with ST-246, a compound under development for treatment of orthopoxviruses [36]. CMX001 is currently in phase II clinical studies for development as a therapeutic agent for human cytomegalovirus, adenovirus, and BK virus infections, as well as for adverse events following smallpox vaccinations.
The purpose of the current studies was to determine the efficacy of CMX001 compared with acyclovir in 2 models of herpes encephalitis and disseminated neonatal herpes. These data were then correlated with the results from a quantitative whole-body distribution study in uninfected mice using orally administered radio-labeled CMX001.
Materials and Methods
Experimental animals. Female BALB/c mice were purchased at 3–4 weeks of age from Charles River Laboratories. Animals were quarantined and acclimated for 3 days prior to use. Mice were group-housed in microisolator cages and used at a quantity of 15 mice per treatment group. They were obtained, housed, used, and killed according to USDA and AAALAC regulatory policies. All animal procedures were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee prior to initiation of studies.
Viruses and cells. The strain of HSV-1 used was E-377, and the strain of HSV-2 was MS. The origin of these viruses has been reported elsewhere [29]. Human foreskin fibroblast (HFF) cells were prepared as primary cultures from freshly obtained newborn human foreskins. Virus pools were prepared and quantified in primary rabbit kidney cells for use in vitro and in vivo. Culture medium for both cell lines was minimum essential medium with Earle salts containing 10% fetal bovine serum and 2 µmol/L L-glutamine, 100 U/mL penicillin, and 25 mmol/L gentamicin.
Antiviral compounds. CMX001 was provided by Chimerix, Inc, and was suspended in 0.4% carboxymethylcellulose for oral delivery to mice. Acyclovir (Sigma) was weighed and suspended in sterile water for oral treatment of mice. Compounds were prepared in a 0.2 mL volume, which was administered once daily (CMX001) or twice daily (acyclovir). Treatments for efficacy evaluations were administered to mice for 7 consecutive days beginning 24–72 h after viral inoculation by oral gavage using doses of 5, 2.5, and 1.25 mg/kg of CMX001 administered once daily or doses of 120, 60, or 30 mg/kg of acyclovir given twice daily at ∼12-h intervals. For pathogenesis experiments, treatments began 24 h post viral inoculation and were administered for 7 consecutive days using 5 mg/kg of CMX001 or 100 mg/kg of acyclovir. These doses were selected on the basis of the results obtained in the mortality experiments.
Experimental infections. Mice were manually restrained for intranasal inoculations using a total volume of 0.04 mL per mouse containing a ∼90% lethal dose of either HSV-1 strain E-377 or HSV-2 strain MS. For these studies, the inoculum contained 4.4 × 104 plaque-forming units (PFU) per mouse for HSV-1 or 1.1 × 105 PFU per mouse for HSV-2. For mortality experiments, animals were evaluated at least once daily for 21 days and 4 times daily during peak occurrence of clinical neurological signs so that mice could be humanely killed. Pathogenesis studies were performed for both HSV-1 and HSV-2 to compare the effect of CMX001 and acyclovir on viral replication of both viral types in target organs of the mice. Three mice each from vehicle-treated and drug-treated groups were killed on days 1, 3, 5, 7, or 10 after inoculation for collection of lung, liver, spleen, kidney, olfactory bulbs, cerebral cortex, pons/medulla, diencephalon, cerebellum, and trigeminal ganglia. Organ samples were pooled by tissue type and homogenized in a 10% wt/vol suspension and frozen until assayed for virus. Virus titers were determined by plating of tissue homogenates on HFF cells, and plaques were enumerated after 3 days incubation. The trigeminal ganglia were collected individually and cocultured directly on primary rabbit kidney cells with N′ N′ dimethylbisacetamide for detection of latent virus as described elsewhere [37]. Briefly, the ganglia were minced, placed onto tissue culture cell monolayers, and monitored for viral cytopathic effects for 3 weeks. Ganglia were transferred weekly onto fresh rabbit kidney cells with new media.
Statistical evaluation. Mortality rates were analyzed by Fisher exact test, and the mean day of death (MDD) results were evaluated using the Mann-Whitney U rank sum test. A P value of ⩽.05 was considered significant when comparing each treated group to the vehicle-treated group. There was no adjustment made for multiple group analysis.
Quantitative drug distribution study. This study was conducted in compliance with USDA Animal Welfare Act regulations, 9CFR 1–4. The procedures in the study were reviewed and approved by the Institutional Animal Care and Use Committee at QPS, LLC. The (C2-14C)-CMX001, monosodium salt (radiochemical purity >98%; Moravek Biochemicals) was administered orally by gavage as a solution (10 mL/kg) to fedstate, male CD-1 albino mice at doses of 2.5, 5, and 10 mg/kg (220–890 µCi/kg). The disposition of radioactivity was quantified using whole-body autoradiography at selected time points up to 72 h (1 animal per time point). After inducing deep anesthesia, each animal was killed at the selected time point. The mouse carcass was imbedded in 2% carboxymethylcellulose, frozen, and sectioned in a Leica CM 3600 cryo-microtome maintained at ∼−20°C. Sagittal animal sections containing all major organs were exposed to Molecular Dynamics phosphor imaging screens along with 14C-blood autoradiographic standards (in triplicate) for subsequent calibration by the image analysis software. Exposed screens were scanned using a Molecular Dynamics Typhoon phosphor imager. The co-exposed autoradiographic standards provided a calibration reference which linked levels of measured density counts to radioactivity per unit mass. Radioactive concentrations in each tissue were converted to µg equivalents/g of tissue, using the known specific activity of the 14C-CMX001 that was administered. When possible, tissues were sampled in representative areas and at several sectioning levels to reduce sampling variance. In these cases, average values were determined and reported. The lower limit of quantitation (LLOQ) was determined for each imaging plate and ranged between 0.013 and 0.016 µg equivalents/g, and the mean LLOQ was 0.015 µg equivalents/g. Reported values below the LLOQ are the mean results obtained using >1 sampling value, of which some were above and below the LLOQ. Values that were below the LLOQ were treated as zero and were included in the mean.
Results
Efficacy studies in vivo. When CMX001 was administered orally to mice infected with HSV-1, mortality was reduced significantly (P ⩽ .001) with all 3 dose levels when treatments were initiated 24 h post viral inoculation (Table 1). When treatments were started 48 h post viral inoculation, 5 and 2.5 mg/kg significantly reduced mortality (P ⩽ .001). If treatments were delayed until 72 h post viral inoculation, CMX001 did not reduce mortality or increase the mean day to death. Although acyclovir was effective at all 3 times of initiation of therapy (P ⩽ .001), ∼10-fold more drug was required.
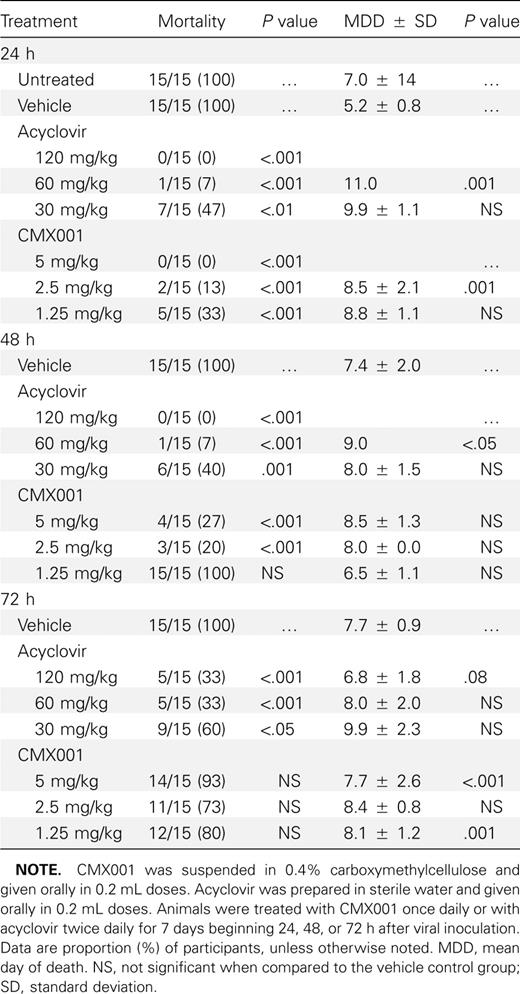
Effect of Treatment with CMX-001 or Acyclovir on the Mortality of BALB/c Mice Inoculated Intranasally with HSV-1 Strain E-377
When CMX001 was administered orally to mice infected with HSV-2,mortality was significantly reduced (P ⩽ .001)with all concentrations when treatments were initiated 24 or 48 h post viral inoculation (Table 2). When treatments were started 72 h post viral inoculation, the 2.5 and 1.25 mg/kg doses significantly reduced mortality (P ⩽ .001). Acyclovir was effective at all 3 dosages and time points (P ⩽ .05) but again required higher dosage levels.
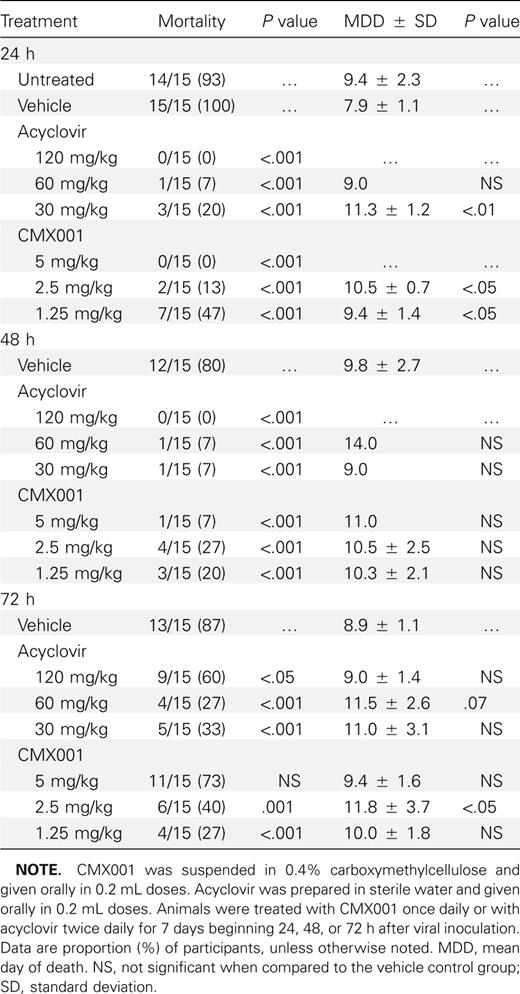
Effect of Treatment with CMX001 or Acyclovir on Mortality of BALB/c Mice Inoculated Intranasally with HSV-2 Strain MS
When mice were infected intranasally with HSV-1 and treatments initiated 24 h post viral inoculation using CMX001 at 5 mg/kg or acyclovir at 100 mg/kg, virus replication in target organs was reduced by both CMX001 and acyclovir when compared with vehicle-treated mice (Figure 1). In lung, liver, kidney, and spleen samples, both CMX001 and acyclovir reduced viral replication below the limits of detection by day 5 post viral inoculation. A 6 log10 PFU/g of tissue reduction in virus titers in the lung, a critical target organ in disseminated disease, was seen when compared with vehicle-treated control mice. In all areas of the brain except the pons/medulla region, treatment with CMX001 reduced viral replication below the limits of detection by day 3 post viral inoculation, which indicated a 3–6 log10 PFU/g of tissue reduction when compared with vehicletreated control mice. Samples taken on day 10 after cessation of treatment also remained below the limits of detection for mice treated with CMX001. In contrast, however, samples obtained from mice treated with acyclovir still had detectable levels of virus present in olfactory bulbs, cerebral cortex, cerebellum, pons-medulla, and diencephalon at various time points during treatment.
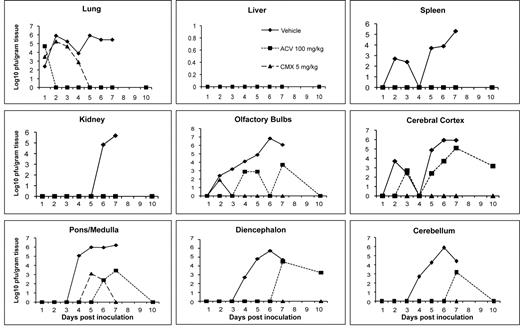
Efficacy of CMX001 or acyclovir (ACV) against HSV-1 replication in target organs of mice. Graphs depict mean viral titers shown as log10 plaqueforming units (PFU)/g of tissue over days 1–10 post viral infection of mice treated 24 h post viral inoculation with vehicle, 5 mg/kg CMX001, or 100 mg/kg ACV. The limit of detection is 1.39 log10 PFU/g of tissue.
There were no statistical differences in viral latency rates as determined by co-cultivation of trigeminal ganglia between groups treated with vehicle, acyclovir, or CMX001 in either HSV-1-infected or HSV-2-infected mice (data not shown). Viral reactivation, as evidenced by the appearance of typical cytopathic effect in rabbit kidney cell monolayers, was observed at an approximately equal rate among treatment groups.
When mice were infected intranasally with HSV-2, and treatments initiated 24 h post viral inoculation using CMX001 at 5 mg/kg or acyclovir at 100 mg/kg, virus titers were reduced in all target organs by both CMX001 and acyclovir when compared with vehicle-treated mice (Figure 2). In lung and liver samples, both CMX001 and acyclovir reduced viral replication below the limits of detection by day 4 post viral inoculation, which indicated a 3 log10 PFU/g of tissue reduction when compared with vehicle-treated control mice. In all areas of the brain, CMX001 reduced viral replication below the limits of detection by day 3 post viral inoculation, which indicated a 4–6 log10 PFU/g of tissue reduction when compared with vehicle-treated control mice. Samples taken on day 10 after cessation of treatment also remained below the limits of detection for mice treated with CMX001. However, samples obtained from mice treated with acyclovir again showed detectable levels of virus present in olfactory bulbs, cerebral cortex, cerebellum, ponsmedulla, and diencephalon at various time points during treatment. In addition, the samples taken on day 10 after cessation of acyclovir treatment had virus quantities that were equal to or exceeded vehicle-treated mice in samples from all 5 regions of the CNS. This result suggested that there may have been a rebound of viral replication in these tissues after cessation of acyclovir treatment that was not observed in mice treated with CMX001.
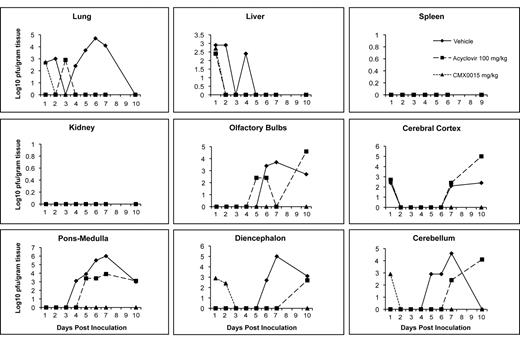
Efficacy of CMX001 or acyclovir (ACV) against HSV-2 replication in target organs of mice. Graphs depict mean viral titers shown as log10 plaque-forming units (PFU)/g of tissue over days 1–10 post viral infection of mice treated 24 h post viral inoculation with vehicle, 5 mg/kg CMX001, or 100 mg/kg ACV. The limit of detection is 1.39 log10 PFU/g of tissue.
Drug distribution studies. After a single oral administration of 5 mg of 14C-CMX001/kg to male mice, drug-associated radioactivity was absorbed from the gastrointestinal tract, and wide distribution to tissues was observed to occur by 2 h post dosing. Drug distribution was widespread at 4 h (Figure 3) and continued through 24 h. The highest levels of drug were detected in the small intestine, and levels in the lung, liver, kidney, and spleen ranged from 0.1 µ to 10 µg equivalents/g of tissue through 24 h post dosing (Figure 4). Moderate to low levels of drug were detected in remaining tissues, and those in which levels were below the limits of quantitation included the brain and spinal cord. However, in mice that received 10 mg of 14C-CMX001/kg, drug concentrations of 0.01–0.02 µg equivalents/g were detected in the CSF, spinal cord, and brain (cerebellum and cerebrum) (Figure 5).
![Whole-body autoradiography showing biodistribution of [C2-14C] CMX001 4 h post oral gavage using 5 mg/kg in male CD mice. The highest concentrations of radioactivity are depicted in red, and in decreasing magnitude in orange, yellow, green, blue, and magenta.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/202/10/10.1086_656717/2/m_202-10-1492-fig003.jpeg?Expires=1716437871&Signature=MyK61-XzP~s2dQCnZUfmJbV6BfygVfAbNZpfqmNy8X6ViNC2EcEGVSFQW~5uT2HLrkp-F4SRVZ1lxsGQPww2OvpdSO4FSelKLT97k-0RVjg21m8jqGkXVDzU9UBiolxpHjGC78Ac~duWniZcsQUZcjt8DE-0uLMkwgBDzLG3-nZnBs~1aZvFzimRbyDtejDMpn5~SmSsZG-JhhFbuHjT~OW2OrY~h4f2Aih73SA3wadyoANUF-Aw8L5xX9FdpzRTJ3DVjezvaeun87OCYex8hg3dy9IW0MG5lxFZkV2EXaP4drIb9xfMkBmUYwNmKpfmW5w2laA6GTlO4KCgqvIaww__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Whole-body autoradiography showing biodistribution of [C2-14C] CMX001 4 h post oral gavage using 5 mg/kg in male CD mice. The highest concentrations of radioactivity are depicted in red, and in decreasing magnitude in orange, yellow, green, blue, and magenta.
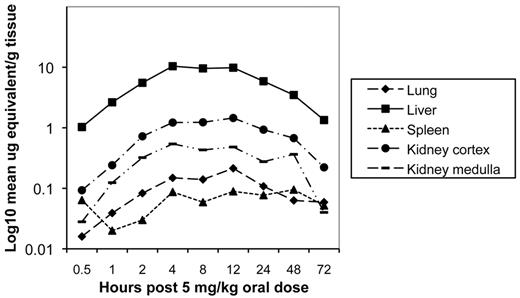
Concentration of radioactive-labeled drug in visceral organs at specified times post dose determined by whole-body autoradiography after a single oral administration of 5 mg of 14C-CMX001/kg in male mice. Quantities depicted as log10 mean mg equivalents/g of tissue.
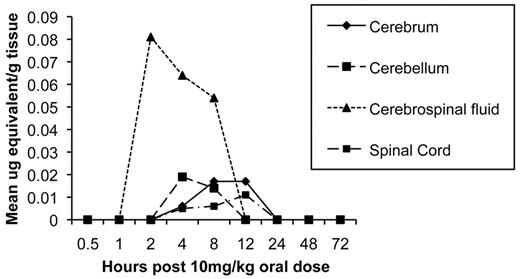
Concentration of radioactive-labeled drug in central nervous system (CNS) tissues at specified times post dose determined by wholebody autoradiography after a single oral administration of 10 mg of 14C-CMX001/ kg in male mice. Quantities depicted as mean µg equivalents/g of tissue.
Discussion
HSV infections of the CNS remain a significant cause of morbidity and mortality in humans, in spite of therapy with acyclovir. For example, with neonatal HSV-2 infections of the CNS, the mortality is low at only 5%, but well over 50% of survivors suffer from significant neurologic sequelea [12]. Similarly, with HSV-1 encephalitis of older children and adults, the mortality increases to 25% and the morbidity to >50%[5]. Thus, new therapeutic approaches are necessary to improve the outcome of these devastating infections. The current studies contained in this communication indicate that CMX001 may provide an alternative to acyclovir either by itself or in combination with acyclovir.
Historically, intranasal infections in mice using HSV-1 have been considered to be an excellent model for human herpes encephalitis because it uses a natural route of infection and has been predictive for the efficacy of both vidarabine and acyclovir in treatment of human HSV infections [20–21, 38]. Viral replication, which first occurs in the nasopharynx and then spreads to the CNS by olfactory and trigeminal nerves, has served to model both adult encephalitis and neonatal CNS disease. Viral replication is first detected in the olfactory bulbs and spreads throughout the CNS by neural routes from the day after inoculation until death by day 7 in untreated mice. Intranasal inoculation of BALB/c mice with HSV-2 has been used as a model of disseminated neonatal herpes with CNS involvement occurring later after infection and an extended mean day to death of ∼9 days. These studies illustrate the importance of using both HSV-1 and HSV-2 in efficacy evaluations because HSV encephalitis in adults is predominantly due to HSV-1, and in neonates ∼70% of cases are due to HSV-2. These models have been predictive for efficacy of vidarabine and acyclovir in treatment of both HSV type-1 and HSV-2 infections in humans.
Despite being a potent inhibitor of HSV replication in vitro and in experimental animal infections, cidofovir has not been used for the treatment of HSV infections in humans due to its lack of oral activity [39] and its nephrotoxicity [40]. However, the drug has been used for the treatment of acyclovir-resistant HSV infections [41]. Several lipid conjugates of cidofovir have been synthesized that are active orally in animals, have reduced toxicity, and have multifold enhanced activity against a variety of viruses including a number of the herpesviruses [24, 30]. The enhanced activity of the lipid conjugates appear to be due to their greater uptake into cells, possibly including the CNS, resulting in a significant increase in the amount of intracellular cidofovir [42]. The conjugate that has received the most interest is the hexadecyloxypropyl analog of cidofovir, originally termed HDP-CDV, which is currently under development as CMX001. The interest in this molecule has been in part due to its being highly active against orthopoxviruses, which are suspected agents of bioterror.
In previous studies using cell cultures, CMX001 was about 100-fold more potent as an inhibitor of HSV replication than acyclovir [30]. In the experimental infections presented in these studies, CMX001 was highly effective in preventing mortality in mice infected with HSV-1 or HSV-2 when treatments were delayed until 48 or 72 h post viral inoculation, respectively, by reducing viral replication in the CNS as well as in lung, liver, spleen, and kidney. Importantly, CMX001 was more efficacious than acyclovir in reducing viral replication in the CNS in mice infected with either HSV-1 or HSV-2. Additionally, no virus replication was detected in tissues after cessation of therapy with CMX001, whereas a rebound in virus replication was observed after cessation of therapy with acyclovir. In these studies, CMX001 given at 5 mg/kg once daily was more efficacious than acyclovir at 100 mg/kg given twice daily and suggests that CMX001 may have potential for use in the treatment of herpes encephalitis, neonatal herpes, or other severe HSV infections in humans.
When CMX001 is delivered orally, it has a very unique tissue distribution profile compared with parenterally administered cidofovir, which does not cross the blood-brain barrier. High levels of drug were found very early in lung, liver, and spleen but not kidney. CMX001 had little effect on the microtubules of the kidney and no apparent toxicity [43]. When mice were dosed with 5 mg of CMX001/kg in a quantitative drug distribution study, the levels in the CNS were below the level of quantitation; however, detectable levels were observed in mice given 10 mg/kg of drug. In the antiviral studies reported in this paper, the highest dose administered was 5 mg/kg once daily for 5 days. Given that the half-life of the activated antiviral form of CMX001, cidofovir-diphosphate, is 6.5 days (unpublished results), the drug may accumulate in the CNS after 7 daily doses and reach levels in excess of those seen in the distribution study after a single 10 mg/kg dose. In these studies, CMX001 had a significant effect on HSV replication in the CNS when therapy was delayed until 24 h post viral inoculation. This would suggest that only a small of amount of drug is needed in the CNS, given its in vitro potency of ∼0.03 µg/mL and/or that viral replication is also being inhibited in peripheral tissues and preventing spread to the CNS. The results obtained in these studies suggest that there is, in fact, virus already replicating in parts of the CNS when therapy was initiated and that deliverable drug can, in fact, inhibit replication in the CNS, resulting in total clearance of virus from the CNS. The lack of viral persistence after treatment with CMX001 would be in contrast to previous studies showing that mice treated with valacyclovir had persistent virus detected in trigeminal ganglia and brain stem tissues [44]. It should also be pointed out that the drug distribution studies were carried out in uninfected mice, and it is certainly feasible that in infected mice additional amounts of drug may have crossed compromised blood-brain or blood-cerebral spinal fluid barriers compared with those observed in uninfected mice. In previous human studies, acyclovir provided as oral valacyclovir using 1 gram 3 times daily provided 20% of the serum concentration entering the CSF with active transport out of the CSF compartment [45].
Considering the continuing mortality and long-term sequelae of adults and infants infected with HSV and treated aggressively with acyclovir, CMX001 holds promise for improved outcomes. Its unique cellular uptake and biodistribution may prove more beneficial in particular with clinical cases of encephalitis. In addition, combination studies with CMX001 and acyclovir both in vitro and in vivo have been carried out, and a synergistic interaction without additive toxicity has been demonstrated [46]. This combination may considerably improve the outcome and long-term well-being of patients infected with these and other herpesviruses and should be considered for use in these infections.
Acknowledgments
The authors thank Dr Richard J. Whitley for his expert review of the manuscript. The authors gratefully acknowledge the expertise of QPS, LLC, in the performance of the whole-body autoradiographic images shown in Figure 3 and on the cover of this issue.
References
Potential conflicts of interest: E.R.K. has an equity interest and serves as a consultant to Chimerix, Inc. The terms of this arrangement have been reviewed and approved by the University of Alabama at Birmingham in accordance with their conflict of interest policies.
Financial support: National Institutes of Health (NIH) National Institute of Allergy and Infectious Diseases (NIAID) (contracts NOI-AI-15439 and NOI-AI-30049) to the University of Alabama at Birmingham.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}