




Abstract
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are both relentlessly progressive and ultimately fatal neurological disorders. ALS is familial in ∼10% of cases and FTD in ∼30%. Inheritance is usually autosomal dominant with variable penetrance. Phenotypic overlap between ALS and FTD can occur within the same kindred. Mutations in copper/zinc superoxide dismutase 1 (SOD1) are found in ∼20% of familial and ∼3% of sporadic ALS cases but are not associated with dementia. Mutations in microtubule associated protein tau (MAPT) are detected in ∼30% of familial FTD kindreds. Dominant ALS with FTD has previously been linked to 9q21 and pure ALS to loci on 16q21, 18q21, 20p13. Here we report the results of a genome-wide linkage study in a large ALS and FTD kindred using Affymetrix 10K GeneChip microarrays. Linkage analysis of single nucleotide polymorphism (SNP) data identified consistently positive log of the odds (LOD) scores across chromosome 9p (maximal LOD score of 2.4). Fine mapping the region with microsatellite markers generated a maximal multipoint LOD score of 3.02 (𝛉 = 0) at D9S1878. Recombination narrowed the conserved haplotype to 12 cM (11 Mb) at 9p13.2–21.3 (flanking markers D9S2154 and D9S1874). Bioinformatic analysis of the region has identified 103 known genes.
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are both relentlessly progressive and ultimately fatal neurological disorders. There is increasing evidence that the two conditions can exist in a phenotypic spectrum within individuals and within autosomal dominant kindreds (Lipton, 2004). In ALS, motoneurons in the motor cortex, brainstem and spinal cord degenerate. Muscle wasting, weakness and spasticity usually starts in one limb but over time becomes generalized, leading to profound global paralysis. Death due to respiratory failure occurs on average only 3 years after symptom onset and there is no therapy that significantly alters the course of the disease (Shaw et al., 2001). The pathological hallmark of ALS is the presence of ubiquitinated inclusions in the perikaryon and proximal axon of surviving motoneurons, but the underlying disease mechanisms are poorly understood (Leigh, 1989). The majority of cases are sporadic, but in ∼10% of patients there is a positive family history (FALS), typically with an autosomal dominant pattern of inheritance and reduced penetrance (Strong, 1991). With few exceptions, familial and sporadic ALS are indistinguishable on clinical and pathological grounds (Shaw et al., 1997; Ince et al., 1998).
FTD is characterized at the outset by a personality change and socially inappropriate behaviour, with relative preservation of memory and cognitive functions. Language defects are common and may present with a semantic dementia or non-fluent aphasia (Hodges, 2001). Eventually, there is a more global cognitive deficit and death often due to immobility and respiratory infection. Survival of pathologically proven FTD is, on average, only 4 years from symptom onset (Rascovsky, 2005). Pathologically, neurons in the superficial frontal cortex and anterior temporal lobes degenerate. The molecular pathology of FTD has been divided into at least three groups: a tauopathy, with neurofibrillary tangles; ALS-like, with ubiquitinated and neurofilamentous inclusions and the remainder lacking distinctive histopathological features (Piguet, 2004). In ∼40% of FTD cases, there is a family history of dementia, indicating a significant genetic contribution (Rosso, 2003). In many sporadic and familial FTD cases there is clinical and pathological evidence of an overlap with ALS (Lipton, 2004).
Linkage to chromosome 21q22 (Siddique et al., 1991) led to the identification of mutations in the Cu/Zn superoxide dismutase (SOD) gene in ALS (SOD1, ALS1 OMIM 105400) (Rosen, 1993). To date, over 109 SOD1 mutations have been reported and can be detected in 20% of FALS and 3% of sporadic ALS cases (Shaw et al., 1997; Andersen et al., 2003). Mice transgenic for several different human SOD1 mutants develop progressive motoneuron degeneration through a toxic gain of function (Gurney et al., 1994) unrelated to catalytic activity (Wang et al., 2003). Although mutant SOD1 is ubiquitously expressed at high levels in many tissues in patients and transgenic mice, degeneration is largely confined to motoneurons. Detergent-resistant SOD1 aggregates are detected in the spinal cord and enriched in their mitochondrial fractions (Jonsson et al., 2004; Liu et al., 2004). It has therefore been postulated that protein misfolding, mitochondrial dysfunction and apoptosis may play a mechanistic role in the pathogenesis of ALS (Liu et al., 2004).
SOD1 has provided only one piece of the molecular jigsaw puzzle, and progress in identifying other autosomal dominant ALS genes has been slow. Linkage to 20q13 was identified in a large Brazilian kindred with a slowly progressive adult-onset form of spinal muscular atrophy (ALS8 OMIM 608627) (Nishimura et al., 2004a). Mutations were subsequently identified in the gene-encoding vesicle-associated membrane protein B (VAPB) in the original and six other families (Nishimura et al., 2004b). One of the VAPB kindreds has a phenotype closer to ALS, with signs of upper and lower motoneuron degeneration and a more rapid disease course.
One large Italian kindred with classical ALS affecting 20 individuals has been confidently linked to a locus on 18q21 with a maximal multipoint log of the odds (LOD) score of 4.5 and haplotype spanning 8 Mb, but a pathogenic mutation has not yet been identified (ALS3, OMIM 606640) (Hand et al., 2002). Another kindred demonstrated linkage to 20p13 with a maximal multipoint LOD score of 3.46 and a shared haplotype of ∼1 Mb (ALS7, OMIM 608031) (Sapp et al., 2003), but the strength of this linkage rests very much on the parameters defining penetrance.
Kindreds with a phenotype encompassing features of FTD, parkinsonism and ALS have been linked to 17q21 and mutations have been identified in the microtubule associated protein tau (MAPT) (OMIM 157041) (Hutton et al., 1998). Tau mutations can be detected in 25–30% of familial FTD kindreds (Rosso, 2003; Stanford, 2004), but pathological mutations in tau have not yet been described in kindreds with pure ALS. Some families with dominant ALS have a phenotype that overlaps with FTD. A locus for ALS–FTD has previously been reported in five families at 9q21 (OMIM 105550) (Hosler, 2000), but no pathogenic mutations have been reported to date. The clinical phenotype and genetic basis of the juvenile-onset motoneuron disorders are also quite distinct, and no linkage or association has been shown with classical adult-onset ALS for ALS2 (2q33, OMIM 205100) (Hentati et al., 1994), ALS4 (9q34, OMIM 602433) (Chance et al., 1998) or ALS5 (15q15–21, OMIM 602099) (Hentati et al., 1998).
We have previously published the results of a genome-wide microsatellite marker scan in a large English family (F1), which demonstrated linkage to chromosome 16q12 with a two-point LOD score of 3.61 (Ruddy et al., 2003). Concurrently, two other unrelated families were published demonstrating linkage to an overlapping region on 16q12 with multipoint LOD scores of 3.29 (Sapp et al., 2003) and 2.06 (Abalkhail, 2003), and the locus has been designated ALS6 (OMIM 608030). In our initial report we described a second family (F2), which showed weaker linkage to the same region with a two-point LOD score of 1.84 (Ruddy et al., 2003). Subsequent to that publication, two members of the F2 kindred have developed ALS. Neither shares the linked chromosome 16 haplotype with the earlier affected individuals, nor the other newly affected person. Here we present data from a genome-wide scan in this kindred using single nucleotide polymorphism (SNP) arrays and fine mapping using microsatellite (STS) markers, which provides convincing evidence of a novel locus on chromosome 9 for autosomal dominant ALS with features of FTD.
Patients and methods
Kindred description and clinical phenotype
A large Dutch kindred with familial ALS was identified and followed over a 20-year period (Fig. 1). There was no consanguinity and the inheritance is consistent with an autosomal dominant pattern with reduced penetrance (∼40% by the age of 70) as several obligate carriers lived until their late 70 s). All but one affected, and many unaffected individuals, were examined by one consultant neurologist (VdeJ). The diagnosis of ALS in the unseen individual (II:5) was made in retrospect by her relatives and from written correspondence, which clearly describes a rapidly progressive muscle wasting illness. Nine individuals in ALS in generations II and III had upper and lower motoneuron signs in at least three clinical regions and fulfilled the El-Escorial Criteria of ‘Clinically Definite ALS’ (Brooks et al., 2000) (Table 1). Electromyography (EMG) confirmed muscle denervation, without evidence of conduction block in all eight individuals who presented with ALS and were tested. The site of disease onset was evenly split between bulbar (five) and limb (five). Variability was seen in the age at onset (mean of 62 years, range 40–72) and survival (mean of 36 months, range 12–90).
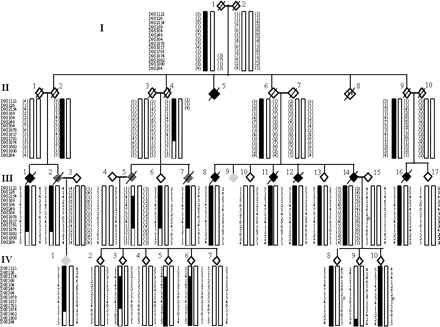
The diagram is a greatly abbreviated depiction of this kindred. Generation II contained 18 individuals (II:8 represents 13 siblings, two of whom died in infancy) and not all siblings are shown in generations III and IV. Four branches of the family in these generations have developed ALS and FTD. Symbols: pure ALS depicted in black, ALS and FTD combined by dark grey and FTD predominant by light grey. Critical recombination events occur in the recreated individual III5. They are robust as the centromeric crossover is also present in two siblings (III:6 and III:7) and the telomeric crossover is also present in three offspring (IV:3, IV:5 and IV:6).
Clinical features and investigation results of affected individuals from Family F2 with ALS–FTD
| Id | Gender | Age at onset (years) | Survival (months) | Site at onset | El Escorial category | Bulbar symptoms | Psychiatric symptoms | Denervation on EMG | ALS on autopsy | FTD on autopsy |
|---|---|---|---|---|---|---|---|---|---|---|
| II:5 | F | 61 | 12 | Bulbar | ? | ? | ? | N/A | N/A | N/A |
| III:1 | F | 70 | 32 | Bulbar | Definite | + | − | Y | +++ | ++ |
| III:2 | M | 72 | 33 | Hand | Definite | + | + | Y | +++ | − |
| III:5 | F | 67 | 19 | Bulbar | Definite | + | + | Y | N/A | N/A |
| III:7 | F | 67 | 36 | Leg | Definite | + | + | Y | N/A | N/A |
| III:8 | F | 63 | 34 | Bulbar | Definite | + | − | Y | +++ | − |
| III:11 | M | 58 | 36 | Bulbar | Definite | + | − | N/A | N/A | N/A |
| III:12 | M | 67 | 36 | Leg | Definite | + | − | Y | N/A | N/A |
| III:14 | F | 40 | 92 | Hand | Definite | + | − | Y | N/A | N/A |
| III:16 | M | 59 | 32 | Hand | Definite | + | − | Y | +++ | − |
| IV:1 | F | 39 | 31 | Behaviour | Possible | + | + | Normal | + | ++ |
| Id | Gender | Age at onset (years) | Survival (months) | Site at onset | El Escorial category | Bulbar symptoms | Psychiatric symptoms | Denervation on EMG | ALS on autopsy | FTD on autopsy |
|---|---|---|---|---|---|---|---|---|---|---|
| II:5 | F | 61 | 12 | Bulbar | ? | ? | ? | N/A | N/A | N/A |
| III:1 | F | 70 | 32 | Bulbar | Definite | + | − | Y | +++ | ++ |
| III:2 | M | 72 | 33 | Hand | Definite | + | + | Y | +++ | − |
| III:5 | F | 67 | 19 | Bulbar | Definite | + | + | Y | N/A | N/A |
| III:7 | F | 67 | 36 | Leg | Definite | + | + | Y | N/A | N/A |
| III:8 | F | 63 | 34 | Bulbar | Definite | + | − | Y | +++ | − |
| III:11 | M | 58 | 36 | Bulbar | Definite | + | − | N/A | N/A | N/A |
| III:12 | M | 67 | 36 | Leg | Definite | + | − | Y | N/A | N/A |
| III:14 | F | 40 | 92 | Hand | Definite | + | − | Y | N/A | N/A |
| III:16 | M | 59 | 32 | Hand | Definite | + | − | Y | +++ | − |
| IV:1 | F | 39 | 31 | Behaviour | Possible | + | + | Normal | + | ++ |
Clinical features and investigation results of affected individuals from Family F2 with ALS–FTD
| Id | Gender | Age at onset (years) | Survival (months) | Site at onset | El Escorial category | Bulbar symptoms | Psychiatric symptoms | Denervation on EMG | ALS on autopsy | FTD on autopsy |
|---|---|---|---|---|---|---|---|---|---|---|
| II:5 | F | 61 | 12 | Bulbar | ? | ? | ? | N/A | N/A | N/A |
| III:1 | F | 70 | 32 | Bulbar | Definite | + | − | Y | +++ | ++ |
| III:2 | M | 72 | 33 | Hand | Definite | + | + | Y | +++ | − |
| III:5 | F | 67 | 19 | Bulbar | Definite | + | + | Y | N/A | N/A |
| III:7 | F | 67 | 36 | Leg | Definite | + | + | Y | N/A | N/A |
| III:8 | F | 63 | 34 | Bulbar | Definite | + | − | Y | +++ | − |
| III:11 | M | 58 | 36 | Bulbar | Definite | + | − | N/A | N/A | N/A |
| III:12 | M | 67 | 36 | Leg | Definite | + | − | Y | N/A | N/A |
| III:14 | F | 40 | 92 | Hand | Definite | + | − | Y | N/A | N/A |
| III:16 | M | 59 | 32 | Hand | Definite | + | − | Y | +++ | − |
| IV:1 | F | 39 | 31 | Behaviour | Possible | + | + | Normal | + | ++ |
| Id | Gender | Age at onset (years) | Survival (months) | Site at onset | El Escorial category | Bulbar symptoms | Psychiatric symptoms | Denervation on EMG | ALS on autopsy | FTD on autopsy |
|---|---|---|---|---|---|---|---|---|---|---|
| II:5 | F | 61 | 12 | Bulbar | ? | ? | ? | N/A | N/A | N/A |
| III:1 | F | 70 | 32 | Bulbar | Definite | + | − | Y | +++ | ++ |
| III:2 | M | 72 | 33 | Hand | Definite | + | + | Y | +++ | − |
| III:5 | F | 67 | 19 | Bulbar | Definite | + | + | Y | N/A | N/A |
| III:7 | F | 67 | 36 | Leg | Definite | + | + | Y | N/A | N/A |
| III:8 | F | 63 | 34 | Bulbar | Definite | + | − | Y | +++ | − |
| III:11 | M | 58 | 36 | Bulbar | Definite | + | − | N/A | N/A | N/A |
| III:12 | M | 67 | 36 | Leg | Definite | + | − | Y | N/A | N/A |
| III:14 | F | 40 | 92 | Hand | Definite | + | − | Y | N/A | N/A |
| III:16 | M | 59 | 32 | Hand | Definite | + | − | Y | +++ | − |
| IV:1 | F | 39 | 31 | Behaviour | Possible | + | + | Normal | + | ++ |
Three individuals, who presented with motor symptoms of ALS, subsequently developed personality and behavioural features during their illness, but none underwent formal psychometric testing. III:2 became progressively apathetic, cognitively impaired and socially isolated and was found to have frontotemporal atrophy at autopsy. Another, III:5, became increasingly self-centred, unreasonable, emotionally labile and uncooperative, which was not attributed to mood disturbance. The third, III:7, developed paranoid visual and auditory hallucinations with inappropriate behaviour. Although mild cognitive and verbal fluency problems are not uncommon in ALS, major behavioural and psychotic symptoms are highly unusual and may reflect frontotemporal dysfunction.
One individual, IV:1, the offspring of the affected III:2, presented at the age of 39 with a progressive dementia and died of respiratory failure after 31 months. Her husband reported that she began behaving in a childish manner, swearing and neglecting her household duties, had lost interest in her surroundings and had taken up smoking—all of these behaviours were uncharacteristic. She was admitted to a psychiatric hospital for investigation. Examination, 18 months after symptom onset, described a ‘wide-eyed, agitated, affectively a childlike woman who perseverates. She has a very short span of attention and repeatedly asks to be allowed to go out and smoke cigarettes. She cries several times briefly but without tears. She lacks insight and thinks that she is in the mental hospital because of her nervousness at home’. Muscle wasting was present in the interossei of both hands and fasciculations were noted in the arms, hands and thighs. Deep tendon reflexes were within normal limits but Hoffmann's sign was strongly positive bilaterally. Plantar responses were flexor. Limited EMG examination at this time was normal. Anti-neuronal autoantibody screen was negative but MRI of the brain revealed striking frontal lobe atrophy (Fig. 2). Examination 6 months later revealed an emaciated woman with little spontaneous speech. Grasp reflexes were negative, but snout, pout and jaw jerk reflexes were positive with tongue and limb fasciculation. Deep tendon reflexes in the limbs were all exaggerated with an extensor left plantar response. The clinical diagnosis was of FTD and ALS.
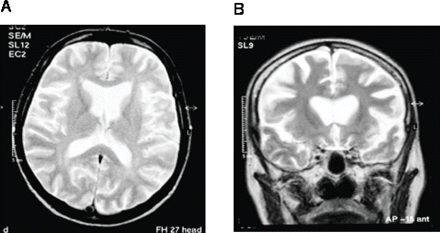
Brain MRI of a 40-year-old woman with an 18-month history of a progressive behavioral, language and cognitive disorder (individual IV:1). (A) Axial T2-weighted image showing symmetrical widening of the frontal lobe sulci, sylvian fissures and anterior horns of the lateral ventricles with relative sparing of the posterior cerebrum. (B) Coronal T2-weighted image showing striking frontal lobe atrophy and an absence of signal change in the white matter of both hemispheres.
One other individual (III:9) died from dementia and may well have had FTD without clinical signs of ALS. He presented with fearfulness, irascibility and violent behaviour, leading to his admission to a psychiatric hospital. He became increasingly withdrawn and unresponsive and eventually died as an inpatient. Until just prior to his death he had been ambulant and could speak, eat and use his hands. As the information could not be verified from medical records and historical details were incomplete, we have not included him in Table 1.
Two-point and multipoint LOD scores between the disease locus and chromosome 9p markers
| Marker | Genetic position | Two-point LOD scores | Multipoint | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (cM) | θ = 0 | θ = 0.1 | θ = 0.2 | θ = 0.3 | θ = 0.4 | LOD scores | |||||
| 1121 | 37.0 | 1.65 | 1.24 | 0.82 | 0.42 | 0.12 | 0.97 | ||||
| 126 | 37.1 | 0.51 | 0.34 | 0.20 | 0.09 | 0.02 | 0.99 | ||||
| 2154 | 42.0 | −0.1 | 0.28 | 0.23 | 0.12 | 0.03 | 0.92 | ||||
| 169 | 42.2 | 1.97 | 1.47 | 0.95 | 0.47 | 0.12 | 2.69 | ||||
| 104 | 42.9 | 2.19 | 1.71 | 1.20 | 0.68 | 0.21 | 2.96 | ||||
| 248 | 44.8 | 1.76 | 1.32 | 0.86 | 0.44 | 0.12 | 3.02 | ||||
| 304 | 48.9 | 2.14 | 1.73 | 1.26 | 0.75 | 0.25 | 3.01 | ||||
| 1878 | 49.6 | 2.41 | 1.9 | 1.34 | 0.76 | 0.24 | 3.02 | ||||
| 1817 | 49.7 | 2.08 | 1.59 | 1.08 | 0.58 | 0.17 | 3.02 | ||||
| 1791 | 51.8 | 2.23 | 1.73 | 1.20 | 0.67 | 0.20 | 2.77 | ||||
| 1874 | 52.3 | 0.17 | 0.65 | 0.50 | 0.27 | 0.08 | 0.70 | ||||
| 1862 | 52.4 | 0.38 | 0.8 | 0.66 | 0.4 | 0.13 | 0.62 | ||||
| 1800 | 60.9 | −0.89 | −0.41 | −0.15 | −0.04 | −0.01 | 0.49 | ||||
| 284 | 67.2 | −0.38 | 0.08 | 0.13 | 0.09 | 0.03 | 0.64 | ||||
| Marker | Genetic position | Two-point LOD scores | Multipoint | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (cM) | θ = 0 | θ = 0.1 | θ = 0.2 | θ = 0.3 | θ = 0.4 | LOD scores | |||||
| 1121 | 37.0 | 1.65 | 1.24 | 0.82 | 0.42 | 0.12 | 0.97 | ||||
| 126 | 37.1 | 0.51 | 0.34 | 0.20 | 0.09 | 0.02 | 0.99 | ||||
| 2154 | 42.0 | −0.1 | 0.28 | 0.23 | 0.12 | 0.03 | 0.92 | ||||
| 169 | 42.2 | 1.97 | 1.47 | 0.95 | 0.47 | 0.12 | 2.69 | ||||
| 104 | 42.9 | 2.19 | 1.71 | 1.20 | 0.68 | 0.21 | 2.96 | ||||
| 248 | 44.8 | 1.76 | 1.32 | 0.86 | 0.44 | 0.12 | 3.02 | ||||
| 304 | 48.9 | 2.14 | 1.73 | 1.26 | 0.75 | 0.25 | 3.01 | ||||
| 1878 | 49.6 | 2.41 | 1.9 | 1.34 | 0.76 | 0.24 | 3.02 | ||||
| 1817 | 49.7 | 2.08 | 1.59 | 1.08 | 0.58 | 0.17 | 3.02 | ||||
| 1791 | 51.8 | 2.23 | 1.73 | 1.20 | 0.67 | 0.20 | 2.77 | ||||
| 1874 | 52.3 | 0.17 | 0.65 | 0.50 | 0.27 | 0.08 | 0.70 | ||||
| 1862 | 52.4 | 0.38 | 0.8 | 0.66 | 0.4 | 0.13 | 0.62 | ||||
| 1800 | 60.9 | −0.89 | −0.41 | −0.15 | −0.04 | −0.01 | 0.49 | ||||
| 284 | 67.2 | −0.38 | 0.08 | 0.13 | 0.09 | 0.03 | 0.64 | ||||
Two-point and multipoint LOD scores between the disease locus and chromosome 9p markers
| Marker | Genetic position | Two-point LOD scores | Multipoint | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (cM) | θ = 0 | θ = 0.1 | θ = 0.2 | θ = 0.3 | θ = 0.4 | LOD scores | |||||
| 1121 | 37.0 | 1.65 | 1.24 | 0.82 | 0.42 | 0.12 | 0.97 | ||||
| 126 | 37.1 | 0.51 | 0.34 | 0.20 | 0.09 | 0.02 | 0.99 | ||||
| 2154 | 42.0 | −0.1 | 0.28 | 0.23 | 0.12 | 0.03 | 0.92 | ||||
| 169 | 42.2 | 1.97 | 1.47 | 0.95 | 0.47 | 0.12 | 2.69 | ||||
| 104 | 42.9 | 2.19 | 1.71 | 1.20 | 0.68 | 0.21 | 2.96 | ||||
| 248 | 44.8 | 1.76 | 1.32 | 0.86 | 0.44 | 0.12 | 3.02 | ||||
| 304 | 48.9 | 2.14 | 1.73 | 1.26 | 0.75 | 0.25 | 3.01 | ||||
| 1878 | 49.6 | 2.41 | 1.9 | 1.34 | 0.76 | 0.24 | 3.02 | ||||
| 1817 | 49.7 | 2.08 | 1.59 | 1.08 | 0.58 | 0.17 | 3.02 | ||||
| 1791 | 51.8 | 2.23 | 1.73 | 1.20 | 0.67 | 0.20 | 2.77 | ||||
| 1874 | 52.3 | 0.17 | 0.65 | 0.50 | 0.27 | 0.08 | 0.70 | ||||
| 1862 | 52.4 | 0.38 | 0.8 | 0.66 | 0.4 | 0.13 | 0.62 | ||||
| 1800 | 60.9 | −0.89 | −0.41 | −0.15 | −0.04 | −0.01 | 0.49 | ||||
| 284 | 67.2 | −0.38 | 0.08 | 0.13 | 0.09 | 0.03 | 0.64 | ||||
| Marker | Genetic position | Two-point LOD scores | Multipoint | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (cM) | θ = 0 | θ = 0.1 | θ = 0.2 | θ = 0.3 | θ = 0.4 | LOD scores | |||||
| 1121 | 37.0 | 1.65 | 1.24 | 0.82 | 0.42 | 0.12 | 0.97 | ||||
| 126 | 37.1 | 0.51 | 0.34 | 0.20 | 0.09 | 0.02 | 0.99 | ||||
| 2154 | 42.0 | −0.1 | 0.28 | 0.23 | 0.12 | 0.03 | 0.92 | ||||
| 169 | 42.2 | 1.97 | 1.47 | 0.95 | 0.47 | 0.12 | 2.69 | ||||
| 104 | 42.9 | 2.19 | 1.71 | 1.20 | 0.68 | 0.21 | 2.96 | ||||
| 248 | 44.8 | 1.76 | 1.32 | 0.86 | 0.44 | 0.12 | 3.02 | ||||
| 304 | 48.9 | 2.14 | 1.73 | 1.26 | 0.75 | 0.25 | 3.01 | ||||
| 1878 | 49.6 | 2.41 | 1.9 | 1.34 | 0.76 | 0.24 | 3.02 | ||||
| 1817 | 49.7 | 2.08 | 1.59 | 1.08 | 0.58 | 0.17 | 3.02 | ||||
| 1791 | 51.8 | 2.23 | 1.73 | 1.20 | 0.67 | 0.20 | 2.77 | ||||
| 1874 | 52.3 | 0.17 | 0.65 | 0.50 | 0.27 | 0.08 | 0.70 | ||||
| 1862 | 52.4 | 0.38 | 0.8 | 0.66 | 0.4 | 0.13 | 0.62 | ||||
| 1800 | 60.9 | −0.89 | −0.41 | −0.15 | −0.04 | −0.01 | 0.49 | ||||
| 284 | 67.2 | −0.38 | 0.08 | 0.13 | 0.09 | 0.03 | 0.64 | ||||
Pathological description
Five individuals underwent detailed pathological assessment; at post-mortem significant upper and lower motoneuron degeneration was detected in all four cases who presented with ALS. Ubiquitinated inclusions were detected in the anterior horn cells in three of these individuals. Bunina bodies were detected in the fourth case and one other who had Hirano bodies and mild frontoparietal atrophy. One individual, who died from ALS at age 74 with no psychiatric symptoms, had occasional neurofibrillary tau tangles, which were consistent with age. Interestingly, in this patient the granular layer of the hippocampus showed ubiquitin-positive (and tau-negative) inclusions that are characteristic for ALS-FTD. No other cases had tau-positive inclusions. Post-mortem examination of the woman who presented with FTD (IV:1) showed striking atrophy of the frontal lobes, precentral gyri and anterior roots of the spinal cord. Light microscopy revealed occasional inclusion bodies in Betz cells of the motor cortex and neuronophagia. Ubiquitinated and nuerofilamentous inclusions were frequent in the granular cell layer of the hippocampi, with features of neurodegeneration and microglial activation. Tau staining revealed no Pick bodies, neuritic tangles or other tau immunoreactive inclusions. Motoneurons in the brainstem nuclei and spinal cord were shrunken with axonal swellings and reactive gliosis, but no inclusions. Muscle pathology confirmed neurogenic atrophy. The findings were reported as being typical of FTD and ALS.
Recruitment and initial mutation screening and linkage to known loci
Family members were contacted by the attending neurologist and nurse. Following informed consent, blood was drawn from 148 family members. Ethical committees in both clinical centres have approved genetic research on ALS. Blood samples were available from eight affected individuals, and two more affected individuals could be recreated by genotyping their unrelated spouses and offspring. DNA was extracted and mutations excluded in two affected individuals by direct sequencing of the coding and flanking sequences of SOD1 (Shaw et al., 1998) and VAPB (Nishimura et al., 2004b). All the exons and intron/exon boundaries of gene encoding MAPT were screened for mutations by direct sequencing in two individuals (Rizzu, 1999). Furthermore, linkage to the MAPT locus at chromosome 17q21 was confidently excluded by SNP linkage analysis, which generated LOD scores of −5.0 (see graphs in Supplementary material). Linkage to the other published loci—ALS3 at 18q21, ALS6 at 16q12, ALS7 at 20p13 and ALS–FTD at 9q21—was excluded by genotyping the affected individuals for informative microsatellite makers spanning these regions (data not shown) and later confirmed by SNP linkage analysis.
Bioinformatics
The most informative members of the family for linkage were identified using Slink (Terwilliger, 1993) and modelled in a genome-wide screen using the following assumptions: autosomal dominant inheritance with age-dependent penetrance, no phenocopies, a disease gene frequency of 0.00001 and equal marker allele frequencies. Liability classes were based on previously published reports (Hand et al., 2001; Ruddy et al., 2002) and refined specifically to account for the age-dependent penetrance of this family: (i) age < 46 years—20% (ii) age > 46 years—40%; and (iii) spouses were given the background population risk. Ages were defined as the age at onset in affecteds and the age at last review of asymptomatic individuals. These assumptions were applied to linkage analysis of STS markers using Mlink (Goldgar, 1992) and Allegro (Gudbjartsson, 2000). Non-parametric linkage (NPL) analysis of SNP genotypes was performed using Merlin (Abecasis et al., 2002). Because of the size of the pedigree, it was initially split by setting individual II:6 (Fig. 1) as a founder and run as two separate pedigrees and the scores were added. LOD scores greater than 0.8 were regarded as requiring further investigation. Genome-wide multipoint analysis of SNP data for a reduced pedigree, containing affected individuals only, was performed also using Merlin. In these regions, STS markers were genotyped and analysed using MLink, and Allegro STS genotypes were imported into PedCheck (O'Connell and Weeks, 1998) to confirm the correct inheritance of alleles. Haplotype analysis was performed initially manually to ensure Mendelian inheritance of alleles. Publicly accessible databases, including NCBI, Ensemble and UCSC, were searched for known and putative genes. Known genes were defined as those for which mRNA, expressed sequence tags (ESTs) or proteins had been confidently identified.
Genotyping using SNP arrays
SNP genotyping was undertaken using the GeneChip® Mapping 10K v2.0 Xba Array containing 10 204 SNP markers following the manufacturer's instructions (Affymetrix, Santa Clara, CA, USA). Briefly, 250 ng of genomic DNA from each sample was digested with the restriction endonuclease XbaI for 2.5 h. Digested DNA was mixed with Xba adapters and ligated using T4 ligase for 2.5 h. Ligated DNA was added to four separate polymerase chain reactions (PCR), amplified, pooled and purified to remove unincorporated dNTPs. The purified PCR product was then fragmented with DNase1, end-labelled with biotin and hybridized to an array for 18 h in an Affymetrix 640 hybridization oven. Arrays were then washed and stained using an Affymetrix Fluidics Station F450. The arrays were scanned using an Affymetrix GeneArray® scanner 2500. The raw microarray feature intensities (RAS scores) were processed using the Affymetrix Genotyping Tools software package GCOS/GDAS (Affymetrix) to derive SNP genotypes, marker order and linear chromosomal location.
Genotyping using microsatellite markers
STS genotyping was performed using fluorescent dye-labelled microsatellite markers (MWG Biotech, Ebersberg, Germany). PCR products were run out on an ABI 3100 Genetic Analyser and analysed using Genotyper software (Applied Biosystems, Foster City, CA, USA). All genotypes that were unclear were repeated until a consistent genotype was obtained. Markers that were uninformative were excluded from further analysis.
Sequencing of candidate genes
Genes within the conserved haplotype were identified from the Ensembl database (http://www.ensembl.org/Homo_sapiens/index.html) and candidate genes were selected. Exons were amplified and sequenced using oligonucleotide primers (MWG Biotech). The fluorescent dideoxy sequencing products were run out on an ABI 3100 and analysed for base pair changes by Sequencher v4.5 (GeneCodes Corp., Ann Arbor, MI, USA).
Results
SNP genotyping
SNP genotyping was performed on 27 individuals. The average SNP genotype call rate was 99.1% (range 98.09–99.88%), providing on average 9957 genotypes per individual. None of the males typed was assigned a heterozygous state for any of the SNPs mapping to the X chromosome. Overall, 0.36% of markers displayed non-Mendelian transmission and a further 0.82% of markers displayed a presumptive transmission error. This implies that the overall genotyping error rate associated with the 10K v2.0 SNP array was in the order of 1.18%.
Linkage analysis and locus identification
SNP genotype data was directly imported into the linkage analysis programme Merlin and analysed initially for NPL. Owing to the size of the family being studied, the SNP chip data was analysed by initially splitting the family into two (individual II:6 and children treated as separate kindred). NPL analysis identified an extensive region with positive LOD scores across chromosome 9p (Fig. 3). The maximum NPL LOD score detected when the family was split into two was 1.31, with the maximum LOD score achievable using this model of 1.54. Although dividing the family was essential if we were to analyse data from all 27 individuals genotyped, we also analysed the data as a single core pedigree as an affected-only study of 18 individuals, which permitted NPL and parametric analyses. There was an NPL peak on chromosome 9p of 1.21 (with a maximum potential LOD score of 1.43) and a parametric peak LOD score of 2.4 (Fig. 4). Only one other region had positive LOD scores of 0.48 (NPL) and 1.29 (parametric) at 21qtel, but linkage was subsequently excluded using STS markers, which demonstrated that the affected individuals lacked a common haplotype and generated negative LOD scores (data not shown, submitted for review). All other chromosomes showed consistently negative LOD scores on parametric analysis using Merlin (data not shown, submitted for review).
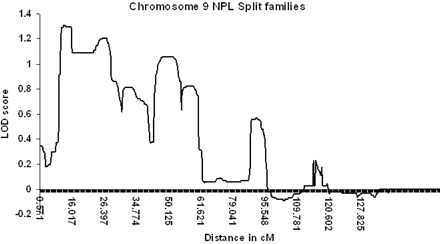
Graphical representation of multipoint LOD scores using the non-parametric linkage analysis programme Merlin in a split family model. The maximum LOD score detected was 1.34 and no other region had a LOD score > 0.8 (maximum achievable = 1.54). No other chromosomes generated LOD scores > 0.8.
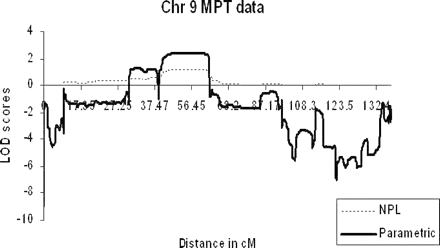
Graphical representation of multipoint LOD scores from an affecteds-only analysis of the kindred comparing non-parametric (NPL) and parametric linkage data generated by Merlin. The maximum NPL LOD score of 1.21 was detected at 9p (with a maximum potential LOD score of 1.43) and a parametric peak of 2.4.
Forty microsatellite markers were used to fine map the region on chromosome 9p, generating a two-point LOD score peak of 2.41 (𝛉 = 0) at D9S1878 (maximum Slink potential LOD score of 2.88). The peak multipoint LOD score of 3.02 occurred between D9S52 and D9S1805 (Table 2). Haplotype analysis revealed a critical recombination event at the centromeric and telomeric end of the haplotype in the recreated individual III:5 (Fig. 1). We believe that the telomeric cross-over event is robust, as it is present in all three offspring (IV:3,5,6) who inherited this chromosome, whereas the centromeric cross-over is present in the same offspring and two siblings (III:6,7). The telomeric flanking marker is D9S2154 and centromeric marker is D9S1874. These recombinant events narrow the conserved haplotype to a 12 cM (11 Mb) region at 9p13.2–21.3.
Candidate gene selection and mutation screening
Bioinformatic analysis of the region has identified 103 known genes. These have been prioritized according to their expression in the brain and possible role in pathogenesis. Three genes were chosen as candidates for initial mutation screening, but no mutation was found by direct sequencing.
Dynactin 3 was selected, as it is a subunit of the multiprotein dynactin complex that is involved in retrograde axonal transport in the motoneuron (Waterman-Storer, 1997). This process is disrupted in motoneuron disease, and recent studies have shown the presence of point mutations in the p150 subunit of the dynactin complex (Puls et al., 2003; Munch, 2004). Mutations in other genes in the complex may also cause disruption to axonal transport in the motoneuron. KIF24 was chosen, as the kinesin protein superfamily is involved in many cellular functions including vesicle transport, axonal transport of neurofilaments and microtubule dynamics (Xia et al., 2003; Serbus, 2005). Mutations in KIF5A have been described in an autosomal dominant hereditary spastic paraplegia (HSP) family (Fichera et al., 2004). This is a motoneuron disorder that indicates that mutations in this superfamily of proteins may lead to disruption of axonal transport in the motoneuron. BAG1 is involved in the ubiquitin proteasome pathway and binds with HSP70, coordinating the chaperoning of proteins targeted for degradation in the 26S proteasome (Takayama, 2001; Alberti, 2003). The presence of ubiquitin positive inclusions in motoneuron disease pathology indicates that this process may be disrupted (Ikeda and Tsuchiya, 2004) and, therefore, singles out BAG1 as a candidate gene.
Conclusions
ALS and FTD are both rapidly fatal late-onset neurodegenerative disorders where the majority of cases are sporadic and families suitable for linkage are extremely rare. Some families with autosomal dominant disease have an overlapping clinical phenotype that includes features of ALS and FTD in a spectrum. Here, we report a large Dutch kindred with late-onset ALS–FTD that demonstrates linkage to chromosome 9p13.2–21.3. Recombination has narrowed the region shared between the families to 11 Mb containing 103 known genes in this region. These will be screened for mutations and prioritized on the basis of their expression in the central nervous system and potential role in the pathogenesis of ALS.
If other ALS or FTD families are linked to this locus, it suggests that mutations in the causative gene may be a significant cause of familial and sporadic ALS and FTD. The identification of another ALS–FTD gene will enable more families to undergo predictive and prenatal testing. It will also allow the development of further transgenic and cellular models of ALS that will hopefully advance our understanding of disease mechanisms and help in developing more effective therapies for ALS.
Subsequent to the completion of this work the authors have learnt that an unrelated Swedish kindred with ALS–FTD has been linked to 9p21.3–p13.3 with a haplotype spanning 14 Mb. This overlaps precisely with our locus and reduces the shared region to 9.8 Mb (Mortia et al., 2006).
Electronic database information
Accession numbers and URLs for data in this article are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for background information on loci)
Center for Medical Genetics, Marshfield Medical Research Foundation, http://research.marshfieldclinic.org/genetics/ (for marker and position)
UK Human Genome Mapping Project Resource Centre, http://www.hgmp.mrc.ac.uk/Bioinformatics/ (for marker and gene position)
UCSC Human Genome Project Working Draft, http://genome.ucsc.edu/
National Centre for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/mapview/ (for marker and gene position)
Genome Database, http://www.gdb.org
Supplementary material
Supplementary data are available at Brain Online.
The authors would like to express their sincere gratitude to the family who contributed so much to this project over many years. Thanks are due to Dr G. A. Hoffland, Radiologist (VieCuri Hospitals, Venlo, Netherlands), for allowing us to reproduce the MRI scans. This work was supported by grants from the Middlemass family, EU contract LSHM-CT-2003-503330 (APOPIS), The Wellcome Trust (J.S.), Medical Research Council, UK (A.A-C.), Motor Neurone Disease Association UK, The Psychiatry Research Trust of the Institute of Psychiatry, the Princess Beatrix Fund (F.B.), Guy's and St Thomas' Charity and ALS Centrum Nederland, which supports the ALS Tissue Bank.
References
Abalkhail HM, Mitchell J, Habgood J, Orrell R, de Belleroche J. A new familial amyotrophic lateral sclerosis locus on chromosome 16q12.1-16q12.2.
Abecasis G, Cherny S, Cookson W, Cardon L. MERLIN—Rapid analysis of dense genetic maps using sparse gene flow trees.
Alberti S, Esser C, Hohfeld J. BAG-1—a nucleotide exchange factor of Hsc70 with multiple cellular functions.
Andersen PM, Sims KB, Xin WW, Kiely R, O'Neill G, Ravits J, et al. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes.
Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis.
Chance PF, Rabin BA, Ryan SG, Ding Y, Scavina M, Crain B, et al. Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34.
Fichera M, Lo Giudice M, Falco M, Sturnio M, Amata S, Calabrese O, et al. Evidence of kinesin heavy chain (KIF5A) involvement in pure hereditary spastic paraplegia.
Goldgar DE, Oniki OR. Comparison of a multipoint identity-by-descent method with parametric multipoint linkage analysis for mapping quantitative traits.
Gudbjartsson DF, Jonasson K, Frigge ML, Kong A. Allegro, a new computer program for multipoint linkage analysis.
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu/Zn superoxide dismutase mutation.
Hand CK, Khoris J, Salachas F, Gros-Louis F, Lopes AA, Mayeux-Portas V, et al. A novel locus for familial amyotrophic lateral sclerosis, on chromosome 18q.
Hentati A, Ouahchi K, Pericak-Vance MA, Nijhawan D, Ahmad A, Yang Y, et al. Linkage of a commoner form of recessive amyotrophic lateral sclerosis to chromosome 15q15-q22 markers.
Hentati A, Pericak-Vance MA, Lennon F, Wasserman B, Hentati F, Juneja T, et al. Linkage of a locus for autosomal dominant familial spastic paraplegia to chromosome 2p markers.
Hodges JR, Miller B. The classification, genetics and neuropathology of frontotemporal dementia. Introduction to the special topic papers: part I.
Hosler BA, Siddique T, Sapp PC, Sailor W, Huang MC, Hossain A, et al. Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17.
Ikeda K, Tsuchiya K. Motor neuron disease group accompanied by inclusions of unidentified protein signaled by ubiquitin.
Ince PG, Tomkins J, Slade JY, Thatcher NM, Shaw PJ. Amyotrophic lateral sclerosis associated with genetic abnormalities in the gene encoding Cu/Zn superoxide dismutase: molecular pathology of five new cases, and comparison with previous reports and 73 sporadic cases of ALS.
Jonsson PA, Ernhill K, Andersen PM, Bergemalm D, Brannstrom T, Gredal O, et al. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis.
Leigh PN, Dodson A, Swash M, Brion JP, Anderton BH. Cytoskeletal abnormalities in motor neuron disease. An immunocytochemical study.
Lipton AM White CL III, Bigio EH. Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration.
Liu J, Lillo C, Jonsson PA, Velde CV, Ward CM, Miller TM, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria.
Morita M, Al-Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia.
Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS.
Nishimura AL, Mitne-Neto M, Silva HC, Oliveira JR, Vainzof M, Zatz M. A novel locus for late onset amyotrophic lateral sclerosis/motor neurone disease variant at 20q13.
Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis.
O'Connell J, Weeks D. PedCheck: a program for identification of genotype incompatibilities in linkage analysis.
Piguet O, Brooks BW, Halliday GM, Schofield PR, Stanford PM, Kwok JB, et al. Similar early clinical presentations in familial and non-familial frontotemporal dementia.
Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, et al. Mutant dynactin in motor neuron disease.
Rascovsky K, Salmon SD, Lipton AM, Leverenz JB, Decarli C, Jagust WJ, et al. Rate of progression differs in frontotemporal dementia and Alzheimer disease.
Rizzu P, Van Swieten J, Joosse M, Hasegawa M, Stevens M, Tibben A, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands.
Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis.
Rosso SM, Donker Kaat L, Baks T, Joosse M, de Koning I, Pijnenburg Y, et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study.
Ruddy DM, Parton MJ, Al-Chalabi A, Lewis CM, Vance C, Smith BN, et al. Two families with familial amyotrophic lateral sclerosis are linked to a novel locus on chromosome 16q.
Sapp PC, Hosler BA, McKenna-Yasek D, Chin W, Gann A, Genise H, et al. Identification of two novel loci for dominantly inherited familial amyotrophic lateral sclerosis.
Serbus LR, Cha B, Theurkauf WE, Saxton WM. Dynein and the actin cytoskeleton control kinesin-driven cytoplasmic streaming in Drosophila oocytes.
Shaw CE, al-Chalabi A, Leigh N. Progress in the pathogenesis of amyotrophic lateral sclerosis.
Shaw CE, Enayat ZE, Chioza BA, Al-Chalabi A, Radunovic A, Powell JF, et al. Mutations in all five exons of SOD-1 may cause ALS.
Shaw CE, Enayat ZE, Powell JF, Anderson VE, Radunovic A, al-Sarraj S, et al. Familial amyotrophic lateral sclerosis: molecular pathology of a patient with a SOD1 mutation.
Siddique T, Figlewicz DA, Pericak-Vance MA, Haines JL, Rouleau G, Jeffers AJ, et al. Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity.
Stanford PM, Brooks WS, Teber ET, Hallupp M, McLean C, Halliday GM, et al. Frequency of tau mutations in familial and sporadic frontotemporal dementia and other tauopathies.
Strong MJ, Hudson A, Alvord WG. Familial amyotrophic lateral sclerosis, 1850–1989: a statistical analysis of the world literature.
Takayama S, Reed JC. Molecular chaperone targeting and regulation by BAG family proteins.
Terwilliger JD, Speer M, Ott J. Chromosome-based method for rapid computer simulation in human genetic linkage analysis.
Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, et al. Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature.
Waterman-Storer CM, Karki SB, Kuznetsov SA, Tabb JS, Weiss DG, Langford GM, et al. The interaction between cytoplasmic dynein and dynactin is required for fast axonal transport.
Author notes
1Department of Neurology, King's College London School of Medicine, Departments of 2Neurology and 3Molecular and Medical Genetics, Institute of Psychiatry, London, UK, 4Davee Department of Neurology and Clinical Neurosciences and 5Department of Cell and Molecular Biology, Northwestern University, Feinberg School of Medicine, Chicago, IL, USA, 6Department of Neurology and 7Department of Neuropathology, Radboud University Medical Centre, Nijmegen, and Departments of 8Neuropathology, 9Neurogenetics and 10Neurology, Academic Medical Centre, Amsterdam, The Netherlands


{kind=link}
{kind=link}
{kind=link}
{kind=link}