




Abstract
Alzheimer's disease poses a looming crisis for the health care system as well as society in general. The low efficacy of current treatments for those already affected with this disease has prompted the suggestion that interventions might be more successful if they were applied before the development of significant pathology, that is, when individuals are clinically asymptomatic. Currently, the field requires a sensitive and specific diagnostic tool for identifying those individuals destined to develop this disease. As a first step, we present here an analysis of cross-sectional data for 95 asymptomatic offspring (50–75 years of age) of autopsy-confirmed late-onset familial Alzheimer's disease cases and 90 age-matched controls, studied with functional magnetic resonance imaging (fMRI) to investigate brain activation patterns. Analysis of activation in response to a paired-associates memory paradigm found significantly different patterns in these groups. At-risk individuals showed more intense and extensive activation in the frontal and temporal lobes including the hippocampus during memory encoding, an increase unrelated to the APOE ε4 allele. They also showed decreased activation particularly in the cingulum and thalamus during both the encoding and recall phases of the task. These results demonstrate that asymptomatic individuals, at genetic risk for development of late-onset Alzheimer's disease by virtue of familial clustering, show functional activation patterns distinct from those without such risk more than a decade before their parent's onset age. While longitudinal study is needed to determine whether these patterns, or a subset of them, are predictive of disease onset, these findings suggest that functional neuroimaging holds promise as a method of identifying pre-clinical Alzheimer's disease.
Introduction
Alzheimer's disease is one of the major health problems in the United States, affecting >2 million Americans above the age of 60 (Brookmeyer et al., 1998), and its impact on the nation's health care system will increase as the proportion of elderly in the population continues to rise, making prevention or delay of disease onset crucial. Alzheimer's disease is a progressive neurodegenerative disorder characterized by global cognitive decline including progressive loss of memory, reasoning and language, with onset of the disease occurring from the fourth to ninth decade of life. At present there is no prevention or cure and the few available pharmacological treatments for those already diagnosed have limited efficacy (Mayeux and Sano, 1999; O'Hara et al., 2001; Tariot and Federoff, 2003). However, preliminary data suggest that the earlier treatments are introduced, the greater their benefit (Tariot and Federoff, 2003) Given that pathological changes begin well before the appearance of clinical symptoms (Troncoso et al., 1998; Ikonomovic et al., 2003), introducing treatments during this asymptomatic period may prove more successful in delaying or even preventing clinical symptoms. Thus, identifying those who will ultimately develop this disease is of paramount importance.
While molecular genetics has identified several genes [presenilin 1 (Sherrington et al., 1995), presenilin 2 (Levy-Lahad et al., 1995) and APP (Goate et al., 1991)] responsible for this disease in families where the age of onset is <60 years of age, these families represent <5% of all Alzheimer's disease cases. The vast majority of cases have an onset after the age of 60, and to date no gene with a clear Mendelian pattern of transmission has been identified. Only the apolipoprotein E (APOE) gene has consistently shown an association with late-onset Alzheimer's disease, but shows no clear familial pattern of disease transmission (Strittmatter et al., 1993; Mullan et al., 1996). While earlier studies reported an association between the ε4 allele and risk for Alzheimer's disease in a dose-dependent manner (Corder et al., 1993; Saunders et al., 1993; Strittmatter et al., 1993; Rebeck et al., 1993; Mullan et al., 1996), more recent reports make it clear that while the ε4 allele advances the age of onset for those who develop the disease, it is not predictive of caseness nor does it increase lifetime incidence (Khachaturian et al., 2004). A recent longitudinal study of healthy elderly did not find the APOE genotype predictive of cognitive decline (Marquis et al., 2002). In addition, among dementia cases or those with mild cognitive impairment, the ε4 allele has low positive predictive value for the diagnosis of Alzheimer's disease (Slooter et al., 1996; Devanand et al., 2005; Wang et al., 2005). This is reflective of the fact that the ε4 allele confers risk for atherosclerosis as well as a number of dementing illnesses (Johnson and Albert, 2000). Thus, APOE allele status appears to be of limited value for identifying future cases.
Currently, only increasing age and the presence of an affected first-degree relative are consistently associated with elevated risk for the development of the disease. Prevalence and incidence estimates vary, but consistently show exponentially increasing rates with age: from an estimated annual incidence between 0.3 and 0.6% among those aged 65–69, to 5.3–7.5% for those aged 85–90 (Gao et al., 1998). The presence of an affected first-degree relative approximately doubles the lifetime risk, while the presence of additional affected family members may increase the risk by 8-fold (van Duijn et al., 1991; Fratiglioni 1993; The Canadian Study of Health and Aging, 1994; Devi et al., 2000). Thus, except for the small percentage of cases linked to the three early-onset genes, there is at present no way to predict the development of the illness for well over 90% of the eventual cases.
Brain imaging holds promise as a technology that may allow us to identify harbingers of subsequent disease. PET and single photon-emission computed tomography (SPECT) imaging studies of asymptomatic carriers of APP or PS1 mutations for early-onset Alzheimer's disease have reported functional differences, with carriers showing metabolic patterns distinct from both non-carriers and diagnosed cases (Perani et al., 1997; Wahlund et al., 1999; Johnson et al., 2001). The Perani study, using [18F]FDG PET, examined resting-state metabolic profiles for seven members of two FAD Italian families with the APP717 Val to Ileu mutation. Of the seven, three were asymptomatic mutation carriers who demonstrated hypometabolism bilaterally in the thalamus as well as in the right parietal–occipital junction. The Wahlund study of nine members of a family with the Swedish APP 670/671 mutation reported that the three asymptomatic mutation carriers had resting-state reduction in glucose metabolism in the right temporal lobe association cortex as compared with the non-carriers. In follow-up over a 2-year period, these reductions became more pronounced especially for the one individual who had developed cognitive problems. Thus, in this individual the metabolic reductions seen with [18F]FDG PET preceded the onset of memory difficulty. The SPECT study of cerebral perfusion reported by Johnson of members of a South American family with the 280/PS1 mutation included 23 members without the mutation, 18 asymptomatic carriers and 16 carriers diagnosed with Alzheimer's disease. Comparison of these groups found that the asymptomatic carriers had decreased perfusion in the hippocampus, anterior and posterior cingulate, and posterior parietal and superior frontal cortices when compared with the non-mutation carriers. The perfusion reductions seen in the posterior cingulate, posterior parietal and superior frontal regions also occurred in the Alzheimer's disease cases but were more pronounced than in the asymptomatic carriers. Thus, these resting-state functional studies show that reductions in glucose metabolism and blood perfusion are evident in mutation carriers in the absence of clinical symptoms. In addition, the structures and regions identified in these studies, particularly the thalamus, posterior cingulate and medial temporal lobe (MTL) including the hippocampus, are brain regions important for memory function, in general the first clinical symptom of cognitive dysfunction.
A fundamental challenge is to identify functional changes or patterns predictive of later disease amongst those who are currently asymptomatic in order to initiate interventions as early as possible. Review of the imaging studies cited above in asymptomatic carriers of early-onset Alzheimer's disease gene mutations suggests that changes in those at elevated risk will most likely occur initially in medial temporal structures, cingulate cortex and thalamus. This study was undertaken to examine brain activation patterns, specifically in these regions, in asymptomatic individuals from familial kindreds already under genetic study for late-onset Alzheimer's disease. These individuals, who are the adult offspring of our autopsy-confirmed cases, along with age-matched controls, were investigated with functional MRI (fMRI), using an episodic memory task, to examine differences in haemodynamic patterns unique to familial risk status.
Material and methods
Sample
At-risk participants in this study were drawn from families enrolled in an ongoing genetic linkage study, developed at the Johns Hopkins University as part of the NIMH Alzheimer's Disease Genetics Initiative (Bassett et al., 2002). Those enrolled have a parent with autopsy-confirmed Alzheimer's disease and at least one additional first-degree relative with a clinical diagnosis of probable Alzheimer's disease, are at least 50 years of age, are free of memory complaints or treatment for cognitive impairments and scored in the normal range on the Telephone Interview for Cognitive Status (TICS) (Brandt et al., 1988). At the time of the study, these offspring were an average of 11 years from the age at which their parent developed Alzheimer's disease. Control participants, who were primarily spouses (65%), were also free of cognitive complaint or treatment, scored in the normal range on the TICS and screened negative on the Alzheimer Dementia Risk Questionnaire (Breitner and Folstein, 1984) and the Dementia Questionnaire (Silverman et al., 1986) for the presence of a first-degree relative with Alzheimer's disease or suspected dementia.
Participants were interviewed regarding personal sociodemographic characteristics, medical and psychiatric history, history of alcohol and tobacco use, and current medications. They also completed a number of self-report inventories focused on nutritional history, environmental exposures, level of exercise, health control, coping styles and personality. All participants travelled to Johns Hopkins Hospital for a day of evaluation that included physical and neurological examination; blood work including serum levels of vitamins, markers of inflammation, trace metals, hormones, thyroid, cholesterol profile and APOE genotyping; cognitive testing focused on memory, attention and language; and brain imaging. The imaging studies included both structural and functional MRI, for 95 at-risk (from a total of 101) and 90 control (from a total of 98) individuals. The study was approved by the Johns Hopkins Institutional Review Board and all participants provided written informed consent.
fMRI scanning protocol
Functional scans were acquired on a 1.5 T Philips Intera-NT scanner (Philips Medical Systems, Best, The Netherlands) at the F.M. Kirby Functional Imaging Research Center (Kennedy Krieger Institute, Baltimore, MD). The system is equipped with Galaxy gradients (66 mT/m at 110 mT/m/s). A standard head coil was used to limit head motion. A sagittal localizer scan was collected to pinpoint the exact location of the brain. Two functional scans were acquired using echo-planar imaging (EPI) and a blood oxygenation level-dependent (BOLD) technique with repetition time (TR) = 1000 ms, echo time (TE) = 39 ms, flip angle (𝛉) = 90°, field of view (FOV) = 230 mm in the xy plane and matrix size = 64 × 64 reconstructed to 128 × 128. Eighteen coronal slices were acquired with a 4.5 mm thickness and an interslice gap of 0.5 mm, oriented perpendicular to the anterior–posterior commissure (AC–PC) line. Slices were acquired sequentially along the z-axis, yielding a total coverage of 90 mm, which allowed for adequate coverage of the MTL, thalamus and cingulum, our primary region of interest. Functional scanning was performed in two sessions, each with 370 time points. Total functional acquisition time was 12 min and 20 s. A high-resolution whole brain scan was obtained using a T1-weighted, 3D MP-RAGE (Magnetization Prepared Rapid Acquisition Gradient Echo) sequence with the following parameters: TR = 8.6 ms, TE = 3.9 ms, FOV = 240 mm, 𝛉 = 8°, matrix size = 256 × 256, slice thickness = 1.5 mm, 124 slices).
Cognitive activation task
Participants were presented with an auditory word-pair-associates learning task. This memory challenge, adapted from Bookheimer et al. (2000), was chosen for its sensitivity to damage in the MTL (Rausch and Babb, 1993), the brain region that is the site of the earliest pathology in Alzheimer's disease (Braak and Braak, 1996). The paradigm, programmed in E-prime (Psychology Software Tools, Inc., Pittsburgh, PA, USA), consisted of two 6 min 10 s sessions, each with six trials. Each trial included an encoding phase, in which seven unrelated word-pairs were presented (e.g. ‘food’ and ‘book’), and a cued recall phase, in which the first word from the pair was presented and the participant was instructed to silently recall the second word of the pair. Activation blocks (encoding and recall) were preceded with rest (baseline) periods (seeFig. 1). At the end of each session, while still in the scanner, participants were asked to recall the 7 word-pairs; thus perfect recall would total 14, that is, the 7 pairs from Session 1 and the 7 pairs from Session 2 (the two sessions utilized unique word-pair sets). Before scanning, all participants were administered the verbal paired-associates test from the Wechsler Memory Scale, third edition, a different paired-associates test than that used for scanning, as part of the neuropsychological test battery, with scores converted to scale scores, in order to evaluate out-of-scanner performance.
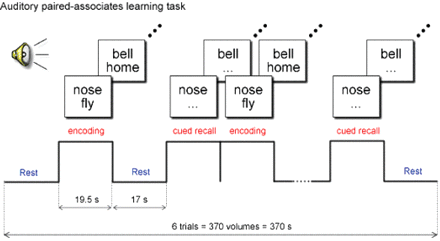
Auditory word-pair-associates paradigm. During the encoding blocks, words are presented in pairs. During the cued recall blocks, subjects are asked to silently recall the second word in the pair, when cued with the first word.
fMRI data pre-processing
Functional data pre-processing was conducted on Windows XP workstations, using Statistical Parametric Mapping (SPM99, Wellcome Department of Imaging Neuroscience, University College, London, UK) running under the MATLAB 6.1 (The Mathworks, Sherborn, MA, USA) programming and run-time environment. Rigid-body registration (motion correction) was performed by realigning all the scans from both sessions to the mean image of all the functionals in both sessions. This was conducted using a six-parameter affine transformation (three translations and three rotations in x-, y-, and z-axes), followed by re-slicing using a ‘windowed’ sinc-interpolation. Realignment output plots and realigned volumes were checked for motion artefacts and size of transformations. Twelve-parameter, affine transformation and non-linear normalization using 7 × 8 × 7 basis functions were used to warp each individual's data into standard stereotaxic space (Talairach and Tournoux, 1988). Template space was defined by SPM's standard EPI template (Montreal Neurologic Institute, McGill University, Montreal, Canada). The template was manually cut to fit each individual scan in order to improve the quality of normalization. Normalized scans were re-sliced to isotropic voxels (2 mm3), using trilinear interpolation and spatially smoothed with a full-width at half-maximum (FWHM) Gaussian kernel of 5 mm.
Sociodemographic and cognitive comparisons
Age, years of education, number of APOE ε4 alleles and performance on the Mini-Mental State Examination, the verbal paired-associates test from the Wechsler Memory Scale and the total word-pairs spontaneously recalled from the fMRI paradigm were compared for the at-risk and control groups by means of two-sample t-tests. The groups were also compared on gender and handedness using chi-square.
Structural data analysis
We conducted a voxel-based morphometry (VBM) study to examine the possibility of anatomical differences between groups. We applied the optimized VBM protocol by Good et al. (2001) to the MP-RAGE scans. This method uses a series of linear and non-linear transformations as well as tissue-type segmentations for cross-participant alignment of grey-matter and white-matter segments. Processed scans were smoothed with a 12 mm kernel. Groups were compared using an analysis of covariance (ANCOVA) with group membership and gender as fixed factors and age, years of education and APOE ε4 status as covariates. SPM contrast maps were generated with significance levels set at P < 0.05, small volume corrected for the left and right hippocampi, parahippocampal gyri, thalamus and cingulum.
fMRI data analysis
Subject-specific time series analyses were conducted using a general linear model within the framework of Statistical Parametric Mapping (SPM99). Data were modelled as epochs (blocks) and convolved with SPM's naïve canonical haemodynamic response function (HRF) (without temporal derivatives) to account for the lag between stimulation and the BOLD signal. High-pass filtering was conducted using a session cut-off of 76 s. No low-pass filtering was conducted. Scaling for global effects across volumes was performed to get rid of global cerebral blood-flow activation effects. The model was estimated using SPM's standard ordinary least squares (OLS). The contrasts of interest subtracted activation during the ‘rest’ condition from the ‘encoding’ and ‘recall’ conditions.
Second-order random effect analyses of whole brain were conducted using subtracted contrast images (‘encoding minus baseline’ and ‘recall minus baseline’ over both sessions) from each individual to investigate both within-group activation (one-sample t-tests), as well as group differences in activation during encoding and recall (separately using two one-way ANOVA analyses). Within-group comparisons used a voxel-wise threshold for inference and visualization using SPM's maximum intensity projections set to P = 0.001, uncorrected, with a spatial extent cluster threshold (k) of 20 voxels. Group differences were examined with two models; the first explored main group effects and the second controlled for the possible confounding effects of gender, handedness, age, education and APOE allele status, with age, education and number of ε4 alleles treated as continuous variables in voxel-specific regression analyses. A voxel-wise threshold for inference and visualization using SPM's maximum intensity projections was set to P = 0.01, uncorrected, with a spatial extent cluster threshold (k) of 20 voxels. The coordinates of activated voxels, which survived the statistical threshold, were produced in MNI space and converted to Talairach space (Talairach and Tournoux, 1988) to facilitate anatomical labelling, which was conducted using the Talairach Daemon software (Lancaster et al., 1997) with an adaptive grey-matter search range of 5 mm3 (Lancaster et al., 2000). Labels were manually checked with the Talairach and Tournoux atlas (Talairach and Tournoux, 1988). Correction for multiple comparisons across the volume of the medial temporal structures was conducted using SPM's Small-volume-Correction (SVC) facility. MTL structural templates were based on the single-subject MNI template segmentation by Tzourio-Mazoyer (Tzourio-Mazoyer et al., 2002).
Sub-sample comparisons were conducted to examine more strictly the impact of the APOE ε4 allele on the activation increases observed in the at-risk group during encoding. The first comparison, restricted to controls, contrasted all individuals with at least one ε4 allele (n = 19) with 19 controls of similar gender and age with the 3/3 genotype. The second comparison, restricted to the at-risk group, contrasted the 38 at-risk individuals with at least one ε4 with the 42 at-risk individuals with the 3/3 genotype. In each case the activation during encoding was compared using a regression model with gender, handedness, age and education included as covariates, setting the threshold at P = 0.01 uncorrected.
Results
The at-risk and control participants, whose groups did not differ in age, education, handedness, gender or APOE allele distribution, also showed no differences on the Mini-Mental State Examination, or word-pair-associates memory testing either before or following scanning (Table 1). Nor did the optimized VBM analysis find any significant volumetric differences in the MTL, cingulum or thalamus. However, the activation patterns during both the encoding and recall phases of the memory tasks were noticeably different. Examining each group independently revealed that control participants showed a statistically significant increase in brain activation during memory encoding bilaterally in the hippocampus and middle frontal gyri, and unilaterally in the left caudate, left middle temporal gyrus, left superior temporal gyrus, right superior frontal gyrus and right parahippocampal gyrus. The at-risk individuals showed activation during encoding in these same areas as well as activation in adjacent regions, including the amygdala and putamen (Table 2). Small-volume-corrected results show that both groups demonstrate significant activation in the left hippocampus (at-risk P = 0.000, controls P = 0.022) and in the right hippocampus (at-risk P = 0.000, controls P = 0.013).
At-risk and control participants: descriptive data
| Variable | At-risk (n = 95) Mean (SD) | Control (n = 90) Mean (SD) | Significance level |
|---|---|---|---|
| Age | 62.01 (6.77) | 62.68 (7.88) | P = 0.563 |
| Education | 15.31 (3.37) | 16.10 (3.17) | P = 0.100 |
| Gender | M = 42%, F = 58% | M = 54%, F = 46% | ξ2 = 0.093 |
| Handedness | Right = 89%, left = 11% | Right = 83%, left = 17% | ξ2 = 0.222 |
| # APOE ε4 alleles | 0 = 61%, 1 = 31%, 2 = 8% | 0 = 78%, 1 = 20%, 2 = 2% | ξ2 = 0.052 |
| MMSE | 28.2 (1.6) | 27.8 (1.7) | P = 0.236 |
| Wechsler Paired-Associates Test pre-scanning | 12.43 (3.2) | 12.19 (3.04) | P = 0.598 |
| Paired-Associates Test recall during fMRI | 5.72 (3.64) | 5.80 (3.76) | P = 0.889 |
| Variable | At-risk (n = 95) Mean (SD) | Control (n = 90) Mean (SD) | Significance level |
|---|---|---|---|
| Age | 62.01 (6.77) | 62.68 (7.88) | P = 0.563 |
| Education | 15.31 (3.37) | 16.10 (3.17) | P = 0.100 |
| Gender | M = 42%, F = 58% | M = 54%, F = 46% | ξ2 = 0.093 |
| Handedness | Right = 89%, left = 11% | Right = 83%, left = 17% | ξ2 = 0.222 |
| # APOE ε4 alleles | 0 = 61%, 1 = 31%, 2 = 8% | 0 = 78%, 1 = 20%, 2 = 2% | ξ2 = 0.052 |
| MMSE | 28.2 (1.6) | 27.8 (1.7) | P = 0.236 |
| Wechsler Paired-Associates Test pre-scanning | 12.43 (3.2) | 12.19 (3.04) | P = 0.598 |
| Paired-Associates Test recall during fMRI | 5.72 (3.64) | 5.80 (3.76) | P = 0.889 |
At-risk and control participants: descriptive data
| Variable | At-risk (n = 95) Mean (SD) | Control (n = 90) Mean (SD) | Significance level |
|---|---|---|---|
| Age | 62.01 (6.77) | 62.68 (7.88) | P = 0.563 |
| Education | 15.31 (3.37) | 16.10 (3.17) | P = 0.100 |
| Gender | M = 42%, F = 58% | M = 54%, F = 46% | ξ2 = 0.093 |
| Handedness | Right = 89%, left = 11% | Right = 83%, left = 17% | ξ2 = 0.222 |
| # APOE ε4 alleles | 0 = 61%, 1 = 31%, 2 = 8% | 0 = 78%, 1 = 20%, 2 = 2% | ξ2 = 0.052 |
| MMSE | 28.2 (1.6) | 27.8 (1.7) | P = 0.236 |
| Wechsler Paired-Associates Test pre-scanning | 12.43 (3.2) | 12.19 (3.04) | P = 0.598 |
| Paired-Associates Test recall during fMRI | 5.72 (3.64) | 5.80 (3.76) | P = 0.889 |
| Variable | At-risk (n = 95) Mean (SD) | Control (n = 90) Mean (SD) | Significance level |
|---|---|---|---|
| Age | 62.01 (6.77) | 62.68 (7.88) | P = 0.563 |
| Education | 15.31 (3.37) | 16.10 (3.17) | P = 0.100 |
| Gender | M = 42%, F = 58% | M = 54%, F = 46% | ξ2 = 0.093 |
| Handedness | Right = 89%, left = 11% | Right = 83%, left = 17% | ξ2 = 0.222 |
| # APOE ε4 alleles | 0 = 61%, 1 = 31%, 2 = 8% | 0 = 78%, 1 = 20%, 2 = 2% | ξ2 = 0.052 |
| MMSE | 28.2 (1.6) | 27.8 (1.7) | P = 0.236 |
| Wechsler Paired-Associates Test pre-scanning | 12.43 (3.2) | 12.19 (3.04) | P = 0.598 |
| Paired-Associates Test recall during fMRI | 5.72 (3.64) | 5.80 (3.76) | P = 0.889 |
Areas of significant activation during encoding and recall in control and at-risk subjects
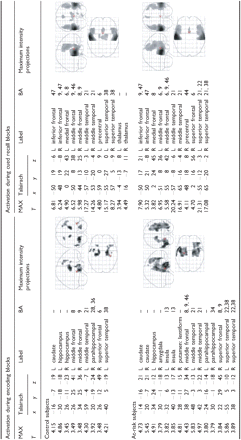
Height threshold (P) = 0.001, uncorrected; cluster-wise voxel threshold k = 20 voxels. All coordinates are in Talairach and Tournoux space (1988). BA = Brodmann area.
Areas of significant activation during encoding and recall in control and at-risk subjects
Height threshold (P) = 0.001, uncorrected; cluster-wise voxel threshold k = 20 voxels. All coordinates are in Talairach and Tournoux space (1988). BA = Brodmann area.
During periods of silent cued recall, controls showed a statistically significant increase in activation bilaterally in the inferior frontal, middle frontal, middle temporal and superior temporal gyri, as well as in the thalamus. Additional activation occurred in the left hemisphere, specifically in the medial frontal and precentral gyrus. The pattern seen in the at-risk participants also showed bilateral activation in the inferior frontal, middle frontal, middle temporal and superior temporal gyri, but none in the thalamus. All areas of unilateral activation were located in the right hemisphere, and included the medial frontal, superior frontal and precentral gyrus. No medial temporal activation was noted in either group (Table 2).
Group comparisons
Separate comparisons of at-risk and control individuals during memory encoding and then during recall were conducted to identify brain regions where these activation patterns differed substantially. Both positive and negative differences were examined. Two analytical models were considered, the first with only main effects and the second with the addition of covariates (age, education, number of APOE ε4 alleles, gender and handedness). The results did not differ in location, spatial extent or height of clusters and therefore only the results from the covariate model are presented.
During memory encoding, at-risk individuals showed statistically significantly greater activation bilaterally in the inferior frontal gyri, and unilaterally in the left superior temporal gyrus, left medial frontal gyrus, right hippocampus and right middle frontal gyrus. When medial temporal differences were more specifically evaluated using small-volume correction for the right hippocampus, the hippocampal difference was found to be significant at P = 0.04, corrected. There was simultaneously less activation in the thalamus and cingulum (Table 3, Figs 2 and 3). Inspection of activation during cued recall also identified areas with significant differences (Table 3, Figs 2 and 3). In this case, the striking difference was the predominance of areas with less activation in the at-risk group. These areas included the thalamus as well as structures in the left hemisphere including the caudate, cingulate, putamen, parahippocampal gyrus and middle frontal gyrus. Groups did not differ on the number of remembered pairs (evaluated by post-scan recall).
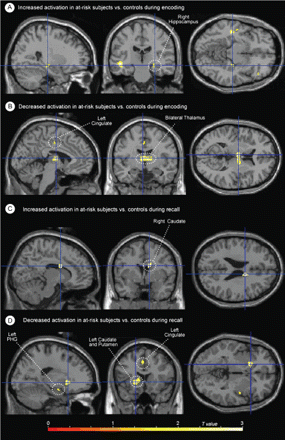
Maximum intensity projections of selected activation differences between groups, mapped on a single-subject template (SPM99 MNI). (A) Locations of increased activation in at-risk subjects versus controls during the encoding phase of the task; (B) locations of decreased activation in at-risk subjects versus controls during the encoding phase of the task; (C) locations of increased activation in at-risk subjects versus controls during the cued recall phase of the task; (D) locations of decreased activation in at-risk subjects versus controls during the cued recall phase of the task. (Review the activation tables for the complete listing of all significant activation differences.)
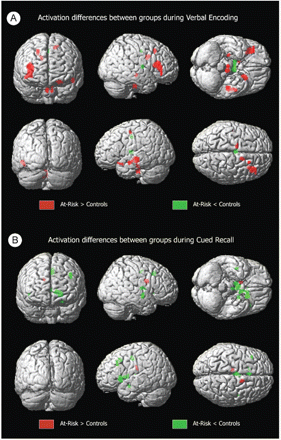
Three-dimensional volume-rendered displays of activation differences between groups (SPM99 MNI). A shows differences in activation between groups during verbal encoding, while B shows differences in activation during recall.
Group differences in activation during encoding and recall
| Max T | Talairach | Label | BA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| x | y | z | ||||||||||
| Encoding | ||||||||||||
| A. Clusters with increased activation (at-risk > controls) | ||||||||||||
| 3.60 | 40 | 28 | 10 | R | Inferior frontal | 13 | ||||||
| 3.20 | 38 | 29 | 4 | R | Inferior frontal | 47 | ||||||
| 2.71 | 46 | 33 | −8 | R | Inferior frontal | 47 | ||||||
| 2.67 | 36 | 33 | −5 | R | Inferior frontal | 47 | ||||||
| 3.54 | −20 | 13 | −16 | L | Inferior frontal | 47 | ||||||
| 3.18 | −24 | 15 | −16 | L | Inferior frontal | 47 | ||||||
| 3.45 | −46 | −24 | −6 | L | Superior temporal | 22 | ||||||
| 2.92 | −50 | −4 | −12 | L | Superior temporal | 38 | ||||||
| 2.42 | −50 | −14 | −6 | L | Superior temporal | 22 | ||||||
| 2.89 | 28 | −22 | −7 | R | Hippocampus | – | ||||||
| 2.81 | −12 | −5 | 48 | L | Medial frontal | – | ||||||
| 2.64 | 22 | 21 | 38 | R | Middle frontal | 8 | ||||||
| 2.64 | 22 | 21 | 41 | R | Middle frontal | 8 | ||||||
| B. Clusters with decreased activation (at-risk < controls) | ||||||||||||
| 3.67 | −4 | −7 | 10 | L | Thalamus | – | ||||||
| 3.40 | 8 | −7 | 8 | R | Thalamus | – | ||||||
| 2.92 | 16 | −7 | 10 | R | Thalamus | – | ||||||
| 2.98 | −2 | −10 | 41 | L | Cingulate | 24 | ||||||
| Recall | ||||||||||||
| A. Clusters with increased activation (at-risk > controls) | ||||||||||||
| 3.10 | 12 | 3 | 22 | R | Caudate | – | ||||||
| B. Clusters with decreased activation (at-risk < controls) | ||||||||||||
| 3.73 | −18 | 17 | −3 | L | Putamen | – | ||||||
| 3.39 | 0 | 6 | −5 | R | Thalamus: ant. nuc. | – | ||||||
| 3.32 | 2 | −5 | 9 | R | Thalamus | – | ||||||
| 2.96 | −8 | 4 | 3 | L | Caudate | – | ||||||
| 3.27 | −2 | −8 | 43 | L | Cingulate | 24 | ||||||
| 3.03 | −2 | 19 | 38 | L | Cingulate | 32 | ||||||
| 3.14 | 44 | −6 | −5 | R | Insula | 13 | ||||||
| 3.08 | −38 | 27 | 34 | L | Middle frontal | 9 | ||||||
| 2.89 | −2 | −15 | 10 | L | Thalamus | – | ||||||
| 2.73 | −2 | −15 | 4 | L | Thalamus | – | ||||||
| 2.79 | −20 | −3 | −13 | L | Parahippocampal | – | ||||||
| Max T | Talairach | Label | BA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| x | y | z | ||||||||||
| Encoding | ||||||||||||
| A. Clusters with increased activation (at-risk > controls) | ||||||||||||
| 3.60 | 40 | 28 | 10 | R | Inferior frontal | 13 | ||||||
| 3.20 | 38 | 29 | 4 | R | Inferior frontal | 47 | ||||||
| 2.71 | 46 | 33 | −8 | R | Inferior frontal | 47 | ||||||
| 2.67 | 36 | 33 | −5 | R | Inferior frontal | 47 | ||||||
| 3.54 | −20 | 13 | −16 | L | Inferior frontal | 47 | ||||||
| 3.18 | −24 | 15 | −16 | L | Inferior frontal | 47 | ||||||
| 3.45 | −46 | −24 | −6 | L | Superior temporal | 22 | ||||||
| 2.92 | −50 | −4 | −12 | L | Superior temporal | 38 | ||||||
| 2.42 | −50 | −14 | −6 | L | Superior temporal | 22 | ||||||
| 2.89 | 28 | −22 | −7 | R | Hippocampus | – | ||||||
| 2.81 | −12 | −5 | 48 | L | Medial frontal | – | ||||||
| 2.64 | 22 | 21 | 38 | R | Middle frontal | 8 | ||||||
| 2.64 | 22 | 21 | 41 | R | Middle frontal | 8 | ||||||
| B. Clusters with decreased activation (at-risk < controls) | ||||||||||||
| 3.67 | −4 | −7 | 10 | L | Thalamus | – | ||||||
| 3.40 | 8 | −7 | 8 | R | Thalamus | – | ||||||
| 2.92 | 16 | −7 | 10 | R | Thalamus | – | ||||||
| 2.98 | −2 | −10 | 41 | L | Cingulate | 24 | ||||||
| Recall | ||||||||||||
| A. Clusters with increased activation (at-risk > controls) | ||||||||||||
| 3.10 | 12 | 3 | 22 | R | Caudate | – | ||||||
| B. Clusters with decreased activation (at-risk < controls) | ||||||||||||
| 3.73 | −18 | 17 | −3 | L | Putamen | – | ||||||
| 3.39 | 0 | 6 | −5 | R | Thalamus: ant. nuc. | – | ||||||
| 3.32 | 2 | −5 | 9 | R | Thalamus | – | ||||||
| 2.96 | −8 | 4 | 3 | L | Caudate | – | ||||||
| 3.27 | −2 | −8 | 43 | L | Cingulate | 24 | ||||||
| 3.03 | −2 | 19 | 38 | L | Cingulate | 32 | ||||||
| 3.14 | 44 | −6 | −5 | R | Insula | 13 | ||||||
| 3.08 | −38 | 27 | 34 | L | Middle frontal | 9 | ||||||
| 2.89 | −2 | −15 | 10 | L | Thalamus | – | ||||||
| 2.73 | −2 | −15 | 4 | L | Thalamus | – | ||||||
| 2.79 | −20 | −3 | −13 | L | Parahippocampal | – | ||||||
Height threshold (P) = 0.01, uncorrected. Cluster-wise voxel threshold k = 20 voxels. All coordinates are in Talairach and Tournoux space (1988). BA = Brodmann area.
Group differences in activation during encoding and recall
| Max T | Talairach | Label | BA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| x | y | z | ||||||||||
| Encoding | ||||||||||||
| A. Clusters with increased activation (at-risk > controls) | ||||||||||||
| 3.60 | 40 | 28 | 10 | R | Inferior frontal | 13 | ||||||
| 3.20 | 38 | 29 | 4 | R | Inferior frontal | 47 | ||||||
| 2.71 | 46 | 33 | −8 | R | Inferior frontal | 47 | ||||||
| 2.67 | 36 | 33 | −5 | R | Inferior frontal | 47 | ||||||
| 3.54 | −20 | 13 | −16 | L | Inferior frontal | 47 | ||||||
| 3.18 | −24 | 15 | −16 | L | Inferior frontal | 47 | ||||||
| 3.45 | −46 | −24 | −6 | L | Superior temporal | 22 | ||||||
| 2.92 | −50 | −4 | −12 | L | Superior temporal | 38 | ||||||
| 2.42 | −50 | −14 | −6 | L | Superior temporal | 22 | ||||||
| 2.89 | 28 | −22 | −7 | R | Hippocampus | – | ||||||
| 2.81 | −12 | −5 | 48 | L | Medial frontal | – | ||||||
| 2.64 | 22 | 21 | 38 | R | Middle frontal | 8 | ||||||
| 2.64 | 22 | 21 | 41 | R | Middle frontal | 8 | ||||||
| B. Clusters with decreased activation (at-risk < controls) | ||||||||||||
| 3.67 | −4 | −7 | 10 | L | Thalamus | – | ||||||
| 3.40 | 8 | −7 | 8 | R | Thalamus | – | ||||||
| 2.92 | 16 | −7 | 10 | R | Thalamus | – | ||||||
| 2.98 | −2 | −10 | 41 | L | Cingulate | 24 | ||||||
| Recall | ||||||||||||
| A. Clusters with increased activation (at-risk > controls) | ||||||||||||
| 3.10 | 12 | 3 | 22 | R | Caudate | – | ||||||
| B. Clusters with decreased activation (at-risk < controls) | ||||||||||||
| 3.73 | −18 | 17 | −3 | L | Putamen | – | ||||||
| 3.39 | 0 | 6 | −5 | R | Thalamus: ant. nuc. | – | ||||||
| 3.32 | 2 | −5 | 9 | R | Thalamus | – | ||||||
| 2.96 | −8 | 4 | 3 | L | Caudate | – | ||||||
| 3.27 | −2 | −8 | 43 | L | Cingulate | 24 | ||||||
| 3.03 | −2 | 19 | 38 | L | Cingulate | 32 | ||||||
| 3.14 | 44 | −6 | −5 | R | Insula | 13 | ||||||
| 3.08 | −38 | 27 | 34 | L | Middle frontal | 9 | ||||||
| 2.89 | −2 | −15 | 10 | L | Thalamus | – | ||||||
| 2.73 | −2 | −15 | 4 | L | Thalamus | – | ||||||
| 2.79 | −20 | −3 | −13 | L | Parahippocampal | – | ||||||
| Max T | Talairach | Label | BA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| x | y | z | ||||||||||
| Encoding | ||||||||||||
| A. Clusters with increased activation (at-risk > controls) | ||||||||||||
| 3.60 | 40 | 28 | 10 | R | Inferior frontal | 13 | ||||||
| 3.20 | 38 | 29 | 4 | R | Inferior frontal | 47 | ||||||
| 2.71 | 46 | 33 | −8 | R | Inferior frontal | 47 | ||||||
| 2.67 | 36 | 33 | −5 | R | Inferior frontal | 47 | ||||||
| 3.54 | −20 | 13 | −16 | L | Inferior frontal | 47 | ||||||
| 3.18 | −24 | 15 | −16 | L | Inferior frontal | 47 | ||||||
| 3.45 | −46 | −24 | −6 | L | Superior temporal | 22 | ||||||
| 2.92 | −50 | −4 | −12 | L | Superior temporal | 38 | ||||||
| 2.42 | −50 | −14 | −6 | L | Superior temporal | 22 | ||||||
| 2.89 | 28 | −22 | −7 | R | Hippocampus | – | ||||||
| 2.81 | −12 | −5 | 48 | L | Medial frontal | – | ||||||
| 2.64 | 22 | 21 | 38 | R | Middle frontal | 8 | ||||||
| 2.64 | 22 | 21 | 41 | R | Middle frontal | 8 | ||||||
| B. Clusters with decreased activation (at-risk < controls) | ||||||||||||
| 3.67 | −4 | −7 | 10 | L | Thalamus | – | ||||||
| 3.40 | 8 | −7 | 8 | R | Thalamus | – | ||||||
| 2.92 | 16 | −7 | 10 | R | Thalamus | – | ||||||
| 2.98 | −2 | −10 | 41 | L | Cingulate | 24 | ||||||
| Recall | ||||||||||||
| A. Clusters with increased activation (at-risk > controls) | ||||||||||||
| 3.10 | 12 | 3 | 22 | R | Caudate | – | ||||||
| B. Clusters with decreased activation (at-risk < controls) | ||||||||||||
| 3.73 | −18 | 17 | −3 | L | Putamen | – | ||||||
| 3.39 | 0 | 6 | −5 | R | Thalamus: ant. nuc. | – | ||||||
| 3.32 | 2 | −5 | 9 | R | Thalamus | – | ||||||
| 2.96 | −8 | 4 | 3 | L | Caudate | – | ||||||
| 3.27 | −2 | −8 | 43 | L | Cingulate | 24 | ||||||
| 3.03 | −2 | 19 | 38 | L | Cingulate | 32 | ||||||
| 3.14 | 44 | −6 | −5 | R | Insula | 13 | ||||||
| 3.08 | −38 | 27 | 34 | L | Middle frontal | 9 | ||||||
| 2.89 | −2 | −15 | 10 | L | Thalamus | – | ||||||
| 2.73 | −2 | −15 | 4 | L | Thalamus | – | ||||||
| 2.79 | −20 | −3 | −13 | L | Parahippocampal | – | ||||||
Height threshold (P) = 0.01, uncorrected. Cluster-wise voxel threshold k = 20 voxels. All coordinates are in Talairach and Tournoux space (1988). BA = Brodmann area.
APOE
As noted in the overall analyses above, the number of APOE ε4 alleles was included in the model and did not prove to be a significant covariate. However, because of the previously reported findings of greater activation among ε4 carriers in response to this paired-associates memory paradigm (Bookheimer et al., 2000), we conducted two additional sub-sample comparisons. The first comparison, restricted to controls (thus removing effects of familial risk), contrasted all individuals with at least one ε4 allele (n = 19) with a set of 19 controls with the 3/3 genotype of similar age and gender. In this comparison, the ε4 group did not show any areas of greater activation during memory encoding. The second comparison, restricted to the at-risk group (holding familial risk status constant), contrasted the 38 at-risk individuals with at least one ε4 with the 42 at-risk individuals with the 3/3 genotype. This analysis also found no regions of significantly greater activation in the ε4 group during encoding.
Discussion
This work presents the only study of brain activation in asymptomatic offspring from confirmed cases of late-onset familial Alzheimer's disease and clearly identifies functional differences in this high-risk sample. In response to the challenge of an episodic memory task, where individuals need to both encode and retrieve new information (in this case unrelated word-pairs), at-risk individuals prove capable of completing this task with the same degree of accuracy as age- and education-matched controls. However, the patterns of activation that accompany each of these phases of the task in those at risk differ markedly from that seen in the control sample. Familial risk is associated primarily with increases in activation during memory encoding, irrespective of APOE allele status, and decreases in activation during cued recall.
Explicit memory impairment is generally considered the earliest clinical symptom of Alzheimer's disease, with studies identifying distinct brain regions involved in the encoding of new information into memory and the retrieval or recall of this stored information. MTL structures, particularly the hippocampus, entorhinal cortex and parahippocampal gyrus, as well as regions in the frontal cortex, have all been shown to be intimately involved in the encoding of new material as demonstrated in both animal and human studies (Henke et al., 1997; Desgranges et al., 1998; Buckner et al., 1999; Rombouts et al., 1999; Schacter and Wagner, 1999; Cabeza and Nyberg, 2000b; Gron et al., 2003; Pihlajamaki et al., 2003). These medial temporal structures, particularly the entorhinal cortex and hippocampus, are the site of initial Alzheimer's disease pathology, which begins while individuals are clinically asymptomatic (Hyman et al., 1984). As shown by the activation maps for the healthy controls, the paired-associates paradigm used in this study has proven robust for examining haemodynamic changes in these important brain regions during encoding, and thus provides a window on the neural processing associated with intentional memory processing. Individuals at familial risk for Alzheimer's disease included in this study evidence more extensive and intense activation in these same brain regions in order to successfully complete the task. This provides support for the compensatory hypothesis that postulates that additional neural resources are recruited to compensate for neuronal loss (Grady et al., 2003; Grossman et al., 2003; Dickerson et al., 2004).
The retrieval of information placed in long-term storage appears to rely on a somewhat different processing network, which varies with the type of stimuli and the manner of retrieval. In this paradigm, recall is initiated with an auditory cue (i.e. the first word of the pair), with individuals asked to think of the second word of the pair. The few studies that have used a similar method of assessing the recall of learned pairs [reviewed by Cabeza and Nyberg (2000a)] find increased activation in the frontal cortex, as well as in the thalamus. Other areas of increased activation are less consistent but can include temporal cortex, cingulate and lateral parietal areas. MTL structures, such as the hippocampus and entorhinal cortex, important during encoding, are less involved in the retrieval of material, with their involvement decreasing over repeated trials (Petersson et al., 1997). While both groups in this study show activation in the frontal and temporal cortices, in contrast to encoding where the at-risk group produced more extensive and intense activation, here, the at-risk show significantly less activation. These less-activated structures include the thalamus and specifically the anterior nucleus, a region important for memory function with major connections to the hippocampal complex and both the anterior and posterior cingulate (Van der Werf et al., 2003; Taber et al., 2004). In fact, the thalamus is consistently less active in the at-risk group across both conditions of this study. Of note, bilateral thalamic hypometabolism was reported as the distinguishing feature of asymptomatic carriers of the APP717 mutation for early-onset Alzheimer's disease (Perani et al., 1997).
The activation patterns seen in these asymptomatic individuals at elevated risk for Alzheimer's disease differ from those reported for individuals already diagnosed with AD, where the most consistent finding across fMRI studies employing episodic memory tasks has been significant reductions in MTL activation including the hippocampus and parahippocampal gyrus (Small et al., 1999; Rombouts et al., 2000; Sperling et al., 2003; Gron and Riepe, 2004). They also differ from those patterns reported for individuals with mild cognitive impairment, where studies report both decreased and increased MTL activation. These studies have not investigated other structures that are probably important, such as the cingulate and thalamus (Small et al., 1999; Machulda et al., 2003; Dickerson et al., 2004). Further work is required to understand whether there is some continuum of dysfunction in specific brain structures and regions or whether there will emerge a picture of temporal alterations that occur in response to changes in the efficiency of brain circuitry, in this case for memory function.
The absence of volumetric differences between the at-risk sample and the controls, in the face of functional differences, is in keeping with previous reports distinguishing functional and structural differences. For example, Reiman, using both resting PET and MRI, contrasted 11 individuals with APOE 4/4 with 22 individuals with APOE 3/3 and found that while the ε4 allele was associated with decreased metabolism, particularly in the cingulum, there were no volumetric differences (Reiman et al., 1996, 1998), demonstrating, as does this study, that functional differences occur in the absence of volumetric loss. It appears that volumetric loss is discernable only in the presence of cognitive decline, which is absent in the at-risk sample included here (Wolf et al., 2001). Recent imaging work suggests that surface deformity of the hippocampus, rather than volume of the hippocampus, may be predictive of subsequent cognitive decline and appearance of Alzheimer's disease (Csernansky et al., 2005).
This study has several methodological shortcomings that must be noted. The scan FOV provided only partial coverage of the brain. This was done to enhance the scan resolution in the MTL; however, it prevented us from investigating other potential differences between groups in regions outside of the FOV. Declarative memory involves structures in the frontal lobe, and thus complete frontal lobe coverage would have been helpful. With the onset of technological advances in fMRI, and the ability to scan at high tesla (3+) with more optimized parameters, it will be possible to use scan parameters that provide adequate coverage of the whole brain, as well as optimal resolution in the MTL in future studies. Secondly, it is possible that there were differences between groups in recall speed, which could have caused the activation differences observed between groups during recall. One could hypothesize that the at-risk sample would be slower, and therefore may show increased activation, reflecting more effort. However, the study finds the opposite. At-risk participants demonstrate decreased activation in several structures during recall, which makes the possibility of a difference in recall speed less likely. It is also unlikely that recall speed was the driving force behind the differences in medial temporal activation, since most of these differences were only found during the encoding phase and not during retrieval. Finally, we did not use a control for auditory stimuli, in order not to interfere with the auditory encoding of test stimuli. Auditory activation, as expected, was present and was reported in the superior temporal gyrus. Since this is anatomically distinct from medial temporal activation, it was not considered a confounding factor in our analysis.
The current study highlights a number of areas for further examination, including the relationship of APOE ε4 to changes in haemodynamic response. This is particularly important since the APOE ε4 allele is associated with a number of chronic medical conditions that impair cognition. For example, while this study demonstrates the lack of association between increases in activation and the presence of the APOE ε4 allele, PET studies have found the ε4/ε4 genotype associated with significantly reduced rates of glucose metabolism in the posterior cingulate, parietal, temporal and prefrontal regions, in both middle-aged and young adults. These are the same areas that have shown metabolic reductions in individuals experiencing memory complaints and those diagnosed with Alzheimer's disease (Reiman et al., 1996, 2004). Thus, an area of further inquiry is the relationship of APOE allele status to decreases in haemodynamic response in this memory task. In addition, it may be important to identify unique activation patterns within this high-risk sample, as these may reflect genetic heterogeneity in late-onset Alzheimer's disease and be linked to specific genes or genetic haplotypes (Kennedy et al., 1995; Rossor et al., 1996; Bassett et al., 2005). Such patterns may also provide information on clinical prognosis as suggested in a recent PET study (Ishii et al., 2003). Both of these may, in turn, ultimately be useful for individualizing treatments. Finally, the consistent reduction in activation in the cingulum and thalamus, which was apparent both in the encoding and cued recall comparisons, will require further investigation. The thalamus and cingulum have been shown to be activated during memory retrieval in a number of studies [seeDesgranges et al. (1998) for a review], and PET studies have reported hypoperfusion in both of these regions in the resting state.
In conclusion, this study finds that individuals at risk for familial late-onset Alzheimer's disease, although clinically asymptomatic, demonstrating normal cognitive performance, present a different pattern of functional brain activity when completing tasks requiring memorization and recall. These functional differences are apparent even though these individuals are more than a decade from the age at which their parent became clinically symptomatic. In addition, these functional differences occur in the absence of volumetric loss, suggesting that compensatory mechanisms may be present before any significant neuronal loss. Indeed, neuropathology studies of individuals showing significant amyloid deposits in the context of normal cognition do not find neuronal loss in temporal lobe structures (Price et al., 2001; West et al., 2004). Finally, the increased brain activation seen in the at-risk subjects during memory encoding is unrelated to the APOE ε4 allele. In summary, this work provides evidence that familial risk for late-onset Alzheimer's disease, related to a yet-unidentified gene or genes, is associated with altered brain function. This work sets the stage for the examination of temporal and spatial patterns of functional alterations in those at genetic risk for this disease. Longitudinal study of these high-risk individuals will enable us to determine whether these patterns, or a subset of them, are ultimately predictive of disease onset.
This work was supported by a grant from the National Institute on Aging, National Institutes of Healths to S.S.B. (AG16324).
References
The Canadian Study of Health and Aging. Risk factors for Alzheimer's disease in Canada.
Bassett SS, Avramopoulos D, Fallin D. Evidence for parent of origin effect in late-onset Alzheimer disease.
Bassett SS, Kusevic I, Cristinzio C, Yassa MA, Avramopoulos D, Yousem DM, et al. Brain activation in offspring of AD cases corresponds to 10q linkage.
Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, et al. Patterns of brain activation in people at risk for Alzheimer's disease.
Braak H, Braak E. Evolution of the neuropathology of Alzheimer's disease.
Brandt J, Spencer M, Folstein M. The telephone interview for cognitive status.
Breitner JC, Folstein MF. Familial Alzheimer dementia: a prevalent disorder with specific clinical features.
Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset.
Buckner RL, Kelley WM, Petersen SE. Frontal cortex contributes to human memory formation.
Cabeza R, Nyberg L. Imaging cognition II: an empirical review of 275 PET and fMRI studies.
Cabeza R, Nyberg L. Neural bases of learning and memory: functional neuroimaging evidence.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families.
Csernansky JG, Wang L, Swank JP, Gado M, McKneel D, Miller MI, et al. Preclinical detection of Alzheimer's disease: hippocampal shape and volume predict dementia onset in the elderly.
Desgranges B, Baron JC, Eustache F. The functional neuroanatomy of episodic memory: the role of the frontal lobes, the hippocampal formation, and other areas.
Devanand DP, Pelton GH, Zamora D, Liu X, Tabert MH, Goodkind M, et al. Predictive utility of apolipoprotein E genotype for Alzheimer disease in outpatients with mild cognitive impairment.
Devi G, Ottman R, Tang MX, Marder K, Stern Y, Mayeux R. Familial aggregation of Alzheimer disease among whites, African Americans, and Caribbean Hispanics in northern Manhattan.
Dickerson BC, Salat DH, Bates JF, Atiya M, Killany RJ, Greve DN, et al. Medial temporal lobe function and structure in mild cognitive impairment.
Fratiglioni L. Epidemiology of Alzheimer's disease. Issues of etiology and validity.
Gao S, Hendrie HC, Hall KS, Hui S. The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta-analysis.
Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease.
Good CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ, Frackowiak RS. A voxel-based morphometric study of ageing in 465 normal adult human brains.
Grady CL, McIntosh AR, Beig S, Keightley ML, Burian H, Black SE. Evidence from functional neuroimaging of a compensatory prefrontal network in Alzheimer's disease.
Gron G, Riepe MW. Neural basis for the cognitive continuum in episodic memory from health to Alzheimer disease.
Gron G, Bittner D, Schmitz B, Wunderlich AP, Tomczak R, Riepe MW. Variability in memory performance in aged healthy individuals: an fMRI study.
Grossman M, Koenig P, Glosser G, DeVita C, Moore P, Rhee J, et al. Neural basis for semantic memory difficulty in Alzheimer's disease: an fMRI study.
Henke K, Buck A, Weber B, Wieser HG. Human hippocampus establishes associations in memory.
Hyman BT, VanHoesen GW, Damascio AR, Barnes CL. Alzheimer's disease: cell-specific pathology isolates the hippocampal formation.
Ikonomovic MD, Mufson EJ, Wuu J, Cochran EJ, Bennett DA, DeKosky ST. Cholinergic plasticity in hippocampus of individuals with mild cognitive impairment: correlation with Alzheimer's neuropathology.
Ishii K, Mori T, Hirono N, Mori E. Glucose metabolic dysfunction in subjects with a clinical dementia rating of 0.5.
Johnson KA, Lopera F, Jones K, Becker A, Sperling R, Hilson J, et al. Presenilin-1-associated abnormalities in regional cerebral perfusion.
Kennedy AM, Newman SK, Frackowiak RS, Cunningham VJ, Roques P, Stevens J, et al. Chromosome 14 linked familial Alzheimer's disease. A clinico-pathological study of a single pedigree.
Khachaturian AS, Corcoran CD, Mayer LS, Zandi PP, Breitner JC, Apolipoprotein E. Epsilon4 count affects age at onset of Alzheimer disease, but not lifetime susceptibility: The Cache County Study.
Lancaster JL, Rainey LH, Summerlin JL, Freitas CS, Fox PT, Evans AE, et al. Automated labeling of the human brain: a preliminary report on the development and evaluation of a forward-transform method.
Lancaster JL, Woldorff MG, Parsons LM, Liotti M, Freitas CS, Rainey L, et al. Automated Talairach atlas labels for functional brain mapping.
Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, et al. A familial Alzheimer's disease locus on chromosome 1.
Machulda MM, Ward HA, Borowski B, Gunter JL, Cha RH, O'Brien PC, et al. Comparison of memory fMRI response among normal, MCI, and Alzheimer's patients.
Marquis S, Moore MM, Howieson DB, Sexton G, Payami H, Kaye JA, et al. Independent predictors of cognitive decline in healthy elderly persons.
Mullan M, Scibelli P, Duara R, Fallin D, Gold M, Schinka J, et al. Familial and population-based studies of apolipoprotein E and Alzheimer's disease.
O'Hara R, Derouesne C, Fountoulakis KN, Yesavage JA. Therapeutic approaches to age-associated neurocognitive disorders.
Perani D, Grassi F, Sorbi S, Nacmias B, Piacentini S, Piersanti P, et al. PET studies in subjects from two Italian FAD families with APP717 Val to Ileu mutation.
Petersson KM, Elfgren C, Ingvar M. A dynamic role of the medial temporal lobe during retrieval of declarative memory in man.
Pihlajamaki M, Tanila H, Hanninen T, Kononene M, Mikkonen M, Jalkanen V, et al. Encoding of novel picture pairs activates the perirhinal cortex: an fMRI study.
Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease.
Rausch R, Babb TL. Hippocampal neuron loss and memory scores before and after temporal lobe surgery for epilepsy.
Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions.
Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, et al. Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E.
Reiman EM, Uecker A, Caselli RJ, Lewis S, Bandy D, de Leon MJ, et al. Hippocampal volumes in cognitively normal persons at genetic risk for Alzheimer's disease.
Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia.
Rombouts SA, Scheltens P, Machielson WC, Barkhof F, Hoogenraad FG, Veltman DJ, et al. Parametric fMRI analysis of visual encoding in the human medial temporal lobe.
Rombouts SA, Barkhof F, Veltman DJ, Machielsen WC, Witter MP, Bierlaagh MA, et al. Functional MR imaging in Alzheimer's disease during memory encoding.
Rossor MN, Kennedy AM, Frackowiak RS. Clinical and neuroimaging features of familial Alzheimer's disease.
Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease.
Schacter DL, Wagner AD. Medial temporal lobe activations in fMRI and PET studies of episodic encoding and retrieval.
Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease.
Silverman JM, Breitner JC, Mohs RC, Davis KL. Reliability of the family history method in genetic studies of Alzheimer's disease and related dementias.
Slooter AJ, Breteler MB, Ott A, Van Broeckhoven C, van Duijn CM. APOE genotyping in differential diagnosis of Alzheimer's disease.
Small SA, Perera GM, DeLaPaz R, Mayeux R, Stern Y. Differential regional dysfunction of the hippocampal formation among elderly with memory decline and Alzheimer's disease.
Sperling RA, Bates JF, Chua EF, Cocchiarella AJ, Rentz DM, Rosen BR, et al. fMRI studies of associative encoding in young and elderly controls and mild Alzheimer's disease.
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease.
Taber KH, Wen C, Khan A, Hurley RA. The limbic thalamus.
Talairach J, Tournoux P. Co-planar stereotaxic atlas of the human brain—3-dimensional proportional system: an approach to cerebral imaging. New York: Thieme,
Tariot PN, Federoff HJ. Current treatment for Alzheimer disease and future prospects. Alzheimer Dis Assoc
Troncoso JC, Cataldo AM, Nixon RA, Barnett JL, Lee MK, Checler F, et al. Neuropathology of preclinical and clinical late-onset Alzheimer's disease.
Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain.
Van der Werf YD, Jolles J, Witter MP, Uylings HB. Contributions of thalamic nuclei to declarative memory functioning.
van Duijn CM, Clayton D, Chandra V, Fratiglioni L, Graves AB, Heyman A, et al. Familial aggregation of Alzheimer's disease and related disorders: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group.
Wahlund LO, Basun H, Almkvist O, Julin P, Axelman K, Shigeta M, et al. A follow-up study of the family with the Swedish APP 670/671 Alzheimer's disease mutation.
Wang PN, Lirng JF, Lin KN, Chang FC, Liu HC. Prediction of Alzheimer's disease in mild cognitive impairment: a prospective study in Taiwan. Neurobiol
West MJ, Kawas CH, Stewart WF, Rudow GL, Troncoso JC. Hippocampal neurons in pre-clinical Alzheimer's disease.
Author notes
1Department of Psychiatry, 2Department of Radiology, School of Medicine and 3Department of Biostatistics, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, USA


{kind=link}
{kind=link}
{kind=link}