




Abstract
Early-onset dystonia is an autosomal dominant movement disorder associated with deletion of a glutamic acid residue in torsinA. We generated four independent lines of transgenic mice by overexpressing human ΔE-torsinA using a neuron specific enolase promoter. The transgenic mice developed abnormal involuntary movements with dystonic-appearing, self-clasping of limbs, as early as 3 weeks after birth. Animals also showed hyperkinesia and rapid bi-directional circling. Approximately 40% of transgenic mice from each line demonstrated these severe behavioral abnormalities. Neurochemical analyses revealed decreases in striatal dopamine in affected transgenic mice, although levels were increased in those that had no behavioral changes. Immunohistochemistry demonstrated perinuclear inclusions and aggregates that stained positively for ubiquitin, torsinA and lamin, a marker of the nuclear envelope. Inclusions were detected in neurons of the pedunculopontine nucleus and in other brain stem regions in a pattern similar to what has been described in DYT1 patients. This transgenic mouse model demonstrates behavioral and pathologic features similar to patients with early-onset dystonia and may help to better understand the pathophysiology of this disorder and to develop more effective therapies.
INTRODUCTION
Early-onset DYT1 dystonia is a potentially disabling form of primary dystonia that is characterized by sustained or repetitive involuntary muscle contractions and abnormal postures ( 1 – 3 ). The disease typically manifests between the ages of 5 and 26 years ( 3 ), and is inherited in an autosomal dominant manner with a penetrance of ∼30–40% ( 4 – 6 ). The reasons for the decreased penetrance in humans are not known. Most cases of early-onset dystonia are caused by a three-base pair (GAG) deletion in the DYT1 gene on chromosome 9q34, resulting in loss of a glutamic acid residue (ΔE) in the protein torsinA ( 7 ). An 18 bp deletion in the DYT1 gene leading to loss of six amino acids (residues 323–328) in torsinA has also been described in a single patient with early-onset dystonia with myoclonic features ( 8 ).
TorsinA mRNA and protein are widely expressed in peripheral tissues ( 7 , 9 ), but are restricted to neurons in the central nervous system ( 9 – 12 ). In developing rats, torsinA expression is maximal at E20 or E21 (postnatal day 1) in the brain and in the peripheral tissues, except in the cerebellum where peak expression is observed at E14 ( 13 ). The function of torsinA is not known, but it has homology to HSP/Clp proteins ( 14 , 15 ), a subclass of AAA + (ATPases associated with cellular activities) proteins, suggesting that it may have chaperone-like function. Indeed, a neuroprotective function for torsinA has recently been demonstrated in both in vitro and in vivo models ( 16 – 19 ) consistent with a chaperone function for this protein.
The pathophysiology of early-onset dystonia is poorly understood and there is no specific therapy for this potentially disabling condition. A Drosophila model of early-onset dystonia has recently been reported ( 20 ). Now, we report, for the first time, that overexpression of ΔE-torsinA generates a transgenic mouse model of early-onset dystonia with a behavioral phenotype and pathologic and neurochemical changes similar to those found in patients with the disease.
RESULTS
Generation of transgenic mice expressing human ΔE-torsinA
We generated multiple (four) transgenic mice overexpressing human ΔE-torsinA using a rat neuron-specific enolase promoter and compared the phenotype with wild-type control mouse. The schema of the DNA construct used for the production of transgenic mice is shown in Fig. 1 A. Genotype analysis performed on DNA isolated from tail biopsies from each of the transgenic mouse lines showed incorporation of ΔE-torsinA cDNA, as demonstrated by the absence of one of the Bse RI restriction sites (Fig. 1 B). All four independent transgenic lines were confirmed by Southern blot analysis. Expression of the transgene was further analyzed by reverse transcription polymerase chain reaction (RT–PCR) on brain samples obtained from wild-type and transgenic animals. Mutant torsinA mRNA was expressed in each of the transgenic lines but not in the wild-type animals (Fig. 1 C). Western blot analysis demonstrated varying levels of torsinA protein expression in the different transgenic lines, but in each case, levels were higher than in the wild-type controls (Fig. 1 D).
Transgenic mice demonstrate abnormal behaviors
Approximately 40% of transgenic mice from each line expressing ΔE-torsinA displayed an abnormal behavior on visual inspection (102 out of a total of 252 transgenic animals). In the large majority of the ‘affected’ animals, behavioral changes were apparent by 3 weeks after birth, although behavioral changes emerged at 10 weeks in one animal. The severity of symptoms increased with age in all animals. The transgenic animals showed normal weight gain and reproduced normally, but affected mothers had difficulty in nursing due to their hyperkinetic behavior.
When affected transgenic mice were hung by their tails, they exhibited dystonic-appearing movements of limbs with self-clasping (Fig. 2 ). This self-clasping behavior was observed in transgenic mice derived from all four independent lines (42 out of 66 animals from TG#13, five out of 14 animals from TG#22, five out of 14 animals from TG#35 and six out of 12 animals from TG#49). Four animals (from line TG#13) also demonstrated intermittent periods of abnormal shaking of the head from side to side and sustained deviation of the head toward one side or the other (Fig. 2 ).
In the computerized activity monitoring system, affected transgenic animals had significantly greater horizontal activity and traveled greater distances than did wild-type animals, although the degree of hyperactivity varied between the different transgenic lines (Fig. 3 A), but differences were not significant between transgenic except for TG#13. Transgenic mice that did not show a behavioral phenotype on visual inspection, nonetheless, had significant hyperactivity in comparison with wild-type controls, although hyperactivity was less than what was observed in animals demonstrating an overt behavioral abnormality (Fig. 3 B).
Transgenic animals also demonstrated abnormal circling behavior, with alternations varying between clockwise and counter-clockwise rotations in the same animal (Fig. 3 C). Circling behavior was noted only in animals that expressed dystonic like movements of the limbs and varied in frequency between the different lines, with some animals turning as frequently as one to two times per second. In order to test whether the circling behavior was due to a vestibular or cochlear malfunction, startle chamber/auditory evoked brain stem responses (ABRs) were performed to assess cochlear function (three affected and three controls) and a swim test was used to assess vestibular function (six affected and six controls). Cochlear and vestibular functions were normal in all animals tested, although affected animals exhibited hyperactivity and circling behaviors on the swim test compared with the wild-type controls (data not shown).
No abnormal behaviors were observed in any of the wild-type control mice ( n =100).
Neuropathology
Brain sections were immunostained with antibodies to torsinA, ubiquitin, MAP2, PDI and lamin to determine whether these proteins accumulate, aggregate and/or form inclusion bodies in transgenic mice as we have described in DYT1 patients ( 21 ). In control animals, cellular immunoreactivity for torsinA and ubiquitin was low and diffuse throughout the cytoplasm of cells in most brain regions. However, in transgenic mice, there were perinuclear aggregates and/or inclusions that stained positively for ubiquitin and torsinA. These were most prominent in cholinergic neurons of the pontine and mesencephalic brain stem consistent with the region of the pedunculopontine nucleus (PPN). They were also noted in pontine neurons and in nerve cells of the peri-aqueductal gray region (Fig. 4 A and B), but were not detected in other brain areas studied including the cerebral cortex, cerebellum, striatum, hippocampus and substantia nigra pars compacta (SNc). A generalized increase in ubiquitinated proteins by immunofluorescence was observed in brain sections of transgenic mice.
Neurochemical analyses show an alteration in striatal dopamine levels in transgenic mice
High-pressure liquid chromatography (HPLC) analysis of brain homogenates obtained from the striata of wild-type control mice and transgenic mice showed alteration in dopamine (DA) levels (Fig. 5 ). In comparison with wild-type control animals, the DA levels were decreased (−39%, P =0.031) in transgenic animals that exhibited an abnormal behavioral phenotype, whereas DA levels were increased (+18%, P =0.035) in transgenic animals that had no obvious behavioral phenotype (unaffected). The striatal 3,4-dihydroxyphenylacetic acid/dopamine (DOPAC/DA) ratio was decreased in both affected (−8%) and unaffected (−33%) transgenic animals when compared with the control mice.
DISCUSSION
We describe, for the first time, the generation of a transgenic mouse model of DYT1 dystonia that has an abnormal behavioral phenotype with dystonia-like contractions in all limbs, and hyperkinetic activities. These features were observed in four independent transgenic mouse lines, indicating that this behavioral phenotype is independent of the site of integration, but a direct result of the ΔE-torsinA protein.
In the present study, we did not express wild-type torsinA in transgenic mice for several reasons. Previously, we and others demonstrated that the overexpression of wild-type, but not mutant, torsinA protected cultured cells from cellular insults ( 16 , 18 , 19 ), and decreased polyglutamine aggregates in Caenorhabditis elegans ( 17 ). Similarly, overexpression of mutant torsinA induced behavioral and pathological changes in Drosophila , which were not seen with wild-type overexpression ( 20 ). Thus, unlike mutant torsinA, overexpression of wild-type torsinA does not have any adverse effects in vitro or in vivo , making it unlikely that expression of wild-type torsinA would cause pathological or behavioral changes in transgenic mice. Therefore, all comparisons of the DYT1 transgenic mice in this study were done with wild-type control mice.
DYT1 dystonia is a clinical disorder that typically manifests during childhood ( 3 ). Approximately 30–40% of patients who carry the DYT1 gene mutation manifest dystonic symptoms. In transgenic mice, behavioral abnormalities developed at a young age (∼21 days postnatal) and the penetrance rate was ∼40%. Mice that did not appear to be affected on routine observation, nonetheless, had hyperkinetic activity when tested in the activity meter. Interestingly, there is evidence that human DYT1 carriers may also not be completely normal as evidenced by abnormal brain networks and learning ( 22 ).
The reason for the reduced penetrance in transgenic animals is not known as they have a similar genetic background and are bred under identical environmental conditions. However, as the founder transgenic mice were derived from F1 hybrid mice, there could be heterogeneity between the founders and their subsequent offspring, even when the founders are mated with an inbred animal. If the incomplete penetrance relates to genetic factors, it is reasonable to suggest that making the mice congenic (by back-crossing with an inbred strain) so as to provide a more homogeneous genetic background, the level of penetrance might increase. An alternative explanation for the incomplete penetrance relates to the putative role of torsinA. The precise function of TorsinA is not known, but it is thought to function as a chaperone protein that promotes the degradation of unwanted proteins ( 14 ). Variability in protein load and/or the activity of degradation enzymes in individual animals could possibly account for the variability in the pathological process and, therefore, for the manifestation of behavioral features.
The molecular mechanisms underlying the neuropathology of DYT1 dystonia are not clearly understood. Most studies have shown no apparent structural abnormalities ( 23 ), but there are isolated reports of an increase in the size of DA neurons in the SNc ( 24 ) and mild neuronal loss with occasional neurofibrillary tangles in the locus coeruleus, SNc, PPN, and dorsal raphe nucleus ( 25 ). More recently, we have used highly sensitive antibodies that detect ubiquitinated-protein conjugates (UPC) to examine brains from human DYT1 patients. These studies demonstrate UPC positive inclusions that also stain positively for torsinA in neurons of the brain stem, notably in the PPN and periaqueductal gray (PAG) ( 21 ). Consistent with human pathology, we found similar inclusion bodies and protein aggregates that were torsinA and ubiquitin positive, in neurons of the PPN and PAG of ΔE-torsinA transgenic animals, but not controls. These inclusions were localized to the perinuclear region of neurons and stained positively for lamin, a marker of the nuclear envelope. This is similar to what has been observed in cultured cells that overexpress ΔE-torsinA and in humans with the DYT1 mutation ( 21 , 26 – 28 ). Although, torsinA is present throughout the central nervous system, relatively high levels are detected in the pons in normal rodents ( 12 ). Thus, the appearance of pathological changes in the PPN, PAG and pontine nuclei in these transgenic animals could be a reflection of increased expression of ΔE-torsinA in these areas. These findings raise the possibility that altered neuronal activity in these brainstem regions could contribute to the pathophysiology of DYT1 dystonia.
Studies in a small number of clinically affected patients with DYT1 dystonia have shown reduced striatal DA levels ( 29 ). In comparison with wild-type control animals, DA levels were significantly decreased in transgenic animals with an abnormal behavioral phenotype (affected), whereas DA levels were significantly increased in transgenic animals that had a normal behavioral phenotype (unaffected). The basis of these changes is not known. There were no changes in DA transporter or tyrosine hydroxylase immunostaining (data not shown) in the brains of transgenic mice. Interestingly, though, recent studies have shown that torsinA can regulate the cellular trafficking of the DA transporter ( 30 ), suggesting that ΔE-torsinA may interfere with DA neurotransmission in affected transgenic animals. Moreover, the high DA levels in unaffected transgenic animals could represent a compensatory response that might account for their relatively normal behavior.
There is evidence that DA plays a role in patients with different forms of dystonia and other hyperkinetic disorders. DA receptor-blocking agents have long been known to be capable of inducing tardive dyskinesia and tardive dystonia ( 31 ). Dystonia is also known to be associated with conditions in which there is reduced dopaminergic function such as Parkinson's disease and dopa-responsive dystonia, which results from a mutation in the GTP cyclohydrolase-1 gene ( 32 ). TorsinA has been shown to be present in Lewy bodies in patients with sporadic Parkinson's disease ( 33 ) and has been shown to be closely associated with α-synuclein ( 34 ). The significance of this observation is not clear, but may relate to Lewy bodies being derived from aggresomes ( 35 , 36 ) and torsinA being recruited because of a chaperone-like function. Additionally, several types of dystonia result from the deficiency of DA resulting in alteration in the components of DA pathway. Reduced levels of DA result in hypofunction of DA D2-receptors and may be a common feature of many of these heritable and secondary dystonic syndromes ( 37 ). In addition, medications that enhance DA function have been reported to improve the symptoms of dystonia ( 38 , 39 ); however, benefits are typically modest, suggesting a more complex pathophysiology.
The PPN has only been recently implicated in dystonia ( 21 ). The PPN is thought to be involved in the initiation and modulation of movements and other stereotypic behavior ( 40 – 42 ). The PPN receives major inputs from the globus pallidus pars interna, subthalamic nucleus (STN) and the substantia nigra pars reticulata, and sends cholinergic and glutamatergic fibers to the SNc. Projections from the PPN also terminate in thalamus, STN, lower brainstem, cerebellar nuclei and spinal cord. The PPN and cuneiform nuclei together constitute the mesencephalic locomotor region, which is thought to regulate muscle tone and rhythmic limb movements during locomotion ( 40 ). Lesions of the PPN are associated with Parkinsonian dysfunction, whereas stimulation evokes striatal DA efflux ( 43 ). These studies illustrate the complex interplay between the PPN, which shows evidence of pathologic protein aggregation in DYT1 dystonia, and structures and transmitters in the brain stem and basal ganglia that are involved in the regulation of motor function. Our studies showing pathology in the brain stem area and alterations in DA levels in the striatum of transgenic mice are consistent with the documented relationship between these brain structures ( 40 – 43 ). Further studies are required to better understand the relationship between these structures and the development of dystonic movements.
In summary, we describe the creation of a transgenic mouse model of DYT1 dystonia caused by overexpressing human ΔE-torsinA. This model has abnormal behavioral phenotype, early-onset, reduced penetrance, and pathological and neurochemical features that resemble the human condition. This mouse model may provide a basis for better understanding of the pathophysiology of DYT1 dystonia and for developing more specific treatments for this potentially disabling condition.
MATERIALS AND METHODS
Generation of DYT1 transgenic mice
ΔE-torsinA cDNA was cloned by RT–PCR using postmortem brain tissue obtained from a patient with DYT1 dystonia and was sequenced. The human ΔE-torsinA cDNA was cloned into the Hind III site of the pNSE-Ex4 vector (gift from Dr Patricia Danielson, Scripps Research Institute, La Jolla, CA, USA). A 7.1 kb Sal I fragment containing the neuron specific enolase promoter, human ΔE-torsinA cDNA, along with the SV40 polyA signal, was excised, purified and used for the microinjection. Transgenic mice were produced on the C57BL/6J×C3H F1 background and bred as heterozygotes onto the C57BL/6J background using standard protocols at the Mount Sinai Mouse Genetics Shared Research Facility. Briefly, pronuclear injections of purified NSE-ΔE-torsinA DNA fragment (2–5 ng/µl) were done with eggs isolated from C57BL/6×C3H (B6C3) F1 hybrid females. All eggs that survive the injections were cultured in KSOM media until surgeries were performed later on the same day. After the microinjections were completed, surviving eggs were reimplanted into the oviducts of pseudopregnant Swiss Webster female mice. After a gestation period of 19 days, the pups were usually born naturally, although a small fraction was delivered by c-section followed by fostering onto lactating mothers. At 10–14 days, the pups were marked by ear notches, and small tail biopsies were performed for the isolation of DNA samples for genotype analysis. After 19–21 days, the pups were weaned. All animal experiments were performed in accordance with the guidelines of National Institutes of Health and with the approval of Institutional Animal Care and Use Committee. We did not generate transgenic mice overexpressing wild-type torsinA in this study.
Genotype analysis
Polymerase chain reaction (PCR) was performed on genomic DNA isolated from tail biopsies of transgenic and wild-type mice. Normal and ΔE-torsinA cDNAs served as positive controls. PCR was performed using the following primer sets: sense primer 5′-CCTGGAATACAAACACCTA-3′ and antisense primer 5′-CAGTGACTCCGGCTGCCAATC. Taq DNA polymerase was used to amplify the DNA with the following conditions: 94°C for 1 min, 55°C for 1 min and 72°C for 1 min, 30 cycles were performed. The PCR products were digested with the restriction enzyme Bse RI to further identify the amplified DNA products for GAG deletion. The GAG deletion in ΔE-torsinA cDNA results in the elimination of a restriction site for the enzyme Bse RI, which recognizes the DNA sequence GAG/GAG.
Reverse transcription and polymerase chain reaction
Total RNA from wild-type and DYT1 transgenic mice brains were isolated using RNA isolation kit from Qiagen (Valencia, CA, USA). A total of 5 µg isolated RNA was reverse transcribed by using one step RT–PCR kit (Qiagen, USA). PCR was performed on the reverse-transcribed sample using the same set of primers that was used for genotyping as described earlier.
Western blotting
Western blotting was performed using tissue homogenates obtained from wild-type and transgenic mice, using rabbit polyclonal antibody to human torsinA, generated against a peptide derived from the C-terminus as described previously ( 9 ). The antibody has been previously characterized and shown to immunoreact with human and rodent torsinA and human ΔE-torsinA ( 9 , 12 ). Protein concentrations were determined using a protein assay kit (Sigma Diagnostics, St Louis, MO, USA). Brain homogenates containing 20 µg of protein were resolved on SDS–PAGE and electroblotted onto a PVDF membrane using 50 m m Tris–HCl buffer (pH 8.4). The blot was incubated with antibody to torsinA for 1 h and detected using an ECL western blot chemiluminescence kit.
Immunohistochemistry
Mice were anesthetized with ketamine (40 mg/kg) and xylazine (5 mg/kg) and perfused transcardially with 1% paraformaldehyde, followed by cold 4% paraformaldehyde (both in 0.1 m PBS). Tissue was embedded in paraffin, and 20 µm sections were cut with a cryostat. Sections were blocked, and washed three times in 3% normal goat serum for 1 h at room temperature. Then they were washed three times in PBS and incubated overnight with antibody to torsinA (1 : 1000 diluted in PBS with 2.5% normal serum); ubiquitin (1 : 1000) from DakoCytomation, Carpinteria, CA, USA; protein disulfide isomerase (PDI, 1 : 1000) from Stressgen Biotechnologies, San Diego, CA, USA; LaminA/C (1 : 500) microtubule associated protein-2 (MAP2,1 : 1000) and choline acetyl transferase (ChAT, 1 : 1000) from Chemicon International, Temecula, CA, USA). Immunolabeling was visualized by using the fluorochrome-conjugated secondary antibodies, Alexa488 and Alexa594 (Molecular Probes, Eugene, OR, USA).
Behavioral studies
Motor function was assessed using a computerized three-dimensional activity monitoring system (AccuScan Instruments, Columbus, OH, USA). The activity monitor has 32 infrared sensor pairs with 16 along each side spaced 2.5 cm apart and 2.3 cm from the floor. This system determines motor activity on the basis of the frequency of interruptions to infrared beams traversing the x, y and z planes. Total distance (in centimeters) traveled and rearing activity and circling were automatically determined from the interruptions of beams in the horizontal and vertical planes, respectively. Mice exhibiting hyperkinetic movements were selected for comparison among different transgenic lines and wild-type controls for horizontal and circling activities. Behavioral data were collected for 30 min and expressed as 5 min blocks. The animals were also videotaped from time to time to record various types of abnormal movements.
Auditory evoked brain stem response
Mice were anesthetized with ketamine (25 mg/kg), and auditory stimuli were independently delivered at a rate of 9.7 Hz to the right and left external ear canals via hollow tubes that were wrapped in sound proofing material. The auditory stimulus was a broadband click (0.1 ms duration) generated by a Grass auditory stimulator (Model S10ASCM). Electrophysiological signals from the active and reference leads were fed to a Grass amplifier (Model 12) with a gain of 50 K and band pass filter settings of 0.1 and 3 kHz. The amplified signals were then routed to an A/D converter [Power 1401, Cambridge Electronic Design (CED), Cambridge, UK] with a sampling frequency of 50 K. Signal 2.1 software (CED) was used to obtain at least two ABRs for each ear by averaging the responses to 300 consecutive stimulus presentations. Averaged ABRs were stored on the computer and analyzed off-line for the latencies of the first five peaks.
Analysis of vestibular function
Vestibular function was analyzed by a swim test in which animals were held by their tails and dropped into a rectangular plastic tank filled with water maintained at 35°C for 1 min ( 44 ). The average swimming time with the head above water was determined for three trials for each mouse.
Striatal content of DA and its metabolites
Striata were homogenized (10% wt/vol) by sonication in icecold 0.4 m perchloric acid with 3,4-dihydroxybenzylamine (DHBA) as internal standard. Homogenates were centrifuged at 25 000 g for 10 min at 4°C and supernatants were collected. The levels of DA, DOPAC and homovanillic acid (HVA) were determined by HPLC with electrochemical detection.
Statistical analysis
ANOVA was used to compare the behavioral activities (horizontal and circling) of the different groups of mice by either taking into account repeated measurements on each animal at various time points (5, 10, 15, 20, 25 and 30 min) or by averaging observations of over all time points, the mean difference ±SE and the P -value between each transgenic line and wild-type controls were determined. Tukey-Kramer post hoc test was used to correct for multiple comparison. Student's t -tests were employed for neurochemical analysis.
ACKNOWLEDGEMENTS
The authors thank Dr Catherine Mytilineou and Dr Kevin Kelley for critical reading of the manuscript and helpful suggestions, and Mr Raymond Johnson for help with HPLC analysis and Ms Pushpanjali Gujjari for technical assistance. This study was supported by grants from National Institute of Neurological disorders and Stroke (RO1 NS43038, P.S.) and Bachmann–Strauss Parkinson and Dystonia Foundation. The transgenic mice used for this study were produced by the Mount Sinai Mouse Genetics Shared Research Facility, which is sponsored in part by NIH (NCI) grant R24 CA88302.
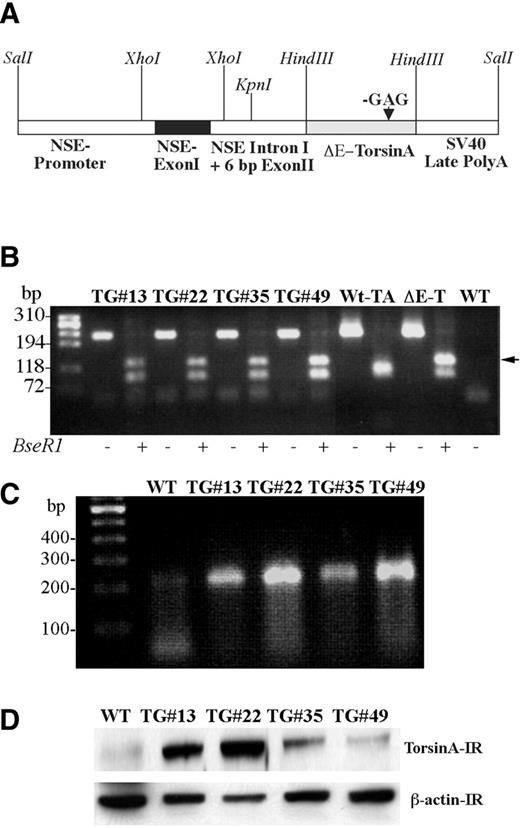
Figure 1. DYT1 transgenic mice analysis. ( A ) Scheme of full-length DNA construct used for the generation of transgenic mouse, showing regions of NSE promoter with exon I, intron I and 6 bp from exon II. The arrow indicates GAG deletion in torsinA cDNA. A SV40 polyadenylation signal sequence is added at the 3′ end of torsinA. ( B ) Genotyping of transgenic mice was done by performing PCR on DNA isolated from tail biopsy of mice followed by digestion of the PCR product with Bse RI restriction enzyme. Cloned cDNAs for wild-type torsinA (Wt-TA) and ΔE-torsinA (ΔE-TA) served as positive controls. PCR amplification of tail and cloned wild-type torsinA and ΔE-torsinA cDNAs yielded a 213 bp DNA product. Digestion of PCR amplified product from the wild-type torsinA cDNA yielded DNA fragments of 24 bp (not seen in figure), 94 and 95 bp fragments (which co-migrate on the agarose gel). In contrast, the mutant cDNA yielded DNA fragments of 95 and 118 bp (arrow) due to the loss of a Bse RI restriction site. − and + indicates the absence or presence of Bse RI restriction enzyme. ( C ) RT–PCR was performed to show the expression of transgene transcript in the brains of transgenic mice. The panel shows that the mRNA for ΔE-torsinA is expressed in all the transgenic lines. ( D ) Western blot analysis of brain homogenates obtained from wild-type control and independent transgenic mouse lines. β-Actin immunoreactivity was used to ensure equal amounts of protein loaded. WT, wild-type control mouse; TG, transgenic mouse; # indicates the line number of mouse. Arrow indicates the additional DNA band seen in transgenic.
![Figure 2. Transgenic mice exhibit self-clasping of limbs and head shaking. Transgenic mice exhibited self-clasping of limbs and abnormal head movements and posturing that were video taped. ( A ) Transgenic mouse showing various self-clasping movements of limbs [(c)–(f)] and the wild-type control mouse is shown in (a) and (b). (TG#13, n =42/66; TG#22, n =5/14 TG#35, n =5/14 and TG#49, n =6/12). Mice aged 6–7 weeks were used for the study. ( B ) Wild-type control with normal head posture (a) and transgenic mouse with abnormal head posture (b).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/1/10.1093_hmg_ddi012/1/m_ddi01202.jpeg?Expires=1716525037&Signature=UJmmk7NMSoskUoX~meGwy0aUA9zEPQs~UM7dI3UmlqSelA3t4uGOs-FDvELF2on0-BEjvV73aWrSLx0-IEVhaamv32tcoIpvfgdFjH9KmxyDgy~DggJpP007Qq59f8OLFZPTc3AEbcg-4sGkNh0behj7md2lH5YW5wHYZLKlALVuK0JD2YpzNeB7JCcVoFzG6-V8NYLTFsGFZjaH-YBeEuOIDzzqGDDjwUMZeGVHzOO6fwRtyzmqiWnwfZRQNPtWyxx3oaIa02sLIBvoM4R2-u-yRtfu-SEkxRqUdiql5EQ1ToP4pq~iSId5B4HDp2MYQLjvmUvemucB5UyiKdYyoA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 2. Transgenic mice exhibit self-clasping of limbs and head shaking. Transgenic mice exhibited self-clasping of limbs and abnormal head movements and posturing that were video taped. ( A ) Transgenic mouse showing various self-clasping movements of limbs [(c)–(f)] and the wild-type control mouse is shown in (a) and (b). (TG#13, n =42/66; TG#22, n =5/14 TG#35, n =5/14 and TG#49, n =6/12). Mice aged 6–7 weeks were used for the study. ( B ) Wild-type control with normal head posture (a) and transgenic mouse with abnormal head posture (b).
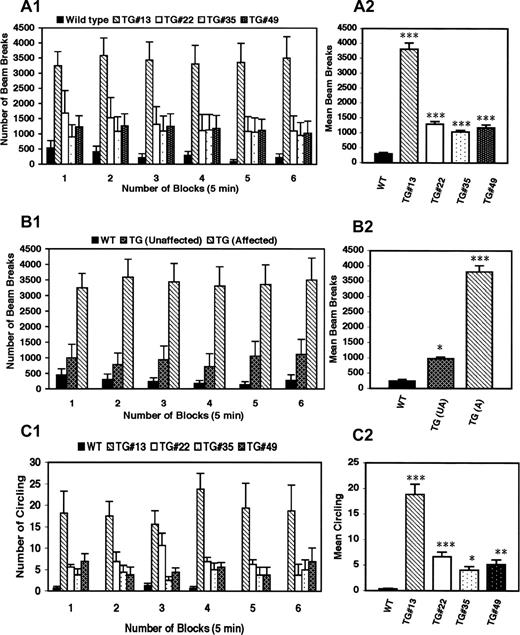
Figure 3. DYT1 transgenic mice exhibit abnormal involuntary hyperkinetic movements. ( A1 ) Horizontal activity was recorded (number of beam breaks/unit time) for wild-type and transgenic lines at various time points (5, 10, 15, 20, 25 and 30 min). ( A2 ) Horizontal activity expressed as average beam breaks over all time points (30 min) for wild-type controls and transgenic littermates. ( B1 ) Comparison of hyperkinetic movements of affected, unaffected transgenic mice and wild-type controls shows that although the unaffected mice do not exhibit self-clasping of limbs and circling, they do have a mild hyperkinesia that is significantly higher than the wild-type control mice. Transgenic mouse line#13 was used to show the difference in activity between affected and unaffected transgenic mice. ( B2 ) Mean beam breaks for repeated observations over a period of 30 min in wild-type controls and unaffected, affected transgenic littermates. ( C1 ) Circling behavior was recorded for wild-type and transgenic lines at various time points. ( C2 ) Circling behavior expressed as average of circling over all time points (30 min) for wild-type controls and transgenic littermates. Vertical bars represent mean±SE at each time point (A1, B1 and C1) or over a period of 30 min (A2, B2 and C2) between each transgenic line and the wild-type controls. For each analysis, 6–7 week-old animals with age-matched controls were used ( n =4–6). Asterisks indicate statistically significant differences between the wild-type control and various transgenic lines: * P <0.05, ** P <0.01 and P <0.001. WT, wild-type control mouse; TG, transgenic mouse; # indicates the line number of mouse.
![Figure 4. Perinuclear accumulation and aggregation of torsinA in the brains of transgenic mice. ( A ) Brain sections from pontine and mesencephalic brain stem of wild-type control and transgenic mice labeled with torsinA and MAP2 [(a) and (b)], torsinA and PDI [(c) and (d)] ubiquitin and PDI (e) and torsinA and ChAT (f). ( B ) Brain sections from wild-type control (WT) and transgenic mice (TG) were labeled with torsinA and lamin A/C [(a) and (b)] and torsinA and ubiquitin [(c)–(f)] demonstrating protein aggregation and neuronal inclusion bodies. Scale bar=10 µ m .](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/1/10.1093_hmg_ddi012/1/m_ddi01204.jpeg?Expires=1716525037&Signature=a5Ce9gRTXhgQzkKFazXfl30-EdCKfmVMmPLWkxlyL1sROIex-sOBG7Nh4rJSddcTuuAUK2reWlSeRjLMaf0-hfTvhNJt2uAVpEKBGBSt90RLKJRM6L73Hs-QbYNxG4BEuUo-i1gxJrOhJLacehLk3SxLMhN9T3ovn4iguda58UMsZjg8eFPCp-dZwedzyXN83VDTZ615-5zaTbEYXKUHawqgAavs-pgTPPwNBW6c1hNvGtIJelTsD6bzgZ8aJ4QQvBCHe8XqS8zso9Xh7DN~l56cYxAEfa0JJCDJqYWzQC00nSfmMN~7e0m6MaZv9maWj31vMyuWtMTG8F1fUIsMVw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 4. Perinuclear accumulation and aggregation of torsinA in the brains of transgenic mice. ( A ) Brain sections from pontine and mesencephalic brain stem of wild-type control and transgenic mice labeled with torsinA and MAP2 [(a) and (b)], torsinA and PDI [(c) and (d)] ubiquitin and PDI (e) and torsinA and ChAT (f). ( B ) Brain sections from wild-type control (WT) and transgenic mice (TG) were labeled with torsinA and lamin A/C [(a) and (b)] and torsinA and ubiquitin [(c)–(f)] demonstrating protein aggregation and neuronal inclusion bodies. Scale bar=10 µ m .
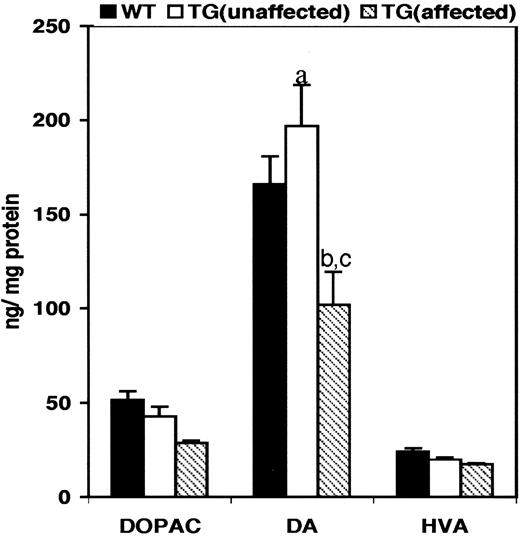
Figure 5. Levels of DA and its metabolite in the striata of transgenic mice. DA and its metabolites in the striatal tissue of transgenic mice were analyzed by HPLC. DOPAC, dihydroxyphenyl acetic acid; DA, dopamine; HVA, homovanilic acid. (a) P =0.031 (wild-type control:unaffected transgenic mice); (b) P =0.035 (wild-type control mice:affected transgenic mice); (c) P =0.015 (unaffected transgenic mice:affected transgenic mice). Bars represent the mean of levels of DOPAC, DA and HVA±SE. A sample size of six animals each was used for the analysis.
References
Bressman, S.B., Fahn, S., Falk, C., Allen, F.H., Jr and Suciu-Foca, N. (
Bressman, S.B. (
Ozelius, L.J., Hewett, J., Kramer, P., Bressman, S.B., Shalish, C., de Leon, D., Rutter, M., Risch, N., Brin, M.F., Markova, E.D. et al. (
Kramer, P.L., Ozelius, L., Brin, M.F., Fahn, S., Kidd, K.K., Gusella, J. and Breakefield, X.O. (
Kramer, P.L., Heiman, G.A., Gasser, T., Ozelius, L.J., de Leon, D., Brin, M.F., Burke, R.E., Hewett, J., Hunt, A.L., Moskowitz, C. et al. (
Ozelius, L.J., Hewett, J.W., Page, C.E., Bressman, S.B., Kramer, P.L., Shalish, C., de Leon, D., Brin, M.F., Raymond, D., Corey, D.P. et al. (
Leung, J.C., Klein, C., Friedman, J., Vieregge, P., Jacobs, H., Doheny, D., Kamm, C., DeLeon, D., Pramstaller, P.P., Penney, J.B. et al. (
Shashidharan, P., Kramer, B.C., Walker, R.H., Olanow, C.W. and Brin, M.F. (
Augood, S.J., Penney, J.B., Jr, Friberg, I.K., Breakefield, X.O., Young, A.B., Ozelius, L.J. and Standaert, D.G. (
Konakova, M., Huynh, D.P., Yong, W. and Pulst, S.M. (
Walker, R.H., Brin, M.F., Sandu, D., Gujjari, P., Hof, P.R., Warren Olanow, C. and Shashidharan, P. (
Xiao, J., Gong, S., Zhao, Y. and LeDoux, M.S. (
Neuwald, A.F., Aravind, L., Spouge, J.L. and Koonin, E.V. (
Ozelius, L.J., Page, C.E., Klein, C., Hewett, J.W., Mineta, M., Leung, J., Shalish, C., Bressman, S.B., de Leon, D., Brin, M.F. et al. (
Hewett, J., Ziefer, P., Bergeron, D., Naismith, T., Boston, H., Slater, D., Wilbur, J., Schuback, D., Kamm, C., Smith, N. et al. (
Caldwell, G.A., Cao, S., Sexton, E.G., Gelwix, C.C., Bevel, J.P. and Caldwell, K.A. (
Kuner, R., Teismann, P., Trutzel, A., Naim, J., Richter, A., Schmidt, N., von Ahsen, O., Bach, A., Ferger, B. and Schneider, A. (
Shashidharan, P., Paris, N., Sandu, D., Karthikeyan, L., McNaught, K.S., Walker, R.H. and Olanow, C.W. (
Koh, Y.H., Rehfeld, K. and Ganetzky, B. (
McNaught, K.S., Kapustin, A., Jackson, T., Jengelley, T.A., JnoBaptiste, R., Shashidharan, P., Perl, D.P., Pasik, P. and Olanow, C.W. (
Ghilardi, M.F., Carbon, M., Silvestri, G., Dhawan, V., Tagliati, M., Bressman, S., Ghez, C. and Eidelberg, D. (
Walker, R.H., Brin, M.F., Sandu, D., Good, P.F. and Shashidharan, P. (
Rostasy, K., Augood, S.J., Hewett, J.W., Leung, J.C., Sasaki, H., Ozelius, L.J., Ramesh, V., Standaert, D.G., Breakefield, X.O. and Hedreen, J.C. (
Hedreen, J.C., Zweig, R.M., DeLong, M.R., Whitehouse, P.J. and Price, D.L. (
Gonzalez-Alegre, P. and Paulson, H.L. (
Naismith, T.V., Heuser, J.E., Breakefield, X.O. and Hanson, P.I. (
Goodchild, R.E. and Dauer, W.T. (
Augood, S.J., Hollingsworth, Z., Albers, D.S., Yang, L., Leung, J.C., Muller, B., Klein, C., Breakefield, X.O. and Standaert, D.G. (
Torres, G.E., Beaulieu, M., Sweeney, A., Shashidharan, P., Caron, M.G. (
Cardoso, F. and Jankovic, J. (
Ichinose, H. and Nagatsu, T. (
Shashidharan, P., Good, P.F., Hsu, A., Perl, D.P., Brin, M.F. and Olanow, C.W. (
Sharma, N., Hewett, J., Ozelius, L.J., Ramesh, V., McLean, P.J., Breakefield, X.O. and Hyman, B.T. (
Olanow, C.W., Perl, D.P., DeMartino, G.N. and McNaught, K.S. (
McNaught, K.S., Shashidharan, P., Perl, D.P., Jenner, P. and Olanow, C.W. (
Todd, R.D. and Perlmutter, J.S. (
Jankovic, J. (
Marsden, C.D., Marion, M.H. and Quinn, N. (
Pahapill, P.A. and Lozano, A.M. (
Lee, M.S., Rinne, J.O. and Marsden, C.D. (
Nandi, D., Stein, J.F. and Aziz, T.Z. (
Forster, G.L. and Blaha, C.D. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}