




Abstract
Neonatal diabetes can either remit and hence be transient or else may be permanent. These two phenotypes were considered to be genetically distinct. Abnormalities of 6q24 are the commonest cause of transient neonatal diabetes (TNDM). Mutations in KCNJ11, which encodes Kir6.2, the pore-forming subunit of the ATP-sensitive potassium channel (KATP), are the commonest cause of permanent neonatal diabetes (PNDM). In addition to diabetes, some KCNJ11 mutations also result in marked developmental delay and epilepsy. These mutations are more severe on functional characterization. We investigated whether mutations in KCNJ11 could also give rise to TNDM. We identified the three novel heterozygous mutations (G53S, G53R, I182V) in three of 11 probands with clinically defined TNDM, who did not have chromosome 6q24 abnormalities. The mutations co-segregated with diabetes within families and were not found in 100 controls. All probands had insulin-treated diabetes diagnosed in the first 4 months and went into remission by 7–14 months. Functional characterization of the TNDM associated mutations was performed by expressing the mutated Kir6.2 with SUR1 in Xenopus laevis oocytes. All three heterozygous mutations resulted in a reduction in the sensitivity to ATP when compared with wild-type (IC50∼30 versus ∼7 µM, P-value for is all <0.01); however, this was less profoundly reduced than with the PNDM associated mutations. In conclusion, mutations in KCNJ11 are the first genetic cause for remitting as well as permanent diabetes. This suggests that a fixed ion channel abnormality can result in a fluctuating glycaemic phenotype. The multiple phenotypes associated with activating KCNJ11 mutations may reflect their severity in vitro.
INTRODUCTION
Neonatal diabetes may be defined as insulin-requiring hyperglycaemia, usually diagnosed within the first 3 months of life (1–3). It may be either transient, resolving within 18 months, or permanent in which case lifelong insulin treatment is required (1–4). A large proportion of transient neonatal diabetes (TNDM) cases subsequently relapse and are diagnosed with diabetes in adolescence (range 4–25 years) (3,4). TNDM and permanent neonatal diabetes (PNDM) are considered to be discrete clinical syndromes, which result from distinct genetic abnormalities of the pancreatic β-cell.
The majority (∼77%) of TNDM cases are caused by an anomaly of the imprinted region on chromosome 6q24 (4,5), including paternal uniparental isodisomy of chromosome 6 (UPD6), paternally inherited duplications of chromosome 6q or a methylation defect in this region (5–8). Two imprinted genes lie in the TNDM minimal region (9,10): ZAC, which encodes a protein involved in cell cycle control, and HYMAI, which generates an untranslated mRNA of unknown function. An animal model over-expressing both genes mimicked the human phenotype when paternally inherited, but the precise gene and the mechanism are still incompletely understood (11). The cause(s) of TNDM when there is not an abnormality of 6q24 are unknown.
The commonest defined causes of PNDM are heterozygous activating mutations in the KCNJ11 gene, which codes for Kir6.2, the inwardly rectifying potassium-channel subunit of the β-cell KATP channel (12–16). Four pore-forming Kir6.2 subunits form an octomeric KATP channel complex with four regulatory sulphonylurea receptor subunits (SUR1) (17). Nucleotides have several effects on KATP channel activity. ATP closes the channel by binding directly to the Kir6.2 subunit, whereas binding and/or hydrolysis of Mg-nucleotides (MgADP, MgATP) at the nucleotide binding domains of SUR1 causes channel opening. Therefore, the net channel activity reflects the balance between these inhibitory and stimulatory influences. In the resting β-cell, the level of KATP channel activity is sufficient to maintain the cell in a hyperpolarized state. Stimulatory glucose concentrations increase the metabolic rate and thereby increase the concentration of ATP at the expense of MgADP. This causes the KATP channels to close, resulting in membrane depolarization, an increase in cytosolic calcium and the triggering of insulin release. The R201H mutation associated with PNDM results in a decreased sensitivity to ATP and hence reduced channel closure, sustained membrane hyperpolarization and permanent severely impaired insulin secretion (12). In addition to causing isolated diabetes, activating mutations in KCNJ11 are also associated with the neurological features of developmental delay, muscle weakness and epilepsy (12). Recently, the functional characterization of these syndromic mutations has shown that they are associated with a greater decrease in sensitivity to ATP than mutations associated with isolated PNDM (18).
Syndromic and non-syndromic PNDM are not the only phenotypes associated with genetic variation in the KCNJ11 gene. The common E23K variant has been shown to predispose to Type 2 diabetes (19–22), probably by causing a small increase in channel activity at physiological nucleotide concentrations (23–25). At the other extreme, mutations that reduce KATP channel activity (either by interfering with the stimulatory actions of Mg-nucleotides or by reducing the number of functional channels at the surface membrane) result in a permanently depolarized β-cell manifesting as congenital hyperinsulinism (26).
We investigated whether activating mutations might also result in TNDM. In this paper, we report the sequencing of the KCNJ11 gene in subjects with relapsing diabetes, who had been clinically defined as having TNDM and who did not have an abnormality of chromosome 6q24. We define three missense KCNJ11 mutations and study the alteration of function associated with these mutations.
RESULTS
Novel KCNJ11 mutations defined in probands with remitting diabetes
We sequenced 11 probands who had been clinically defined as having TNDM, on the basis of remitting diabetes, which presented in the first 6 months of life, in whom abnormalities in the imprinted region of 6q24 had been excluded. We identified the three novel heterozygous mutations (G53S, G53R, I182V) in probands from families ISPAD 42, ISPAD 79 and ISPAD 50. These missense mutations resulted in the substitution of glycine at codon 53 by serine G53S (GGC>AGC) or arginine G53R (GGC>CGC) and in the substitution of isoleucine at codon 182 by valine I182 V (ATC>GTC). In all families, diabetes was only seen in subjects with KCNJ11 mutations and all subjects without these mutations were non-diabetic (Fig. 1). There was transmission of diabetes from an affected parent to their offspring in ISPAD 42 and ISPAD 79. There was no family history of diabetes in ISPAD 50, and analysis of DNA from the unaffected parents showed the mutation in the affected proband arose de novo. In family ISPAD 79, the mutation was shown to arise de novo in the proband's mother (Fig. 1). None of the mutations was present in 100 non-diabetic UK Caucasian subjects. All the mutations affect residues that are conserved among Homo sapiens, Mus musculus, Rattus norvegicus, Cavia porcellus and Oryctolagus cuniculus.
Patients with KCNJ11 mutations can show evidence of remission and relapse of diabetes
The clinical characteristics of the patients with KCNJ11 mutations are shown in Table 1. The probands were diagnosed with severe hyperglycaemia between the ages of 1 and 16 weeks and required insulin (>0.6 U insulin/kg/day). All three cases started to reduce their doses of insulin between the ages of 6 and 10 months and were able to discontinue their insulin completely and maintain excellent glycaemia as shown by normal blood glucose values and normal HbA1c. In one case, the proband from family ISPAD 79, there was a subsequent relapse of the diabetes. He was insulin treated until 17 months of age and then remained off insulin until 28 months. At 28 months, he became diabetic again and returned to low dose insulin treatment (0.2 U/kg/day), with good glycaemic control. His control has subsequently deteriorated, and at 10 years, he is on 0.6 U/Kg/day of insulin and had his highest HbA1c value to-date (8.4%). The sibling in family ISPAD 42 also had insulin-requiring diabetes that went into remission at the age of 20 months. In this case, at the age of 2 years, this child now has an HbA1c above normal and requires occasional short acting insulin, suggesting that he is likely to relapse. These probands and sibling fit clinical criteria for TNDM.
The two affected parents in families ISPAD 42 and ISPAD 79 both have young–onset diabetes and low birth weight, which is likely to result from the KCNJ11 mutation. These subjects were diagnosed at 5 years and 11 weeks, respectively, but no periods of remission were detected and they have remained diabetic in adult life.
There are no consistent features except for diabetes in the mutation carriers, although intra-uterine growth retardation (small for gestational age) below the 10th percentile is seen in at least one affected member of each family. In family ISPAD 79, both mother and child have learning difficulties.
Functional analysis of the novel mutations
Figure 2 maps the location of the novel TNDM mutations and the PNDM R201H mutation onto a homology model of Kir6.2 (27). I182 and R201 both lie within the predicted binding site for ATP, whereas G53 is not directly involved in nucleotide binding but lies in a region linking the binding site to the transmembrane domain. Mutagenesis studies have been carried out previously on both TNDM residues in the truncated Kir6.2 subunit, Kir6.2ΔC (27,28). Some of the mutations resulted in impaired inhibition by ATP without altering intrinsic gating. However, the specific amino acid changes identified in our patients were not investigated.
The functional impact of the Kir6.2 mutations was assessed in Xenopus oocytes by co-expressing SUR1 with either wild-type or mutant human Kir6.2 alone or a 1 : 1 mixture of wild-type and mutant Kir6.2 to mimic the heterozygous state. Wild-type KATP channels were silent in intact oocytes but opened following application of the metabolic inhibitor, azide, which lowers intra-cellular ATP. These currents were fully blocked by 0.5 mm tolbutamide, indicating that they flowed through open KATP channels. In contrast, significant resting whole-cell K+ currents were present in oocytes expressing homozygous (hom) or heterozygous (het) I182 V, G53S and G53R channels (Fig. 3). In all cases, the TNDM mutant currents were increased by azide and substantially blocked by tolbutamide. We also measured currents in oocytes expressing the PNDM mutation R201H in human Kir6.2 for comparison. Hom and hetR201H currents were larger than wild-type currents in resting oocytes, were activated by azide and were largely blocked by tolbutamide (Fig. 3).
To examine the functional effects of these mutations in further detail, we recorded KATP currents in giant inside-out oocyte membrane patches. This was performed first in Mg2+-free solution to investigate the ATP sensitivity of the Kir6.2 subunit without the confounding stimulatory action of MgATP mediated by SUR1. Wild-type Kir6.2/SUR1 currents were blocked by addition of low concentrations of ATP to the intra-cellular (bath) solution, as described previously (IC50 ∼7 µm; Table 2). HomG53S, homG53R or homI182V currents exhibited reduced sensitivity to block by ATP, the IC50 values for each being increased to ∼30 µm (Fig. 4A and Table 2). Although hetG53S, hetG53R and hetI182V currents appeared to show a small right-shift in the ATP concentration-response relationship, this did not reach statistical significance. In comparison, the ATP sensitivity of homozygous channels containing the PNDM mutation R201H was shifted further to the right (IC50 ∼300 µm).
We next examined the ATP sensitivity of wild-type and mutant KATP channels in the presence of Mg2+, to mimic more closely the normal cytoplasmic environment (Fig. 4B). Under these conditions, the MgATP concentration–response curves of all homozygous and heterozygous mutant channels were shifted further to the right and heterozygous channels became clearly distinguishable from wild-type channels. The estimated IC50 values of het and hom TNDM channels were ∼50 and 300 µm, respectively, when compared with corresponding values of ∼150 µm and 2 mm for R201H (Table 2). The relative current at a representative physiological MgATP concentration (1 mm) is also given in Table 2 and clearly shows that G53S, G53R and I182V channels would be more open than wild-type, but less open than R201H channels under these conditions, both in the heterozygous and homozygous states.
Although we were able to estimate apparent IC50 values for ATP for the TNDM channels in the presence of Mg2+, the data were not well described by a single inhibitory Hill equation. Instead, we used a function that represents an inhibitory effect of ATP (similar to that observed in Mg2+-free solution), competing with an activatory action of the Mg-nucleotide (Eq. 2). This enabled very good fits of the wild-type and mutant channel data, when the inhibitory Ki values were fixed to the IC50 values measured in Mg2+-free solution, with only minimal adjustments necessary in the activation parameters (Fig. 4B). This equation also describes the net activation by ATP in the presence of Mg2+ as previously seen with Kir6.2 mutations, which were designed to identify the inhibitory ATP-site and which shifted the inhibition by ATP into the millimolar range (29).
We also examined the effect of the sulphonylurea tolbutamide (100 µm) on wild-type and mutant channels in excised patches. No significant difference was observed between the extent of channel inhibition of wild-type, homG53S, homG53R and homI182V channels (56±6, 48±7, 64±7 and 56±5% block, respectively, n=5–7). The extent of block is less than that observed in intact cells, because sulphonylureas also abolish channel activation by MgADP, and therefore have a greater effect when the channels are exposed to cytoplasmic Mg-nucleotides.
DISCUSSION
We have identified three novel heterozygous missense-activating mutations in the KCNJ11 gene, which are associated with the remitting diabetes phenotype. This is the first report of a gene mutation associated with TNDM. Channel dysfunction was not previously expected to be associated with the fluctuating glycaemia seen in TNDM, especially as it caused PNDM, which was thought to be genetically distinct. We have shown that the mechanism for the reduced insulin secretion and diabetes with these mutations is likely to be due to a moderate reduction in KATP channel ATP sensitivity, thereby reducing insulin secretion.
The aetiological role of the novel heterozygous mutations G53S, G53R and I182V in the diabetes in these families is established by, their absence in 200 normal chromosomes, conservation of the altered amino acids across species and the segregation with diabetes in families. The strongest genetic evidence is the presence of de novo mutations in two diabetic subjects born to unaffected parents. The diabetes of the three probands and the sibling from family ISPAD 42 meet clinical criteria for TNDM; they have insulin-requiring diabetes diagnosed in the first 4 months of life with subsequent remission with normoglycaemia achieved in the absence of insulin. One carrier of a TNDM mutation in KCNJ11, the proband from family ISPAD 79 also shows a clear subsequent relapse of their diabetes. Relapse is seen in a sizeable proportion of TNDM due to abnormalities of 6q (3,4).
We have, therefore, shown TNDM is genetically heterogeneous. The commonest cause is an abnormality of the imprinted region of 6q24 that includes ZAC and HYMA1, which accounts for over 70% of cases in a large UK series (4). KCNJ11 mutations account in our series for ∼27% of chromosome 6q24 negative cases and ∼8% of all cases. However no KCNJ11 mutations were found in a French study examining seven 6q24 negative TNDM subjects (14). The genetic heterogeneity in TNDM is reflected in phenotypic heterogeneity. The TNDM patients with KCNJ11 mutations differ from subjects with 6q24 imprinting defects, as previously described (4): the birth weight is higher, the mean age of diagnosis is later (mean 5.2 versus 1.0 weeks) and the time to remission is longer (mean 76 versus 16 weeks). This suggests that the Kir6.2-TNDM subjects have a less severe β-cell defect in utero and early life, but compensatory mechanisms take longer to achieve euglycaemia than that in 6q24-TNDM. This would fit with the idea of a relatively constant but mild β-cell defect associated with a KCNJ11 mutation. Larger numbers of individuals with this genetic aetiology are required for a more detailed comparison of the differences in phenotype of individuals with remitting diabetes due to KCNJ11 defects when compared with chromosome 6q24 abnormalities.
The moderately activating mutations G53S, G53R or I182V are not invariably associated with TNDM. Despite inheriting the same mutation as their children with TNDM, the diabetes in two mothers did not remit and one was not diagnosed until 5 years. Although the mother in family ISPAD 42 did not have TNDM, she does have a milder phenotype than that associated with PNDM mutations (12–16); she was diagnosed at 5 years (the only patient diagnosed after 6 months) and only requires a low dose of insulin (0.36 U/kg) to achieve good glycaemic control. Although the apparently different clinical presentations could represent remission not being diagnosed, it is likely that there is modification of the phenotype by other genetic or environmental factors. The E23K polymorphism does not explain the more severe phenotype in the mothers: in family ISPAD 79, both the proband and his mother were heterozygous, whereas in family ISPAD 42, the proband was heterozygous but the mother was homozygous for the wild-type allele. In light of recent evidence, it is not surprising that there is variability in the phenotype in our families. Patients with the PNDM R201H mutation, for example, have been reported to have varying levels of C-peptide, ranging from undetectable to the normal range (12,30); although the KCNJ11 V59M mutation has been identified in a total of 10 individuals (12,14–16), seven of these show a striking genotype–phenotype correlation with the presentation of developmental delay in addition to diabetes (12,14–16), whereas three have only diabetes. This, therefore, exemplifies the variability in phenotype that can result from the same mutation in Kir6.2 (12,16). Moreover, it is well recognized with other genetic subtypes of monogenic diabetes that there is variability in the phenotype within and between families. In families with HNF1α MODY, there is variation within families of age of onset of diabetes (31), and in families with HNF1β MODY, there is variability within families in the renal, pancreatic and uterine phenotype observed (32). The milder functional effect of G53S, G53R and I182V is consistent with the higher birth weights of these babies when compared with those carrying PNDM KCNJ11 mutations (12). This work shows that the classification of neonatal diabetes needs to involve molecular genetic aetiology, as well as the clinically based subcategories of TNDM and PNDM.
Functional characterization
Homozygous channels containing the TNDM mutations G53S, G53R and I182V or the PNDM R201H mutation exhibited reduced ATP sensitivity. In the absence of Mg2+, the IC50 for ATP was shifted ∼4-fold for all TNDM mutations when compared with an ∼40-fold shift observed with R201H. Neither heterozygous TNDM nor PNDM channels exhibited significantly shifted IC50 values when compared with wild-type channels under these conditions. In the presence of Mg2+, however, more profound and significant shifts in the apparent ATP sensitivities of both homozygous and heterozygous channels were observed. However, the ATP-sensitivities of hom. and hetR201H channels were still shifted more severely than those of channels containing the TNDM mutations. This is clearly seen in the relative magnitudes of the KATP currents at 1 mm MgATP, a concentration that might be found in a resting pancreatic β-cell (33). The reduced ATP sensitivity accounts for the greater resting whole cell currents observed for both het and hom mutant channels when compared with wild-type. In the β-cells, the greater the KATP current at raised glucose concentrations, the more difficult it would be to depolarize the β-cell and to trigger insulin release.
The functional characterization of these mutations, therefore, supports the concept that there is a range of diabetes phenotypes associated with activating mutations of KCNJ11 and that the severity of reduction in ATP sensitivity determines the phenotype of the diabetes. We show that all three novel mutations associated with remitting diabetes exhibit a remarkably consistent moderate reduction in the sensitivity to ATP, which would explain the reduced insulin secretion seen in these subjects. The reduction in ATP sensitivity of the TNDM associated mutations (Fig. 4) is less marked than that seen with R201H, the commonest mutation seen in PNDM (Fig. 4) (12).
The identification of mutations in the KCNJ11 gene that cause TNDM has implications for β-cell physiology. We considered a gain of function defect in Kir6.2 might be expected to produce a fixed β-cell deficit. Thus, we had not anticipated a fluctuating phenotype. A fluctuating phenotype could occur for several reasons such as because of a reduced insulin requirement at the time of remission or because there is compensation at the level of the β-cell, pancreas, or whole body, which can over come this fixed defect. Another possibility is that the mutated KCNJ11 gene also alters β-cell turnover. An intriguing support for the latter is that animal models lacking the KCNJ11 gene have suggested that the KATP channel is critical for normal islet architecture and β-cell survival (34). In man where direct examination of the pancreas is impossible, the mechanism of the restoration of euglycaemia cannot be defined. To answer this question, it will be necessary to establish an animal model of a moderately activating KCNJ11 mutation (35), similar to that developed for the imprinted region on 6q24 (11).
The finding that the mutations associated with TNDM did not prevent the action of tolbutamide in excised patches or whole oocytes is encouraging, as it suggests that patients carrying these mutations might respond clinically to sulphonylureas. This is most important in the patients who remit and are likely to remain diabetic in the long term.
Our finding supports the hypothesis that there is a spectrum of clinical presentations of diabetes associated with activating mutations of KCNJ11 (Fig. 5). TNDM being an intermediate phenotype between susceptibility to Type 2 diabetes (E23K) and mutations that cause PNDM. More severe activating mutations than those causing PNDM alone may cause neurological features such as developmental delay, muscle weakness and epilepsy (12,18). Therefore, functional characterization has shown a graded decrease in ATP sensitivity, which fits with this spectrum of clinical presentations of diabetes.
In conclusion, we present the first report of activating mutations in KCNJ11 causing a remitting diabetic phenotype, typical of TNDM. These mutations impair ATP sensitivity of the KATP channel, but do not have as profound an effect as the mutations reported recently in cases of PNDM (12,18). It, therefore, appears that activating mutations in Kir6.2 can produce a spectrum of disease, ranging from an increased risk of Type 2 diabetes (E23K) through TNDM to non-syndromic and syndromic PNDM, depending on the degree to which the mutation alters KATP channel ATP sensitivity.
MATERIALS AND METHODS
Patients
DNA was sequenced from 11 probands with TNDM. All these patients had insulin-treated diabetes diagnosed before 6 months (median 14 days, range 1–105 days) and subsequently had normoglycaemia when completely off insulin. Patients were recruited through the British Paediatric Association Surveillance Unit, clinical geneticists, neonatologists, paediatric endocrinologists, Diabetes UK and the authors of TNDM reports, during the last 20 years. These 11 cases include the seven negative cases from the previously reported study (4). Imprinted abnormalities of chromosome 6q24, as previously described (5–8), mutations in the glucokinase (GCK) gene, exocrine pancreatic insufficiency and pancreatic agenesis were excluded. Informed consent was obtained from all participants or their parents.
Mutation analysis
The coding region and the intron–exon boundaries of the ATP-sensitive channel subunit Kir6.2 (KCNJ11) gene were amplified from genomic DNA by PCR, as previously described (12). The products were sequenced by standard methods on an ABI 3100 (Applied Biosystems, Warrington, UK). All polymorphisms identified, including the E23K variant, were recorded in all individuals sequenced. Family relationships were confirmed using a panel of 10 microsatellite markers (36). KCNJ11 was also sequenced in 100 non-diabetic individuals.
Functional studies
Molecular biology.
Human Kir6.2 (GenBank accession no. NM000525) containing the common polymorphisms E23, I377 and rat SUR1 (GenBank accession no. L40624) were used in this study. Site-directed mutagenesis of Kir6.2 was performed using the QuickChange™XL system (Stratagene). Wild-type and mutant cDNAs were cloned in the pBF vector, and capped mRNA prepared using the mMESSAGE mMACHINE large scale in vitro transcription kit (Ambion, Austin, TX, USA), as previously described (37,38).
Oocyte collection.
Female Xenopus laevis were anaesthetized with MS222 (2 g/l added to the water). One ovary was removed via a mini-laparotomy, the incision sutured and the animal allowed to recover. Immature stage V–VI oocytes were incubated for 60 min with 1.0 mg/ml collagenase (Roche, type A) and manually defolliculated. Oocytes were injected with 0.8 ng wild-type or mutant Kir6.2 mRNA and ∼4 ng of SUR1 mRNA (giving a 1 : 5 ratio). For each batch of oocytes, all mutations were injected, to enable direct comparison of their effects. Isolated oocytes were maintained in Barth's solution and studied 1–6 days after injection (37).
Electrophysiology
Two-electrode voltage-clamp method. Currents were recorded from Xenopus oocytes 1–3 days after mRNA injection. Whole-cell currents were recorded from intact oocytes using the two-electrode voltage-clamp method, filtered at 1 kHz and digitized at 4 kHz. Oocytes were constantly perfused at 20–24°C with a solution containing (in mm): 90 KCl, 1 MgCl2, 1.8 CaCl2 and 5 HEPES ( pH7.4 with KOH). Metabolic inhibition was produced by 3 mm Na-azide. Whole-cell currents were monitored in response to voltage steps of ±20 mV from a holding potential of −10 mV. Tolbutamide (0.5 mm) was tested on all oocytes to confirm the observed current flowed through KATP channels.
Patch-clamp analysis. Macroscopic currents were recorded from giant excised inside-out patches at a holding potential of 0 mV and at 20–24°C using either an Axopatch 200B patch-clamp amplifier and pclamp software (Axon Instruments, Foster City, CA, USA) (37) or an EPC10 amplifier with HEKA patchmaster software (Digitimer Ltd, Welwyn Garden City, UK). The pipette (external) solution contained (in mM): 140 KCl, 1.2 MgCl2, 2.6 CaCl2 and 10 HEPES (pH 7.4 with KOH). The 1.3 mm Mg2+ intra-cellular (bath) solution contained (in mm): 107 KCl, 2 MgCl2, 1 CaCl2, 10 EGTA and 10 HEPES (pH 7.2 with KOH; final [K+] ∼140 mm). The Mg-free solution contained (in mm): 107 KCl, 2.6 CaCl2, 10 EDTA and 10 HEPES (pH 7.2 with KOH; final [K+] ∼140 mm. K2ATP was added to the earlier-mentioned solutions to prepare a 10 mm stock, and in the case of the Mg2+-containing solutions, MgCl2 was added to maintain the free [Mg2+] at ∼1.3 mm. Tolbutamide was prepared as a 100 mm stock solution in 0.1 m KOH. The pH of all solutions was readjusted as required. Rapid exchange of solutions was achieved by positioning the patch in the mouth of one of a series of adjacent inflow pipes placed in the bath.
Currents were recorded in response to repetitive 3 s voltage ramps from −110 to +100 mV. The slope conductance was measured by fitting a straight line to the current–voltage relation between−20 and −80 mV: the average response to five consecutive ramps was calculated in each solution.
ACKNOWLEDGEMENTS
We thank all the patients, their families and their clinicians for their help with these studies. The authors wish to thank Dr Shozeb Haider for providing the structural model of KirBac1.1 and Kir3.1, Drs Alan Jaap and Frank Hind for their assistance with family ISPAD 79 and Dr Robinson for his assistance with family ISPAD 50. This work was funded by the Wellcome Trust and Diabetes UK. A.L.G. and F.R. are Diabetes UK RD Lawrence Research Fellows. A.T.H. is a Wellcome Trust Research Leave Clinical Fellow, F.M.G. is a Wellcome Trust Senior Research Fellow in Clinical Science, E.R.P. is a Wellcome Trust Clinical Research Fellow and F.M.A. is the Royal Society GlaxoSmithKline Research Professor.
The authors wish it to be known that, in their opinion, the first three authors should be regarded as joint First Authors.
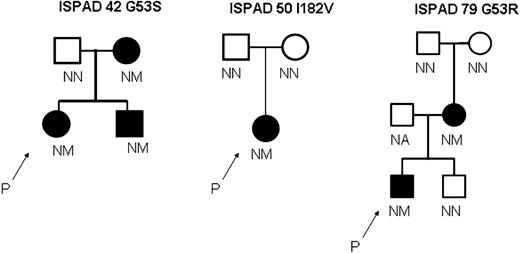
Figure 1. Diabetes status and mutations in the gene encoding Kir6.2 in three families. These partial pedigrees show families with G53S, G53R and I182V mutations. In all pedigrees, spontaneous mutations explain the absence of diabetes in the parents and its presence in a child. Squares represent males, and circles represent females. Filled circles and squares represent persons with diabetes. A two-letter code for allele status is shown underneath each symbol: M denotes mutation, N no mutation and NA not available for testing. P and an arrow denote the proband in each family (the first affected member recruited for this study). Amino acids are denoted by their single-letter codes.
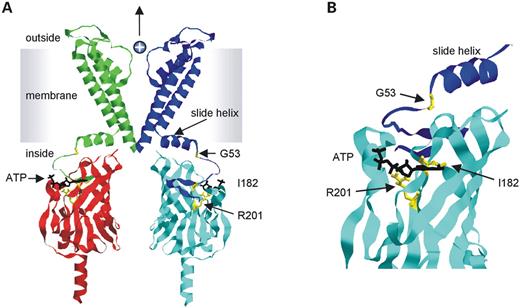
Figure 2. Location of mutated residues in a structural model of Kir6.2. (A) Homology model of Kir6.2, based on the crystal structures of KirBac1.1 (39) and Kir3.1 (40), viewed from the side. For clarity, each subunit is shown in a different colour and only two transmembrane domains and two cytosolic domains are illustrated. Residues mutated in TNDM (G53, I182) and PNDM (R201) are coloured yellow and shown in ball-and-stick format. ATP is shown in black. (B) Close-up of the ATP-binding site showing the location of the residues mutated in TNDM and PNDM. Both I182 and R201 lie within the ATP-binding pocket, whereas G53 lies some distance away.
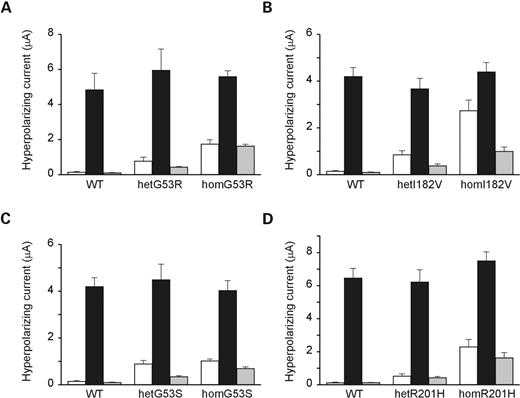
Figure 3. Effects of mutations on whole-cell KATP currents. Mean steady state whole-cell currents evoked by a voltage step from −10 to −30 mV before (white bar) and after (black bar) application of 3 mm azide and in the presence of 3 mm azide and 0.5 mm tolbutamide. Currents were recorded from Xenopus oocytes co-expressing SUR1 and either wild-type Kir6.2 (WT) or (mutant Kir6.2 (hom) alone or a 1 : 1 mixture of WT and mutant Kir6.2 (het), for the TNDM mutations G53R (A), G53S (C), I182V (B) or the PNDM mutation R201H (D). The number of oocytes is 8–12 in each case.
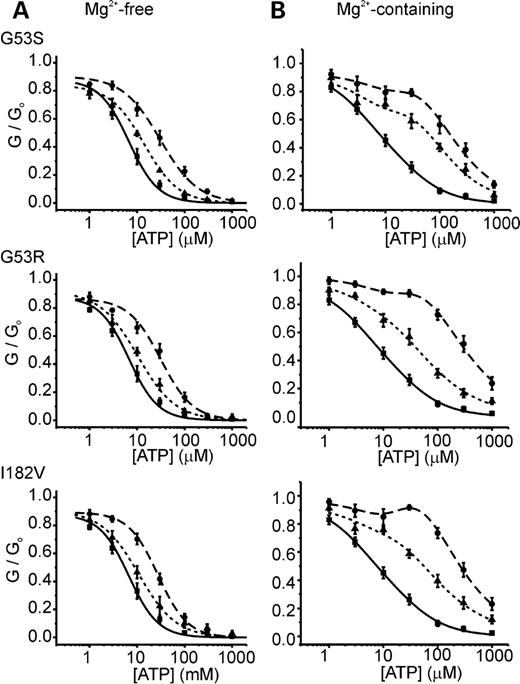
Figure 4. ATP concentration–response relationships for wild-type and mutant KATP channels. ATP dose–response relationships for channels comprising SUR1 with wild-type Kir6.2, Kir6.2-G53S, Kir6.2-G53R or Kir6.2-I182V. Macroscopic currents were recorded in giant inside out patches excised from Xenopus oocytes expressing homozygous or heterozygous channels (1 : 1 mixture of wild-type and mutant Kir6.2), in the absence (A) or presence (B) of Mg2+ in the bath solution. The wild-type data is replicated between graphs. Black squares, wild-type; black diamonds, homozygous mutant; black triangles, heterozygous mutant. MgATP concentration–response curves are shown with the best fit of the data to Eq. (2), with the Ki fixed to the IC50 obtained in the absence of Mg2+ and the following additional parameters (Ka, hi, ha, A): wt (10.0, 1.0, 2.4, 0.8), homG53S (12.2, 0.7, 3.5, 1.3), homG53R (20.0, 0.6, 2.7, 1.4), homI182V (11.8, 0.7, 3.5, 1.8), hetG53S (14.8, 0.8, 2.7, 2.3), hetG53R (11.5, 0.6, 2.0, 0.6) and hetI182V (10.0, 0.6, 2.5, 0.9).
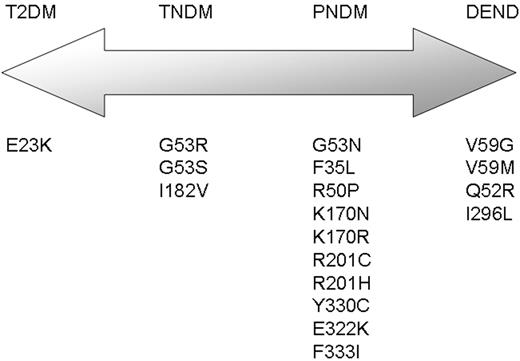
Figure 5. Schematic representation of the spectrum of clinical presentations of diabetes associated with mutations in the KCNJ11 gene. T2DM, type 2 diabetes; TNDM, transient neonatal diabetes; PNDM, permanent neonatal diabetes; DEND, developmental delay, muscle weakness, epilepsy, dysmorphic features and neonatal diabetes. Mutations shown are taken from Refs. (12–16).
Clinical characteristics of patients with TNDM and KCNJ11 mutations
| Family number | ISPAD 42 | ISPAD 79 | ISPAD 50 | |||
| Subject | Proband (II:1) | Brother (II:2) | Mother (I:2) | Proband (III:1) | Mother (II:2) | Proband (II:1) |
| Gender (male/female) | F | M | F | M | F | F |
| Mutation | G53S | G53S | G53S | G53R | G53R | I182V |
| Birth weight kg/percentile | 2.02/85 | 3.37/92 | 2.81/9 | 3.57/94 | 2.3/0.2 | 1.535/0.1 |
| Gestation weeks (birth details) | 32 (Caesarean section, maternal pre-eclampsia) | 36.5 (induced, maternal pre-eclampsia) | 40 | 38.5 (induced, maternal diabetes) | 41 | 36 |
| Diabetes presentation | ||||||
| Age diagnosed (weeks) | 2 | 3 | 260 | 16 | 11 | 1 |
| Presentation | Hyperglycaemia | Hyperglycaemia | Routine urine test | Polyuria and polydipsia | Hyperglycaemia | Hyperglycaemia |
| Glucose (mmol/l) | 16.8 | 15.2 | 11.2 | 25.8 | — | 16.2 |
| C-peptide (pmol/l) | — | — | — | 439 | — | <165 |
| Pancreatic autoantibodies | — | — | — | GAD negative | — | GAD and ICA negative |
| Early diabetes | ||||||
| Maximum insulin dose (U/kg/day) | 1.6 | 0.73 | 1.0 | 1.5 | 0.8 | |
| Age at maximum insulin dose (months) | 8 | 9 | 10 | 16 | 6 | |
| HbA1c at maximum insulin dose (%) | 7.0 | 8.7 | 4.4% | — | ||
| Diabetes remission? | Yes | Yes | No | Yes | No | Yes |
| Age when insulin stopped (months) | 14 months | 20 months | No remission | 17 months | No remission | 7 months |
| Lowest HbA1c of treatment (reference range) | 5.3% (<6.5) | 6.4% (4.2–6.3) | — | 4.3% (<6.5) | — | 5.8%(4.1–6.5) |
| Present | ||||||
| Diabetes relapse? | No | ? | Not applicable | Yes | Not applicable | No |
| Age of relapse (years) | 3.4 | |||||
| Current age (years) | 4.5 | 2.1 | 10.7 | 42.4 | 5.7 | |
| Current insulin dose (U/kg/day) | Nil | Occasional insulin if glucose >12 mmol/l | 0.36 | 0.6 | 0.8 | Nil |
| Current HbA1c (%) | 6.3 | 7.1 | 6.0 | 8.4 | 7.0 | — |
| Neurological features | None | None | None | Learning difficulties | Learning difficulties | None |
| Family number | ISPAD 42 | ISPAD 79 | ISPAD 50 | |||
| Subject | Proband (II:1) | Brother (II:2) | Mother (I:2) | Proband (III:1) | Mother (II:2) | Proband (II:1) |
| Gender (male/female) | F | M | F | M | F | F |
| Mutation | G53S | G53S | G53S | G53R | G53R | I182V |
| Birth weight kg/percentile | 2.02/85 | 3.37/92 | 2.81/9 | 3.57/94 | 2.3/0.2 | 1.535/0.1 |
| Gestation weeks (birth details) | 32 (Caesarean section, maternal pre-eclampsia) | 36.5 (induced, maternal pre-eclampsia) | 40 | 38.5 (induced, maternal diabetes) | 41 | 36 |
| Diabetes presentation | ||||||
| Age diagnosed (weeks) | 2 | 3 | 260 | 16 | 11 | 1 |
| Presentation | Hyperglycaemia | Hyperglycaemia | Routine urine test | Polyuria and polydipsia | Hyperglycaemia | Hyperglycaemia |
| Glucose (mmol/l) | 16.8 | 15.2 | 11.2 | 25.8 | — | 16.2 |
| C-peptide (pmol/l) | — | — | — | 439 | — | <165 |
| Pancreatic autoantibodies | — | — | — | GAD negative | — | GAD and ICA negative |
| Early diabetes | ||||||
| Maximum insulin dose (U/kg/day) | 1.6 | 0.73 | 1.0 | 1.5 | 0.8 | |
| Age at maximum insulin dose (months) | 8 | 9 | 10 | 16 | 6 | |
| HbA1c at maximum insulin dose (%) | 7.0 | 8.7 | 4.4% | — | ||
| Diabetes remission? | Yes | Yes | No | Yes | No | Yes |
| Age when insulin stopped (months) | 14 months | 20 months | No remission | 17 months | No remission | 7 months |
| Lowest HbA1c of treatment (reference range) | 5.3% (<6.5) | 6.4% (4.2–6.3) | — | 4.3% (<6.5) | — | 5.8%(4.1–6.5) |
| Present | ||||||
| Diabetes relapse? | No | ? | Not applicable | Yes | Not applicable | No |
| Age of relapse (years) | 3.4 | |||||
| Current age (years) | 4.5 | 2.1 | 10.7 | 42.4 | 5.7 | |
| Current insulin dose (U/kg/day) | Nil | Occasional insulin if glucose >12 mmol/l | 0.36 | 0.6 | 0.8 | Nil |
| Current HbA1c (%) | 6.3 | 7.1 | 6.0 | 8.4 | 7.0 | — |
| Neurological features | None | None | None | Learning difficulties | Learning difficulties | None |
Clinical characteristics of patients with TNDM and KCNJ11 mutations
| Family number | ISPAD 42 | ISPAD 79 | ISPAD 50 | |||
| Subject | Proband (II:1) | Brother (II:2) | Mother (I:2) | Proband (III:1) | Mother (II:2) | Proband (II:1) |
| Gender (male/female) | F | M | F | M | F | F |
| Mutation | G53S | G53S | G53S | G53R | G53R | I182V |
| Birth weight kg/percentile | 2.02/85 | 3.37/92 | 2.81/9 | 3.57/94 | 2.3/0.2 | 1.535/0.1 |
| Gestation weeks (birth details) | 32 (Caesarean section, maternal pre-eclampsia) | 36.5 (induced, maternal pre-eclampsia) | 40 | 38.5 (induced, maternal diabetes) | 41 | 36 |
| Diabetes presentation | ||||||
| Age diagnosed (weeks) | 2 | 3 | 260 | 16 | 11 | 1 |
| Presentation | Hyperglycaemia | Hyperglycaemia | Routine urine test | Polyuria and polydipsia | Hyperglycaemia | Hyperglycaemia |
| Glucose (mmol/l) | 16.8 | 15.2 | 11.2 | 25.8 | — | 16.2 |
| C-peptide (pmol/l) | — | — | — | 439 | — | <165 |
| Pancreatic autoantibodies | — | — | — | GAD negative | — | GAD and ICA negative |
| Early diabetes | ||||||
| Maximum insulin dose (U/kg/day) | 1.6 | 0.73 | 1.0 | 1.5 | 0.8 | |
| Age at maximum insulin dose (months) | 8 | 9 | 10 | 16 | 6 | |
| HbA1c at maximum insulin dose (%) | 7.0 | 8.7 | 4.4% | — | ||
| Diabetes remission? | Yes | Yes | No | Yes | No | Yes |
| Age when insulin stopped (months) | 14 months | 20 months | No remission | 17 months | No remission | 7 months |
| Lowest HbA1c of treatment (reference range) | 5.3% (<6.5) | 6.4% (4.2–6.3) | — | 4.3% (<6.5) | — | 5.8%(4.1–6.5) |
| Present | ||||||
| Diabetes relapse? | No | ? | Not applicable | Yes | Not applicable | No |
| Age of relapse (years) | 3.4 | |||||
| Current age (years) | 4.5 | 2.1 | 10.7 | 42.4 | 5.7 | |
| Current insulin dose (U/kg/day) | Nil | Occasional insulin if glucose >12 mmol/l | 0.36 | 0.6 | 0.8 | Nil |
| Current HbA1c (%) | 6.3 | 7.1 | 6.0 | 8.4 | 7.0 | — |
| Neurological features | None | None | None | Learning difficulties | Learning difficulties | None |
| Family number | ISPAD 42 | ISPAD 79 | ISPAD 50 | |||
| Subject | Proband (II:1) | Brother (II:2) | Mother (I:2) | Proband (III:1) | Mother (II:2) | Proband (II:1) |
| Gender (male/female) | F | M | F | M | F | F |
| Mutation | G53S | G53S | G53S | G53R | G53R | I182V |
| Birth weight kg/percentile | 2.02/85 | 3.37/92 | 2.81/9 | 3.57/94 | 2.3/0.2 | 1.535/0.1 |
| Gestation weeks (birth details) | 32 (Caesarean section, maternal pre-eclampsia) | 36.5 (induced, maternal pre-eclampsia) | 40 | 38.5 (induced, maternal diabetes) | 41 | 36 |
| Diabetes presentation | ||||||
| Age diagnosed (weeks) | 2 | 3 | 260 | 16 | 11 | 1 |
| Presentation | Hyperglycaemia | Hyperglycaemia | Routine urine test | Polyuria and polydipsia | Hyperglycaemia | Hyperglycaemia |
| Glucose (mmol/l) | 16.8 | 15.2 | 11.2 | 25.8 | — | 16.2 |
| C-peptide (pmol/l) | — | — | — | 439 | — | <165 |
| Pancreatic autoantibodies | — | — | — | GAD negative | — | GAD and ICA negative |
| Early diabetes | ||||||
| Maximum insulin dose (U/kg/day) | 1.6 | 0.73 | 1.0 | 1.5 | 0.8 | |
| Age at maximum insulin dose (months) | 8 | 9 | 10 | 16 | 6 | |
| HbA1c at maximum insulin dose (%) | 7.0 | 8.7 | 4.4% | — | ||
| Diabetes remission? | Yes | Yes | No | Yes | No | Yes |
| Age when insulin stopped (months) | 14 months | 20 months | No remission | 17 months | No remission | 7 months |
| Lowest HbA1c of treatment (reference range) | 5.3% (<6.5) | 6.4% (4.2–6.3) | — | 4.3% (<6.5) | — | 5.8%(4.1–6.5) |
| Present | ||||||
| Diabetes relapse? | No | ? | Not applicable | Yes | Not applicable | No |
| Age of relapse (years) | 3.4 | |||||
| Current age (years) | 4.5 | 2.1 | 10.7 | 42.4 | 5.7 | |
| Current insulin dose (U/kg/day) | Nil | Occasional insulin if glucose >12 mmol/l | 0.36 | 0.6 | 0.8 | Nil |
| Current HbA1c (%) | 6.3 | 7.1 | 6.0 | 8.4 | 7.0 | — |
| Neurological features | None | None | None | Learning difficulties | Learning difficulties | None |
ATP IC50 values of wild-type and mutant KATP channels
| Mutation (n) | IC50 in the absence of Mg [n] | Apparent IC50 in the presence of Mg [n] | Current in 1 mm MgATP |
|---|---|---|---|
| Wild-type | 6.7 (5.5, 8.2) [11] | 10.3 (8.9, 12.0) [8] | 2.4±0.9 |
| homG53S | 30.8 (24.7, 38.4) [10] | 175 (136, 225) [6] | 14±2 |
| homG53R | 31.6 (24.9, 40.0) [5] | 330 (255, 427) [8] | 24±5 |
| homI182V | 27.5 (24.6, 30.7) [9] | 302 (243, 376) [7] | 23±4 |
| homR201H | 299 (286, 313) [7] | 1960 (1830, 2100) [5] | 68±2 |
| hetG53S | 12.4 (11.4, 13.5) [5] | 60 (44, 83) [5] | 6.1±1.0 |
| hetG53R | 10.2 (8.2, 12.8) [6] | 43 (32, 58) [6] | 11±2 |
| hetI182V | 10.8 (8.1, 14.5) [5] | 61 (50, 74) [5] | 12±3 |
| hetR201H | 11.1 (9.5, 12.9) [7] | 143 (118, 173) [5] | 22±5 |
| Mutation (n) | IC50 in the absence of Mg [n] | Apparent IC50 in the presence of Mg [n] | Current in 1 mm MgATP |
|---|---|---|---|
| Wild-type | 6.7 (5.5, 8.2) [11] | 10.3 (8.9, 12.0) [8] | 2.4±0.9 |
| homG53S | 30.8 (24.7, 38.4) [10] | 175 (136, 225) [6] | 14±2 |
| homG53R | 31.6 (24.9, 40.0) [5] | 330 (255, 427) [8] | 24±5 |
| homI182V | 27.5 (24.6, 30.7) [9] | 302 (243, 376) [7] | 23±4 |
| homR201H | 299 (286, 313) [7] | 1960 (1830, 2100) [5] | 68±2 |
| hetG53S | 12.4 (11.4, 13.5) [5] | 60 (44, 83) [5] | 6.1±1.0 |
| hetG53R | 10.2 (8.2, 12.8) [6] | 43 (32, 58) [6] | 11±2 |
| hetI182V | 10.8 (8.1, 14.5) [5] | 61 (50, 74) [5] | 12±3 |
| hetR201H | 11.1 (9.5, 12.9) [7] | 143 (118, 173) [5] | 22±5 |
IC50 values are presented as geometric mean (1 standard error range). The current in 1 mm MgATP is expressed as a percentage of the control current in the absence of nucleotide.
ATP IC50 values of wild-type and mutant KATP channels
| Mutation (n) | IC50 in the absence of Mg [n] | Apparent IC50 in the presence of Mg [n] | Current in 1 mm MgATP |
|---|---|---|---|
| Wild-type | 6.7 (5.5, 8.2) [11] | 10.3 (8.9, 12.0) [8] | 2.4±0.9 |
| homG53S | 30.8 (24.7, 38.4) [10] | 175 (136, 225) [6] | 14±2 |
| homG53R | 31.6 (24.9, 40.0) [5] | 330 (255, 427) [8] | 24±5 |
| homI182V | 27.5 (24.6, 30.7) [9] | 302 (243, 376) [7] | 23±4 |
| homR201H | 299 (286, 313) [7] | 1960 (1830, 2100) [5] | 68±2 |
| hetG53S | 12.4 (11.4, 13.5) [5] | 60 (44, 83) [5] | 6.1±1.0 |
| hetG53R | 10.2 (8.2, 12.8) [6] | 43 (32, 58) [6] | 11±2 |
| hetI182V | 10.8 (8.1, 14.5) [5] | 61 (50, 74) [5] | 12±3 |
| hetR201H | 11.1 (9.5, 12.9) [7] | 143 (118, 173) [5] | 22±5 |
| Mutation (n) | IC50 in the absence of Mg [n] | Apparent IC50 in the presence of Mg [n] | Current in 1 mm MgATP |
|---|---|---|---|
| Wild-type | 6.7 (5.5, 8.2) [11] | 10.3 (8.9, 12.0) [8] | 2.4±0.9 |
| homG53S | 30.8 (24.7, 38.4) [10] | 175 (136, 225) [6] | 14±2 |
| homG53R | 31.6 (24.9, 40.0) [5] | 330 (255, 427) [8] | 24±5 |
| homI182V | 27.5 (24.6, 30.7) [9] | 302 (243, 376) [7] | 23±4 |
| homR201H | 299 (286, 313) [7] | 1960 (1830, 2100) [5] | 68±2 |
| hetG53S | 12.4 (11.4, 13.5) [5] | 60 (44, 83) [5] | 6.1±1.0 |
| hetG53R | 10.2 (8.2, 12.8) [6] | 43 (32, 58) [6] | 11±2 |
| hetI182V | 10.8 (8.1, 14.5) [5] | 61 (50, 74) [5] | 12±3 |
| hetR201H | 11.1 (9.5, 12.9) [7] | 143 (118, 173) [5] | 22±5 |
IC50 values are presented as geometric mean (1 standard error range). The current in 1 mm MgATP is expressed as a percentage of the control current in the absence of nucleotide.
References
Shield, J.P. (
Polak, M. and Shield, J. (
von Muhlendahl, K.E. and Herkenhoff, H. (
Temple, I.K., Gardner, R.J., Mackay, D.J., Barber, J.C., Robinson, D.O. and Shield, J.P. (
Temple, I.K., James, R.S., Crolla, J.A., Sitch, F.L., Jacobs, P.A., Howell, W.M., Betts, P., Baum, J.D. and Shield, J. (
Gardner, R.J., Robinson, D.O., Lamont, L., Shield, J.P. and Temple, I.K. (
Temple, I.K., Gardner, R.J., Robinson, D.O., Kibirige, M.S., Ferguson, A.W., Baum, J.D., Barber, J.C.K., James, R.S. and Shield, J.P.H. (
Gardner, R.J., Mackay, D.J., Mungall, A.J., Polychronakos, C., Siebert, R., Shield, J.P., Temple, I.K. and Robinson, D.O. (
Kamiya, M., Judson, H., Okazaki, Y., Kusakabe, M., Muramatsu, M., Takada, S., Takagi, N., Arima, T., Wake, N., Kamimura, K. et al. (
Arima, T., Drewell, R.A., Oshimura, M., Wake, N. and Surani, M.A. (
Ma, D., Shield, J.P.H., Dean, W., Leclerc, I., Knauf, C., Burcelin, R., Rutter, G.A. and Kelsey, G. (
Gloyn, A.L., Pearson, E.R., Antcliff, J.F., Proks, P., Bruining, G.J., Slingerland, A.S., Howard, N., Srinivasan, S., Silva, J.M., Molnes, J. et al. (
Gloyn, A.L., Cummings, E.A., Edghill, E.L., Harries, L.W., Scott, R., Costa, T., Temple, I.K., Hattersley, A.T. and Ellard, S. (
Vaxillaire, M., Populaire, C., Buisiah, K., Cave, H., Gloyn, A.L., Hattersley A, T., Czernichow, P., Froguel, P. and Polak, M. (
Sagen, J.V., Raeder, H., Hathout, E., Shehadeh, N., Gudmundsson, K., Baevre, H., Abuelo, D., Phornphutkul, C., Molnes, J., Bell, G. et al. (
Massa, O., Iafusco, D., D'Amato, E., Gloyn, A.L., Hattersley, A.T., Pasquino, B., Tonini, G., Dammacco, F., Zanette, G., Meschi, F. et al. (
Clement, J.P.I., Kunjilwar, K., Gonzalez, G., Schwanstecher, M., Panten, U., Aguilar-Bryan, L. and Bryan, J. (
Proks, P., Antcliff, J.F., Lippiat, J., Gloyn, A.L., Hattersley, A.T. and Ashcroft, F.M. (
Gloyn, A.L., Weedon, M.N., Owen, K., Turner, M.J., Knight, B.A., Hitman, G.A., Walker, M., Levy, J.C., Sampson, M., Halford, S. et al. (
Nielsen, E.M., Hansen, L., Carstensen, B., Echwald, S.M., Drivsholm, T., Glumer, C., Thorsteinsson, B., Borch-Johnsen, K., Hansen, T. and Pedersen, O. (
Love-Gregory, L., Wasson, J., Lin, J., Skolnick, G., Suarez, B. and Permutt, M.A. (
Florez, J., Burtt, N., de Bakker, P., Almgren, P., Tuomi, T., Holmkvist, J., Gaudet, D., Hudson, T., Schaffner, S., Daly, M. et al. (
Riedel, M.J., Boora, P., Steckley, D., de Vries, G. and Light, P.E. (
Schwanstecher, C., Neugebauer, B., Schulz, M. and Schwanstecher, M. (
Schwanstecher, C., Meyer, U. and Schwanstecher, M. (
Thomas, P.M., Yuyang, Y. and Lightner, E. (
Trapp, S., Haider, S., Jones, P., Sansom, M.S. and Ashcroft, F.M. (
Li, L., Wang, J. and Drain, P. (
Gribble, F.M., Tucker, S.J, Haug, T and Ashcroft, F.M. (
Zung, A., Glaser, B., Nimri, R. and Zadik, Z. (
Klupa, T., Warram, J.H., Antonellis, A., Pezzolesi, M., Nam, M., Malecki, M.T., Doria, A., Rich, S.S. and Krolewski, A.S. (
Bingham, C. and Hattersley, A.T. (
Detimary, P., Dejonghe, S., Ling, Z., Pipeleers, D., Schuit, F. and Henquin, J.C. (
Seino, S., Iwanaga, T., Nagashima, K. and Miki, T. (
Koster, J.C., Marshall, B.A., Ensor, N., Corbett, J.A. and Nichols, C.G. (
Frayling, T.M., McCarthy, M.I., Walker, M., Levy, J.C., O'Rahilly, S., Hitman, G.A., Rao, P.V., Bennett, A.J., Jones, E.C., Menzel, S. et al. (
Gribble, F., Ashfield, R., Ammala, C. and Ashcroft, F.M. (
Trapp, S., Proks, P., Tucker, S.J. and Ashcroft, F.M. (
Kuo, A., Gulbis, J.M., Antcliff, J.F., Rahman, T., Lowe, E.D., Zimmer, J., Cuthbertson, J., Ashcroft, F.M., Ezaki, T. and Doyle, D.A. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}