




Abstract
McKusick–Kaufman syndrome (MKS) is an autosomal recessive disorder characterized by post-axial polydactyly, congenital heart defects and hydrometrocolpos, a congenital structural abnormality of female genitalia. Mutations in the MKKS gene have also been shown to cause some cases of Bardet–Biedl syndrome (BBS) which is characterized by obesity, pigmentary retinopathy, polydactyly, renal abnormalities and hypogenitalism with secondary features of hypertension and diabetes. Although there is overlap in clinical features between MKS and BBS, MKS patients are not obese and do not develop retinopathy or have learning disabilities. To further explore the pathophysiology of BBS and the related disorder MKS, we have developed an Mkks−/− mouse model. This model shows that the absence of Mkks leads to retinal degeneration through apoptosis, failure of spermatozoa flagella formation, elevated blood pressure and obesity. The obesity is associated with hyperphagia and decreased activity. In addition, neurological screening reveals deficits in olfaction and social dominance. The mice do not have polydactyly or vaginal abnormalities. The phenotype of the Mkks−/− mice closely resembles the phenotype of other mouse models of BBS (Bbs2−/− and Bbs4−/−). These observations suggest that the complete absence of MKKS leads to BBS while the MKS phenotype is likely to be due to specific mutations.
INTRODUCTION
McKusick–Kaufman syndrome (MKS) is an autosomal recessive disorder characterized by post-axial polydactyly, congenital heart defects and hydrometrocolpos, a congenital structural abnormality of female genitalia (1,2). The MKKS gene was initially mapped and identified in a large Old Order Amish kindred. The predicted MKKS protein shows homology to the alpha subunit of the Thermoplasma acidophilum thermosome (1,3), a prokaryotic chaperonin complex with structural similarity to a eukaryotic chaperonin called ‘tailless complex polypeptide ring complex’ (TRiC), a hetero-oligomeric chaperonin involved in the folding of many proteins including tubulin and actin (4). Mutations in the MKKS gene have also been shown to cause some cases of Bardet–Biedl syndrome (BBS) which is characterized by obesity, pigmentary retinopathy, polydactyly, renal malformations, functional abnormalities and hypogenitalism with secondary features of hypertension and diabetes (5–9). Although there is significant overlap in clinical features between MKS and BBS, MKS syndrome patients are not obese and do not develop retinopathy or learning disabilities.
To date, eight BBS loci have been mapped and the BBS gene at each of these loci has been identified including the MKKS gene (also referred to as BBS6) (3,10–18). With the exception of MKKS, the other known BBS proteins have no significant similarity to chaperonins. However, BBS4 and BBS8 contain tetratricopeptide repeat domains indicating that they may interact with other proteins. The recently identified BBS3 gene is an ADP-ribosylation factor-like gene (17,18).
Recent data indicate that BBS genes are involved in cilia function and intracellular transport. BBS genes are conserved in ciliated organisms, but not in non-ciliated organisms (16,17). Some BBS proteins have been shown to localize to the basal body of ciliated cells and to be involved in intraflagellar transport (15,19). We recently demonstrated that Bbs4- and Bbs2-knockout mice have features of the human disorder and that absence of these proteins leads to failure of spermatozoa flagella formation, but not the formation of cilia in general. Furthermore, we demonstrated that absence of Bbs2 or Bbs4 proteins does not completely disrupt initial photoreceptor cell development, although aberrant disc morphogenesis is apparent. Connecting cilia are also present in animals deficient for Bbs2 or Bbs4; but photoreceptors undergo cell death due to apoptosis (20,21). To further explore the pathophysiology of BBS and the related disorder MKS, we have now developed and characterized an Mkks−/− mouse model.
RESULTS
Generation of Mkks-knockout mice
Mkks-deficient mice were generated using a gene-targeting construct designed to remove exon 3 of the Mkks gene in its entirety (Fig. 1A). The targeting construct was transfected into embryonic stem (ES) cells and ∼1000 ES cell lines were genotyped to identify five (0.5%) homologous recombinants. Two targeted clones were injected into blastocysts and each produced chimeras. Chimeric animals were used to generate Mkks heterozygous (Mkks+/−) mice on mixed and inbred genetic backgrounds by mating with C57BL/6J and 129/SvEv mice, respectively. Mice were genotyped using polymerase chain reaction (PCR) (Fig. 1B). Mkks−/− mice were generated less frequently than predicted by Mendelian inheritance (P<0.05). Of 600 mice genotyped, the proportion of Mkks+/+, Mkks+/− and Mkks−/− was 26.5, 54.4 and 19.1%, respectively. The relative paucity of Mkks−/− mice is likely due to either the in utero demise of homozygous knockout embryos or the death of homozygous knockout animals shortly after birth. We have carefully monitored newborn mice and have found nine instances of post-natal death, where we were able to recover the animal for genotyping. Two of these nine mice were Mkks−/−, three were homozygous normal and four were heterozygotes, a distribution that is not different from Mendelian ratios. We have also sacrificed three pregnant female mice at 14 days gestation and genotyped the fetuses. Three fetuses were homozygous normal, nine were heterozygotes and six were Mkks−/−. All fetuses were grossly normal on examination with the exception of one Mkks−/− fetus which was significantly smaller than the other fetuses. These data suggest that some Mkks−/− mice die late in gestation.
Mkks targeting resulted in a null allele as demonstrated by the absence of Mkks mRNA by northern blot analysis (Fig. 1C).
Mkks−/− mice become obese and have high leptin levels
Mkks-deficient mice were grossly normal with respect to morphology, although they appeared to be smaller at birth than their littermates. At the time of weaning (3 weeks of age), Mkks−/− mice weighed significantly less than their wild-type and heterozygous littermates. By ∼8–12 weeks of age, Mkks-null mice weighed the same as Mkks+/+ and Mkks+/− animals. After 28 weeks, Mkks−/− mice were significantly larger than both wild-type and heterozygotes (Fig. 2A). The difference in weight between Mkks−/− and controls was greater in females than in male mice.
Longitudinal feeding studies indicated that Mkks−/− mice eat more than their heterozygous and wild-type littermates (Fig. 2B) and show a significant decrease in activity as measured by 24 h telemetry when compared with wild-type mice (Fig. 2C). Mkks−/− mice had higher average serum leptin levels than wild-type controls (27±15.3 SD and 2.3±2.6 SD ng/ml, respectively, N=4).
Mkks−/− mice have elevated arterial pressure
Mkks−/− mice had significantly higher diastolic, mean and systolic blood pressure than controls as determined by 24 h telemetry. The average difference in mean arterial pressure between knockout and control animals was 12 mmHg (Table 1).
Mkks−/− mice do not exhibit polydactyly
Although polydactyly is a common feature of both MKS and BBS in humans, no Mkks−/− mice had polydactyly or other notable limb abnormalities. This was determined by counting the digits of all four extremities of 115 Mkks−/− mice. In addition, X-ray analysis was performed on 20 adult Mkks−/− mice. X-ray analysis of these animals revealed that none had limb anomalies including polydactyly, syndactyly, brachydactyly and missing or duplicated bones of the extremities.
Mkks−/− mice possess neurosensory and behavioral phenotypes
Behavioral and functional analyses of the mice was assessed using a modified SHIRPA protocol (22). The data from 38 separate observational measurements were compared between Mkks+/+, Mkks+/− and Mkks−/− mice. Those tests showing significant differences among genotypes are summarized in Table 1. The average age of the mice at the time of evaluation was 23 weeks. The age range was 16–34 weeks. There were no significant differences when comparing the data from Mkks+/− with Mkks+/+ mice of either sex. Similarly, there were no significant differences between knockout and control mice in muscle and lower motor neuron functions such as body position, balance and co-ordination, gait, positional passivity, tail elevation, grip strength or body tone, nor were there significant differences in sensory functions such as toe pinch and reflexes. Finally, there was no significant effect of genotype on visual placing (P=0.43), a test in which the mouse is lowered down onto a wire grid and the extension of the forearms is observed.
Mkks−/− female mice were less vocal during handling when compared with controls (P<0.05). Mkks−/− mice showed diminished olfaction as measured by the ability to find hidden food (Table 2).
As the Mkks−/− mice were observed to be more docile to handling, a social dominance test was performed. In 33/46 trials (72%; P<0.05), the Mkks+/+ mice were found to be dominant over Mkks−/− in a paired social dominance test. When heterozygous mice were tested against Mkks−/− mice, the heterozygous mice were also dominant (35/46 trials; 76%; P<0.05). This finding was true for both male and female animals. There was no difference in social dominance between wild-type and heterozygous animals (P>0.05).
Mkks−/− mice display photoreceptor degeneration
Mkks−/− mice appeared to have normal retinas early in life but subsequently undergo retinal degeneration. Eyes from Mkks−/− mice of ages 2–11 months were evaluated histologically (Fig. 3). At 2–3 months of age, inner and outer segments were clearly differentiated, the outer nuclear layer was slightly reduced and cell nuclei were present in the subretinal space. The inner and outer segments were reduced in height (Fig. 3A). At 4–5 months of age, the outer nuclear layer showed a moderate reduction and contained three to seven rows of nuclei. A remnant of material corresponding to poorly differentiated inner and outer segments was still present at this age between the outer nuclear layer and the RPE (Fig. 3B). At 6 months of age, the outer nuclear layer was greatly reduced, with only one to two rows of nuclei remaining and a variable amount of inner/outer segment material; numerous cells were present in the subretinal space (Fig. 3C). By 8 months, the outer nuclear layer was completely degenerated and no remnants of inner or outer segments are detectable (data not shown).
Anti-rhodopsin staining of the retinas of Mkks−/− animals with monoclonal antibody RetP1 prior to complete loss of photoreceptors revealed normal opsin immunoreactivity in outer segments at 2 months of age. In addition, some rhodopsin was localized to cell bodies in the outer nuclear layer in Mkks−/− mice (Fig. 3D).
Mkks−/− mice fail to form spermatozoa flagella
Mkks−/− male mice failed to impregnate heterozygous or wild-type female animals. In order to investigate the reason for their infertility, we examined the testes of Mkks−/−. Absence of flagella was noted in the seminiferous tubule lumens in the testes of Mkks−/− mice (Fig. 4A and B). The absence of flagella was noted in Mkks−/− mice of all ages examined. Nuclei with highly condensed chromatin, resembling spermatozoa heads were present, but flagella were absent.
Cells from Mkks−/− mice develop cilia
Kidneys from young mice were examined by scanning electron microscopy to evaluate renal primary cilial morphology in Mkks−/− mice. Cilia in Mkks−/− specimens appeared to be of normal size and density (Fig. 4C). To further investigate whether the MKKS gene product plays a role in cilia biogenesis, we cultured tracheal epithelial cells from young and old wild-type and Mkks−/− mice. After seeding, mouse tracheal epithelial cells (MTEC) rapidly proliferated and became polarized. Some tracheal epithelial cells developed into ciliated cells under culture conditions that exposed the cells to an air–liquid interface (23). The cilia were evaluated by immunofluorescent labeling with an anti-β-tubulin-IV antibody. Epithelial cells derived from both wild-type and Mkks−/− mice developed a similar number of cilia and the morphology of the cilia appeared normal when visualized with confocal microscopy (Fig. 4C).
DISCUSSION
We show that MKKS-deficient mice develop obesity and retinal degeneration and that males fail to form flagella. In addition, these animals have an increased arterial pressure, olfactory dysfunction and a behavioral phenotype suggesting abnormal social interaction. The findings of obesity, retinal degeneration and failure of flagella formation are similar to the features of Bbs2- and Bbs4-null mice, but the retinal degeneration has a later onset than that seen in Bbs2−/− and Bbs4−/− mice (20,21).
The results reported here for Mkks−/− animals and previously for Bbs2−/− and Bbs4−/− mice indicate that Bbs proteins are not absolutely required for the formation of either motile or primary cilia. This is demonstrated by the fact that young Mkks-, Bbs2- and Bbs4-null mice produce photoreceptor cells that are grossly normal morphologically but subsequently undergo complete degeneration. In the current study, we provide evidence that tracheal epithelial cells from Mkks−/− mice are capable of generating morphologically normal cilia in culture. However, in each of the three mouse models we have examined to date (Mkks−/−, Bbs4−/− and Bbs2−/−), the assembly of spermatozoa flagella is compromised. The observed lack of flagella in the seminiferous tubules may be due to a complete failure to form these structures or, alternatively, may result from a rapid degeneration of flagella following their synthesis. However, in the animals we have studied, we have not observed any spermatozoa with flagella, indicating that the flagella are never formed. The failure of flagella formation or maintenance during spermiogenesis indicates that there are differences between the processes of cilia compared with flagella assembly.
Hydrometrocolpos, vaginal agenesis and other genitourinary malformations have been reported in both MKS and BBS patients (24). Approximately 3% of BBS patients have given birth (24). No vaginal anomalies have been observed in the Mkks−/− mice, although they appear to have decreased fertility. Three of five Mkks−/− females mated have become pregnant.
Despite the initial normal assembly of cilia in Mkks-deficient mice, evidence of abnormal cilia function exists. For example, although the outer segments of the photoreceptors appear to develop normally, they become disorganized, and eventually the photoreceptor cells undergo degeneration. Further study of these animal models will help to clarify the precise role of BBS proteins in these intracellular processes. The observation that a subset of outer nuclear layer cell bodies appeared to have mislocalized opsin is consistent with the notion that cilia function may be impaired in photoreceptor cells. The observation that Mkks−/− mice develop retinal degeneration leading to complete blindness also implies that the null state more closely resembles BBS-associated mutations in the MKS gene than the missense mutations in MKS.
Our data indicate that Mkks-null mice develop obesity associated with increased food consumption similar to the findings in Bbs2- and Bbs4-knockout mice. The increase in food consumption begins prior to the onset of obesity during the period of catch-up growth and continues after the Mkks−/− mice weigh significantly more than wild-type littermates. Mkks−/− mice also have a decrease in activity as measured by 24 h telemetry. On the basis of these data, it would appear that the obesity is caused by a combination of hyperphagia and decreased activity.
The hormone leptin is produced in adipose tissue and circulates to the hypothalamus where it acts on its receptor. Leptin is an important component in long-term weight regulation. When active, it signals a decrease in food intake and an increase in energy metabolism thereby limiting the degree of weight gain (25). Leptin levels are highly correlated with BMI of mice and humans (26). The state of elevated leptin levels that occurs with obesity is termed leptin resistance. Leptin resistance has been identified in a variety of obese mouse models (27). When comparing normal littermates with four models of genetically overweight mice, Maffei et al. (26) found the leptin levels were 10 times greater in diabetic and yellow agouti mutant mice, 5-fold more in fat mutant mice and 2-fold as high in tubby mutant mice. In another obese mouse model that lacks the peptide neuromedin U, the leptin levels are <2-fold higher than the wild-type controls (28). This model has been shown to be independent of the leptin signaling pathway. Our Mkks−/− mice have serum leptin levels 10 times the control level. It has been reported that aging results in a leptin resistant state in obese rodents (29) but in our Mkks-null mice, elevated leptin levels are seen in mice as young as 9–12 weeks old, before the onset of obesity. This would seem to indicate that leptin resistance develops before obesity.
The increased arterial pressure observed in Mkks−/− mice is consistent with the clinical reports of hypertension in BBS patients (9,30) as well as with the strong association between obesity and hypertension (30). High circulating leptin levels may contribute to the increased arterial pressure observed in the obese Mkks−/− mice. Indeed, besides its effect on appetite and metabolism, leptin acts in the hypothalamus to increase blood pressure through activation of the sympathetic nervous system (31).
The Mkks-null mouse shares many of the clinical findings found in human BBS patients (Table 3). The Mkks−/− mouse model recapitulates the phenotype observed in the BBS2- and BBS4-knockout mouse models, though some aspects of the phenotype may be less severe. These observations suggest that the complete absence of MKKS leads to BBS, whereas the MKS phenotype is likely to be due to the functional consequence of the compound His84Tyr/Ala24Ser allele reported in MKS patients. (3). It has been hypothesized that the MKS mutation is a hypomorph, whereas mutations in MKKS causing BBS represent complete loss of function (24). Although the results observed in our MKKS mouse model are consistent with this hypothesis, the functional status of the numerous putative BBS6 missense mutations remains to be characterized. It will be of interest to determine how some Mkks mutations lead to photoreceptor degeneration and obesity whereas others do not result in these phenotypes.
MATERIALS AND METHODS
Generation of MKKS-knockout mice
129/SvJ genomic DNA was used as template to clone PCR-derived fragments into the targeting vector pOSDUPDEL (a gift from O. Smithies, University of North Carolina, Chapel Hill, NC, USA). A 3.6 kb fragment spanning exons 2–3 of the MKKS gene was cloned into the KpnI/SalI site, followed by a neomycin resistance gene flanked by LoxP sites for positive selection. The 3′ flanking region was a 1.9 kb NheI/ClaI fragment spanning exons 3–4. A thymidine kinase cassette distal to the 3′-arm was used for negative selection. The vector was linearized with NotI and electroporated into R1 ES cells (129X1/SvJ3 129S1/Sv). G418 resistant clones, in which the targeting vector had recombined with the MKKS gene, were identified using PCR analysis with primers located outside both the 5′- and 3′-targeted regions and within the neomycin resistance gene. Two ES cell lines were used to generate mouse lines on a C57BL/6 background.
Genotyping of mice
Mice were genotyped by PCR using genomic DNA prepared from tail biopsies or embryonic tissue. We generated a 4.1 kb 5′-fragment specific to heterozygous and homozygous knockout mice using a primer outside the targeted region (5′-TGCAGTTTTCAGGTAAGTTCCA-3′) and within the neomycin resistance gene (5′-TCGCCTTCTTGACGAGTTCT-3′). A 2.9 kb fragment was generated with a primer outside of the 3′-targeted region (5′-GCAAAGATGAGACCCTTAAAACA-3′) and within the neomycin resistance gene (5′-AGCCAACGCTATGTCCTGAT-3′). Homozygous knockout mice were identified using two primer pairs within the deleted region (sense 5′-TCGAAACCTTTCAGTCACCC-3′, antisense 5′-GCAAAGATGAGACCCTTAAAACA-3′; and sense 5′-TTCGAATCCCAGTTGACTTCAG-3′, antisense 5′-GCAAAGATGAGACCCTTAAAACA-3′). Long template PCR was done at 94°C for 1 min, followed by 35 cycles at 94 C for 15 s, 60 C for 30 s, 68 C for 5 min and a final 7 min extension at 68 C.
RNA isolation and northern blot analysis
Tissues from adult mice were rapidly frozen in liquid nitrogen and stored at −80°C until use. Total cellular RNA extraction, gel electrophoresis, blotting, hybridization and autoradiography were carried out as previously described (32). A partial mouse Mkks cDNA probe was generated and hybridized with the northern blot. The blot was stripped of radioactivity and re-hybridized with a cDNA probe for β-actin to verify equal loading of RNA.
Weight studies
For evaluating initial weights and weight gain, animals were weighed at 3 and 4 weeks of age and on a monthly basis thereafter. For food consumption studies, a fixed weight of food was added to each cage containing a single animal and the remaining food was weighed on 5 days of each week. The average food consumption was then calculated for each week.
Leptin serum levels
Serum was collected from female Mkks−/− mice (two at 11 weeks and two at 26 weeks) and their age-matched controls. Leptin levels in the serum were measured using a Multi-Analyte Profile (Charles River Lab).
Primary behavioral observation screen
The primary observation screen is a modification of the SHIRPA protocol (22). Complete details of the experimental procedure can be found on the website for mouse mutagenesis consortium partners: the MRC Mammalian Genetics Unit Harwell, SmithKline Beecham Pharmaceuticals, Imperial College, London and Queen Mary and Westfield College, London, UK (http://www.mgc.har.mrc.ac.uk/mutabase/). Data from 38 separate observations were quantified and recorded for each animal. Assessment of each animal began with the observation of undisturbed behavior in a cylindrical clear viewing jar (height 20 cm, diameter 11 cm). The mice were then transferred to an arena (45×25 cm2) for the observation of motor behavior. This was followed by a sequence of manipulations which assessed vision, grip strength, righting reflex, negative geotaxis, body tone, reflexes, limb tone, provoked biting and salivation. Throughout this procedure, incidences of abnormal behavior, fear, irritability, aggression or vocalization were recorded. The procedure was preformed on a total of 57 adult mice, 10 male and nine female Mkks-knockout mice and their age-matched heterozygote and wild-type controls. A similar number of Bbs4-null, and Bbs2-null, heterozygous and wild-type mice were evaluated. Primary observers were masked to the genotyping data.
Social dominance tube test
Twenty-three Mkks wild-type, heterozygote and knockout mice (14 male and nine females each) were tested as previously described (33) in a 30 cm long×3.5 cm diameter (3.0 cm diameter for smaller mice, 4.5 cm diameter for larger mice) tube. Two age-matched same gender mice, a wild-type, a heterozygote or a knockout, were placed at opposite ends of the tube and released. A subject was declared a ‘winner’ when its opponent backed out of the tube. Each pairing was performed twice in random order for a total of 138 match ups. Thirty Bbs4 mice and 33 Bbs2 mice were tested in a similar manner.
Olfactory test
Twelve wild-type, heterozygote and knockout mice (five male and seven females each) were tested as previously described (34). Olfactory ability was assessed immediately after a 3 h period of food deprivation. A small piece of Doritos Nacho Corn Chip was hidden beneath 1–3 cm of bedding in a clean mouse cage. Each mouse was released into a cage and the time required to locate the chip was recorded for a maximum of 20 min. Similarly, 16 Bbs4-null mice (10 female and six male) and 13 Bbs2 mice (six male and seven female) were tested with their heterozygote and wild-type controls.
Blood pressure and activity
A radio-telemetric system was used to record arterial pressure, heart rate and activity. Mice were anesthetized with Ketamine (91 mg) and Xylazine (9.1 mg) cocktail intraperitoneally. The left common carotid artery was isolated and the catheter was inserted and tied securely using silk. The transmitter was slipped under the skin and down into a dissected free ‘pocket’ along the flank as close to the right hindlimb as possible. The neck incision was closed using silk and further sealed with tissue adhesive. After 1 week of recovery from surgery, arterial pressure, heart rate and activity were recorded in the conscious unrestrained state for 5 days.
Morphological analysis of Mkks−/− mice
Morphological studies were conducted as described previously (19,20). Sections from at least two knockout and wild-type animals were examined per time point. Tissues examined included eyes, testes and kidneys. Acrylamide embedded sections were stained with hematoxylin–eosin stain and photomicrographs were collected using an Olympus BX-41 microscope with a SPOT-RT digital camera. Kidneys from young 2-week-old knockout mice were fixed by immersion in half strength Karnovsky's fixative overnight and cortical fragments were critical point dried and sputter coated prior to photomicrography using a Hitachi S4000 scanning electron microscope (Central Microscopy Research Facility, The University of Iowa).
MTEC culture
Culture of MTEC was performed (22). Briefly, tracheal cells from either 6-week-old or 8-month-old wild-type and Bbs6-knockout mice were cultured at 37 C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM)/Ham's F12 medium supplemented with 4 mm glutamine, 10 U/ml penicillin, 10 µg/ml streptomycin, 5% fetal bovine serum (all Gibco BRL), 10 µg/ml insulin (Roche), 5 µg/ml human transferrin, 62 µg/ml cholera toxin, 10 nm retinoic acid (all Sigma), 5 ng/ml epidermal growth factor and 0.03 mg/ml bovine pituitary extract (both BD Biosciences), on rat-tail-collagen (BD Biosciences) coated Transwell-membranes (CoStar). Once the culture reached confluency, an air–liquid interface was created by removing medium from the upper compartment and adding DMEM/F12 medium with only 2% NuSerum (BD) and 10 nm retinoic acid to the lower compartment. Culture medium in the lower chamber was changed twice per week. To visualize cilial microtubules, cells were simultaneously fixed and permeabilized for 5 min in PHEM buffer (60 mm PIPES, 25 mm HEPES, 10 mm EGTA, 2 mm MgSO4, pH 7.0) containing 0.5% formaldehyde, 0.1% glutaraldehyde and 0.5% Triton X-100, followed by fixation for 10 min in PHEM buffer containing 1% formaldehyde. Cilia were visualized by immunocytochemical labeling with β-tubulin IV antibody (Biogenex) and were viewed using a Bio-Rad multiphoton microscope.
Data analysis
Significance was determined using Student's t-test. Where appropriate, for example with scaled data from the primary behavioral observations, non-parametric analyses were conducted using the Fisher's exact test.
ACKNOWLEDGEMENTS
We thank P. Karp, K. Bugge, M. Olvera, C. Eastman, R. Swiderski, R. Burry, A. Fischer and K. Wolken for technical assistance and D. Aguiar Crouch for administrative assistance. We also acknowledge The University of Iowa, Department of Pathology and the Central Electron Microscopy Research Facility at the University of Iowa for assistance. This work was supported by the following grants and organizations: NIH grants P50-HL-55006 (V.C.S.) and R01-EY-11298 (V.C.S. and E.M.S.), Carver Endowment for Molecular Ophthalmology (E.M.S. and V.C.S.), Research to Prevent Blindness, New York, NY (Department of Ophthalmology, University of Iowa). V.C.S. and E.M.S. are investigators of the Howard Hughes Medical Institute.
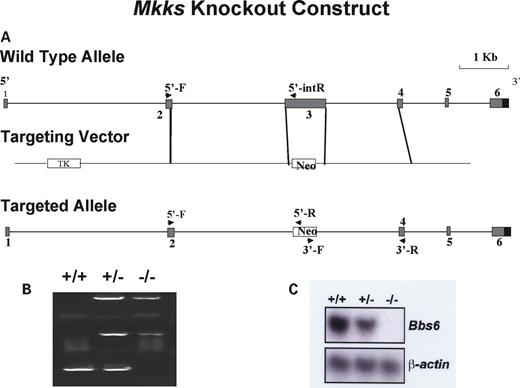
Figure 1. Schematic representation of MKKS targeted disruption strategy. (A) Map of the MKKS locus (top); gene targeting vector (middle); expected structure of the mutated locus (bottom). Arrows indicate primers used for the 5′- and 3′-homologous recombination test and internal knockout test. (B) PCR genotyping of wild-type, heterozygous (+/−) and Bbs null (−/−) mice. The top and middle amplimers correspond to 5′- and 3′-regions of the neomycin resistance gene. The bottom band is derived from amplification using MKKS exon 3 internal primers. (C) Northern blot analysis of Mkks expression in kidney total cellular RNA from wild-type (+/+), heterozygous (+/−) and homozygous (−/−) animals. The probe is Mkks partial cDNA. (Bottom) The same blot re-probed with β-actin as a loading control.
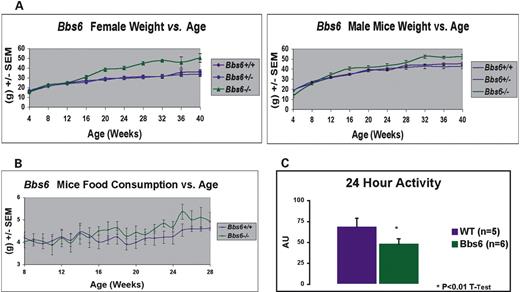
Figure 2. Weight gain and food consumption of wild-type, Mkks+/− and Mkks−/− mice. (A) Weight gain in female and male mice versus age. A minimum of seven animals was included in each group. MKKS−/− mice had lower birth weights than Mkks+/− or Mkks+/+ mice but by week 16 were significantly heavier. (B) Food consumption for male and female mice, averaged every 7 days. A minimum of six animals was included in each group. Mkks−/− mice consume significantly more food than Mkks+/+ and Mkks+/− mice. (C) Twenty-four hour activity data as measured by telemetry. Mkks−/− mice exhibit significantly less locomotion when compared with wild-type.
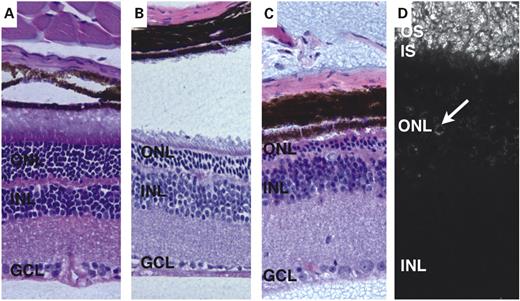
Figure 3. Hematoxylin–eosin staining of Mkks−/− (A–C) mouse retinas. Mice were examined at 2–3 months of age (A), 4–5 months (B) and 6 months (C). Some degenerative changes in the outer nuclear layer (ONL) are apparent in 2- to 3-month-old mice, although inner and outer segments are clearly distinguishable. By 5 months of age (B), the distinction between inner and outer segments is less apparent and the outer nuclear layer is significantly thinner than in Mkks+/+ eyes. At 6 months of age (C), the outer nuclear layer has largely degenerated and no inner or outer segments are present. Anti-rhodopsin staining is present in the outer segment by 2 months of age (D). IS, inner segments; OS, outer segments; INL, inner nuclear layer; GCL, ganglion cell layer.
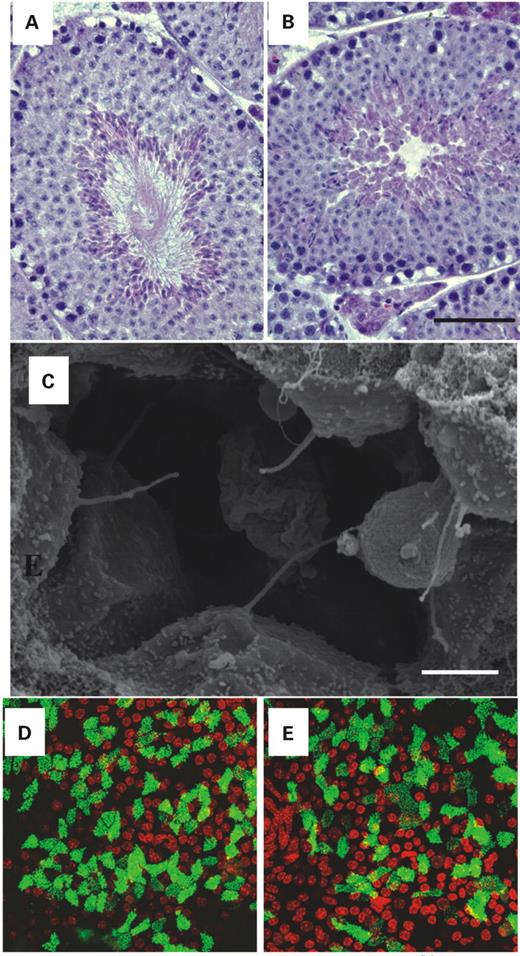
Figure 4. Cilia containing structures in Mkks−/− mice. Sections through the seminiferous tubules of wild-type (A) and Mkks−/− (B) mice reveal that mice deficient in the MKKS gene fail to produce normal flagella (Hematoxylin–eosin stain, scale bar=90 µm). Scanning electron microscopy of the renal distal tubule (C) of Mkks−/− mice reveals normal formation of renal cilia. Scale bar=2 µm. Projected Z-series of cultured tracheal epithelial cells of wild-type (D) and Mkks−/− (E) mice following differentiation with an air–fluid interface. The morphology of the cilia in the Mkks−/− mice is similar to that of wild-type mice. Cells were labeled with an antibody directed against β-tubulin-IV to visualize microtubule (green). Nuclei were counter-stained with TO PRO3 (red).
Blood pressure (mmHg) and heart rate (beats per min) of Mkks−/− mice and controls
| Parameter | Wild-type (N=5) | Mkks−/− (N=6) | P-value |
|---|---|---|---|
| Mean blood pressure | 120±2 | 132±3 | 0.0014 |
| Systolic BP | 134±2 | 146±3 | 0.0006 |
| Diastolic BP | 105±3 | 117±4 | 0.0091 |
| Heart rate (beats per min) | 471±11 | 419±7 | 0.478 |
| Parameter | Wild-type (N=5) | Mkks−/− (N=6) | P-value |
|---|---|---|---|
| Mean blood pressure | 120±2 | 132±3 | 0.0014 |
| Systolic BP | 134±2 | 146±3 | 0.0006 |
| Diastolic BP | 105±3 | 117±4 | 0.0091 |
| Heart rate (beats per min) | 471±11 | 419±7 | 0.478 |
Data are presented as mean±SEM. P-value is calcualted using Student's t-test.
Blood pressure (mmHg) and heart rate (beats per min) of Mkks−/− mice and controls
| Parameter | Wild-type (N=5) | Mkks−/− (N=6) | P-value |
|---|---|---|---|
| Mean blood pressure | 120±2 | 132±3 | 0.0014 |
| Systolic BP | 134±2 | 146±3 | 0.0006 |
| Diastolic BP | 105±3 | 117±4 | 0.0091 |
| Heart rate (beats per min) | 471±11 | 419±7 | 0.478 |
| Parameter | Wild-type (N=5) | Mkks−/− (N=6) | P-value |
|---|---|---|---|
| Mean blood pressure | 120±2 | 132±3 | 0.0014 |
| Systolic BP | 134±2 | 146±3 | 0.0006 |
| Diastolic BP | 105±3 | 117±4 | 0.0091 |
| Heart rate (beats per min) | 471±11 | 419±7 | 0.478 |
Data are presented as mean±SEM. P-value is calcualted using Student's t-test.
Summary of significant differences among MKKS-, Bbs2- and Bbs4-null mice compared with their wild-type littermates in SHIRPA primary screen behavioral observations
| BBS2−/− | BBS4−/− | Mkks−/− | Wild-type | |
|---|---|---|---|---|
| Touch escape | 0 (P<0.0001) | 0 (P<0.000005) | 0 (P=0.07) | 1 |
| Pelvic elevation | 2 (P<0.02) | 2 (P<0.01) | 1 (P<0.001) | 2 |
| Vocalization | 0 (P<0.0001) | 0 (P<0.01) | 0 (P<0.05)a | 1 |
| Olfaction | 11.8 min | 11.4 min | 9.8 min | 2.6 min |
| BBS2−/− | BBS4−/− | Mkks−/− | Wild-type | |
|---|---|---|---|---|
| Touch escape | 0 (P<0.0001) | 0 (P<0.000005) | 0 (P=0.07) | 1 |
| Pelvic elevation | 2 (P<0.02) | 2 (P<0.01) | 1 (P<0.001) | 2 |
| Vocalization | 0 (P<0.0001) | 0 (P<0.01) | 0 (P<0.05)a | 1 |
| Olfaction | 11.8 min | 11.4 min | 9.8 min | 2.6 min |
See http://www.mgc.har.mrc.ac.uk/mutabase/ for description. Data is expressed as mode followed by P-values from Fisher's exact test in parentheses.
aSignificant in females only.
Summary of significant differences among MKKS-, Bbs2- and Bbs4-null mice compared with their wild-type littermates in SHIRPA primary screen behavioral observations
| BBS2−/− | BBS4−/− | Mkks−/− | Wild-type | |
|---|---|---|---|---|
| Touch escape | 0 (P<0.0001) | 0 (P<0.000005) | 0 (P=0.07) | 1 |
| Pelvic elevation | 2 (P<0.02) | 2 (P<0.01) | 1 (P<0.001) | 2 |
| Vocalization | 0 (P<0.0001) | 0 (P<0.01) | 0 (P<0.05)a | 1 |
| Olfaction | 11.8 min | 11.4 min | 9.8 min | 2.6 min |
| BBS2−/− | BBS4−/− | Mkks−/− | Wild-type | |
|---|---|---|---|---|
| Touch escape | 0 (P<0.0001) | 0 (P<0.000005) | 0 (P=0.07) | 1 |
| Pelvic elevation | 2 (P<0.02) | 2 (P<0.01) | 1 (P<0.001) | 2 |
| Vocalization | 0 (P<0.0001) | 0 (P<0.01) | 0 (P<0.05)a | 1 |
| Olfaction | 11.8 min | 11.4 min | 9.8 min | 2.6 min |
See http://www.mgc.har.mrc.ac.uk/mutabase/ for description. Data is expressed as mode followed by P-values from Fisher's exact test in parentheses.
aSignificant in females only.
Comparison of Mkks mouse phenotype to Bbs2 and Bbs4 KO mice and to human MKS and BBS patient phenotypes
| Phenotype | ||||
|---|---|---|---|---|
| MKKS−/− mice | Bbs4−/− and Bbs2−/− mice | Human BBS | Human MKS | |
| RP | + | + | + | − |
| Obesity | + | + | + | − |
| Lack of flagella | + | + | + | − |
| Polydactyly | − | − | + | + |
| Olfaction deficit | + | + | + | ? |
| Phenotype | ||||
|---|---|---|---|---|
| MKKS−/− mice | Bbs4−/− and Bbs2−/− mice | Human BBS | Human MKS | |
| RP | + | + | + | − |
| Obesity | + | + | + | − |
| Lack of flagella | + | + | + | − |
| Polydactyly | − | − | + | + |
| Olfaction deficit | + | + | + | ? |
Comparison of Mkks mouse phenotype to Bbs2 and Bbs4 KO mice and to human MKS and BBS patient phenotypes
| Phenotype | ||||
|---|---|---|---|---|
| MKKS−/− mice | Bbs4−/− and Bbs2−/− mice | Human BBS | Human MKS | |
| RP | + | + | + | − |
| Obesity | + | + | + | − |
| Lack of flagella | + | + | + | − |
| Polydactyly | − | − | + | + |
| Olfaction deficit | + | + | + | ? |
| Phenotype | ||||
|---|---|---|---|---|
| MKKS−/− mice | Bbs4−/− and Bbs2−/− mice | Human BBS | Human MKS | |
| RP | + | + | + | − |
| Obesity | + | + | + | − |
| Lack of flagella | + | + | + | − |
| Polydactyly | − | − | + | + |
| Olfaction deficit | + | + | + | ? |
References
Stone, D.L., Slavotinek, A., Bouffard, G.G., Banerjee-Basu, S., Baxevanis, A.D., Barr, M. and Biesecker, L.G. (
Robinow, M. and Shaw, A. (
Slavotinek, A.M., Stone, E.M., Mykytyn, K., Heckenlively, J.R., Green, J.S., Heon, E., Musarella, M.A., Parfrey, P.S., Sheffield, V.C. and Biesecker, L.G. (
Frydman, J., Nimmesgern, E., Erdjument-Bromage, H., Wall, J.S., Tempst, P. and Hartl, F.U. (
Green, J.S., Parfrey, P.S., Harnett, J.D., Farid, N.R., Cramer, B.C., Johnson, G., Heath, O., McManamon, P.J., O'Leary, E. and Pryse-Phillips, W. (
Biedl, A. (
Bardet, G. (
Elbedour, K., Zucker, N., Zalzstein, E., Barki, Y. and Carmi, R. (
Harnett, J.D., Green, J.S., Cramer, B.C., Johnson, G., Chafe, L., McManamon, P., Farid, N.R., Pryse-Phillips, W. and Parfrey, P.S. (
Mykytyn, K., Braun, T., Carmi, R., Haider, N.B., Searby, C.C., Shastri, M., Beck, G., Wright, A.F., Iannaccone, A., Elbedour, K. et al. (
Nishimura, D.Y., Searby, C.C., Carmi, R., Elbedour, K., Van Maldergem, L., Fulton, A.B., Lam, B.L., Powell, B.R., Swiderski, R.E., Bugge, K.E. et al. (
Katsanis, N., Beales, P.L., Woods, M.O., Lewis, R.A., Green, J.S., Parfrey, P.S., Ansley, S.J., Davidson, W.S. and Lupski, J.R. (
Mykytyn, K., Nishimura, D.Y., Searby, C.C., Shastri, M., Yen, H.J., Beck, J.S., Braun, T., Streb, L.M., Cornier, A.S., Cox, G.F. et al. (
Badano, J.L., Ansley, S.J., Leitch, C.C., Lewis, R.A., Lupski, J.R. and Katsanis, N. (
Ansley, S.J., Badano, J.L., Blacque, O.E., Hill, J., Hoskins, B.E., Leitch, C.C., Kim, J.C., Ross, A.J., Eichers, E.R., Teslovich, T.M. et al. (
Li, J.B., Gerdes, J.M., Haycraft, C.J., Fan, Y., Teslovich, T.M., May-Simera, H., Li, H., Blacque, O.E., Li, L., Leitch, C.C. et al. (
Chiang, A.P., Nishimura, D., Searby, C., Elbedour, K., Carmi, R., Ferguson, A.L., Secrist, J., Braun, T., Casavant, T., Stone, E.M. and Sheffield, V.C. (
Fan, Y., Esmail, M.A., Ansley, S.J., Blacque, O.E., Boroevich, K., Ross, A.J., Moore, S.J., Badano, J.L., May-Simera, H., Compton, D.S. et al. (
Kim, J.C., Badano, J.L., Sibold, S., Esmail, M.A., Hill, J., Hoskins, B.E., Leitch, C.C., Venner, K., Ansley, S.J., Ross, A.J. et al. (
Mykytyn, K., Mullins, R.F., Andrews, M., Chiang, A.P., Swiderski, R.E., Yang, B., Braun, T., Casavant, T., Stone, E.M. and Sheffield, V.C. (
Nishimura, D., Fath, M., Mullins, R., Searby, C., Andrews, M., Davis, R., Andorf, J., Mykytyn, K., Swiderski, R., Yang, B. et al. (
Rogers, D., Fisher, E., Brown, S., Peters, J., Hunter, A. and Martin, J. (
You, Y., Richer, E.J., Huang, T. and Brody, S.L. (
Slavotinek, A.M. and Biesecker, L.G. (
Friedman, J.M. and Halaas, J.L. (
Maffei, M., Halaas, J., Ravussin, E., Pratley, R.E., Lee, G.H., Zhang, Y., Fei, H., Kim, S., Lallone, R., Ranganathan, S. et al. (
Halaas, J.L., Boozer, C., Blair-West, J., Fidahusein, N., Denton, D.A. and Friedman, J.M. (
Hanada, R., Teranishi, H., Pearson, J.T., Kurokawa, M., Hosoda, H., Fukushima, N., Fukue, Y., Serino, R., Fujihara, H., Ueta, Y. et al. (
Scarpace, P.J., Matheny, M. and Tumer, N. (
Beales, P.L., Elcioglu, N., Woolf, A.S., Parker, D. and Flinter, F.A. (
Rahmouni, K., Correia, M.L., Haynes, W.G. and Mark, A.L. (
Swiderski, R.E., Ying, L., Cassell, M.D., Alward, W.L., Stone, E.M. and Sheffield, V.C. (
Lijam, N., Paylor, R., McDonald, M.P., Crawley, J.N., Deng, C.X., Herrup, K., Stevens, K.E., Maccaferri, G., McBain, C.J., Sussman, D.J. et al. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}