



Abstract
Defects of mitochondrial polymerase γ (POLG) underlie neurological diseases ranging from myopathies to parkinsonism and infantile Alpers syndrome. The most severe manifestations have been associated with mutations of the ‘spacer’ region of POLG, the function of which has remained unstudied in humans. We identified a family, segregating three POLG amino acid variants, A467T, R627Q and Q1236H. The first two affect the spacer region and the third is a polymorphism, allelic with R627Q. Three grades of disease severity appeared to correlate with the genotypes. The patient with the most severe outcome, cerebellar ataxia syndrome, had all three variants, those with R627Q and Q1236H had juvenile-onset ptosis and gait disturbance and those with a single A467T allele had late-onset ptosis. To evaluate the molecular pathogenesis of these spacer defects, we expressed and purified the mutant proteins and studied their catalytic properties in vitro. The A467T substitution resulted in clearly decreased activity, DNA binding and processivity of the polymerase. Our biochemical data, the dominant manifestation of A467T and its previously reported high frequency in the Belgian population (0.6%), emphasize the role of this mutation as a common cause of neurological disease. Further, biochemical evidence that a polymorphic variant may modify the function of a mutant POLG, if occurring in the same polypeptide, is shown here. Finally, and surprisingly, other pathogenic spacer mutants showed DNA-binding affinities and processivities similar to or higher than the controls, suggesting that the disease-causing mechanisms of spacer mutations extend beyond the basic catalytic functions of POLG.
INTRODUCTION
Defects in nuclear-encoded proteins involved in mitochondrial DNA (mtDNA) replication or maintenance have recently been identified as important causes of neurodegeneration and muscle disease. The common result of these defects is either mtDNA depletion or accumulation of multiple deletions of mtDNA in the postmitotic tissues of the patient (1–3). Currently, mutations in four genes are known to cause mtDNA instability: the heart, muscle and brain-specific adenine nucleotide (nt) translocator 1 (OMIM no. 103220; ANT1, also known as AAC2 or hANC1) (4), the replicative mtDNA helicase Twinkle (OMIM no. 606075) (5), the thymidine phosphorylase (OMIM no. 131222), taking part in nucleoside pool regulation (6) and the catalytic subunit of the mtDNA polymerase (OMIM no. 174763, POLG, POLG1, POLGα) (7).
MtDNA replication requires the functional DNA polymerase (POLG), helicase Twinkle and mitochondrial single-stranded DNA-binding protein (8) and proceeds in an asymmetric (9,10) or strand-synchronous fashion (11,12). POLG is the only known DNA polymerase in the mitochondrion, making up ∼1% of the total cellular DNA polymerase activity. The human POLG holoenzyme comprises a 140 kDa catalytic subunit (POLGα) and a 55 kDa accessory subunit (POLGβ). POLGα exhibits both DNA polymerase and 3′–5′ exonuclease activities, whereas POLGβ increases DNA-binding affinity, stimulates the catalytic activities and enhances the processivity of the holoenzyme (8). The region of POLGα that lies between the exonuclease and polymerase has been termed the spacer (Fig. 1). Its size and sequence differ substantially from other members of the family-A DNA polymerases, and it harbors four POLGα-specific sequence blocks (γ1–γ4) conserved in species from fruit fly to man. Mutagenesis of these conserved blocks in the fruit fly protein affected differentially the activity, processivity and DNA-binding affinity of the enzyme, as well as the balance between the polymerase and exonuclease activities, suggesting that the spacer maintains enzyme function by several mechanisms (13). The large interdomain region is likely to participate in DNA-template binding and guidance, as well as in subunit interactions. To date, human disease mutations occurring in the spacer region have not been biochemically characterized.
Mutations of POLG1 represent the most common cause of autosomal recessive and dominant progressive external ophthalmoplegia (ar/adPEO) (7,14,15) (http://dir-apps.niehs.nih.gov/polg/ and references therein). Dominant mutations affecting the polymerase domain result in PEO (7,14), and often parkinsonism, peripheral neuropathy or early menopause (15,16). However, mutations in the proof reading exonuclease domain may result in severe, recessively inherited peripheral neuropathy, sometimes accompanied by late-onset PEO (17). Recently, we and collaborators described POLG1 spacer mutations in a pure central nervous system (CNS) phenotype, manifesting as sensory neuropathy and adult-onset spinocerebellar ataxia (18). Furthermore, spacer mutations were found frequently in the infantile Alpers syndrome, affecting most severely the brain and the liver (19,20). These recent reports emphasize the exceptional variability of POLGα-associated neurological phenotypes and the specific role for spacer mutations in the most severe neurological manifestations.
We report here a pedigree with central and peripheral nervous system, as well as muscle involvement. We found three POLGα amino acid variants segregating in the family and studied their clinical manifestations, as well as their molecular genetic features. Furthermore, we describe the first biochemical characterization of human POLGα spacer-region mutations.
RESULTS
Case reports
Figure 2 shows the pedigree of the family.
Case III/1 (index case)
This 46-year-old female patient developed progressive blepharoptosis at the age of 20 and ophthalmoplegia during subsequent years. Blepharoplasty was performed twice at the ages of 37 and 38, with only transient relief. At the age of 35, the patient developed progressive unsteadiness of gait. She noted fatigue, myalgia and cramps, which were triggered by moderate exercise. At the age of 44, restless legs syndrome was successfully treated with 100 mg levodopa and 300 mg gabapentin. The patient developed deficits in memory and concentration, and suffered from recurrent episodes of depression, which were treated with antidepressive drugs. Neurological examination at the age of 46 showed bilateral blepharoptosis, limited eye movements in all directions except downwards, lateral rotatory nystagmus and slight upbeat nystagmus in upward gaze, but no saccades. She had facial muscle weakness, dysarthria and slight proximal muscle weakness. She had glove and stocking-like hypoesthesia, distally impaired vibration sense, absent ankle reflexes and weak other tendon reflexes, as well as mild dysmetria. Her gait was broad-based, with further impairment with eyes closed. Tandem gait was impaired. Nerve conduction studies showed normal compound muscle action potential latencies and amplitudes, and motor nerve conduction velocities were within the normal range. Compound sensory action potentials could not be recorded from the sural nerves, and median nerve sensory action potentials were reduced with normal conduction velocity. Electromyography revealed an increased number of small polyphasic potentials, with early recruitment in deltoid and biceps muscles. Magnetic resonance imaging revealed slightly enlarged subarachnoidal space in the vermal region and above hemispheres. Spectroscopy showed elevated brain lactate in the cerebral white matter, basal ganglia and cerebrospinal fluid. Elevated lactate was not seen in the cerebellum, but N-acetyl-l-aspartate/creatine ratio was reduced, suggesting neuronal loss. In routine blood tests, serum lactate and creatine kinase were within normal range on several occasions. Biopsy of the deltoid muscle showed myopathic features, with <1%, ragged red fibers and no COX-negative fibers. Electron microscopy showed abundant structurally abnormal mitochondria with para-crystalline inclusions (Fig. 3).
Case II/9
This 73-year-old man, the father of the index patient, was the youngest of a sibship of nine. According to family history, his mother had ptosis. This patient developed bilateral blepharoptosis in his youth. Neurological examination at the age of 72 showed severe blepharoptosis, lids almost covering his entire pupils. Eye movements were normal, his muscle strength and sensation were good and tendon reflexes were normal, but his gait was unsteady.
Case II/13
This 64-year-old woman, the mother of the index patient, developed slowly progressive blepharoptosis at the age of 45. She underwent blepharoplasty at the age of 58. In the neurological examination at the age of 63, she had ptosis, but no other neurological deficits.
Case II/10
This 66-year-old woman developed progressive blepharoptosis slowly at the age of 50. She underwent blepharoplasty at the age of 62, and in the same year, she suffered a transitory ischemic attack. At the age of 66, no clinical findings except ptosis were detectable.
Case II/16
This 50-year-old woman developed progressive blepharoptosis slowly at the age of 40. She underwent blepharoplasty at the age of 47. Her neurological examination at the age of 50 was normal.
Case III/2
This 45-year-old man displayed severe bilateral blepharoptosis similar to his father but has refused clinical or genetic examinations.
Case III/3, IV/1
These subjects, 43 and 25 years old, respectively, are healthy by history and by clinical examination.
Case IV/2, IV/3
These young men are 23 and 19 years old, respectively. IV/2 has probable mild blepharoptosis and IV/3 displays mild bilateral blepharoptosis and slight unsteadiness during tandem gait. Otherwise, their neurological examinations were normal.
Genetic findings
We analyzed the complete coding sequences of the genes encoding ANT1 (NM_002693), Twinkle (NM_021830) and POLG1 (NM_002693), as well as the 22 transfer RNA genes of mtDNA in the index patient. The sequences of the genes encoding ANT1 and Twinkle were identical to those of controls. However, in POLG1 alleles, the patient carried three nt changes (Fig. 4), which resulted in an amino acid change: a G→A transition at nt 1399 of exon 7, a G→A transition at nt 1880 of exon 10 and a G→T transition at nt 3708 of exon 23. These changes predicted amino acid substitutions from alanine to threonine at position 467 (A467T), arginine to glutamine at position 627 (R627Q) and glutamine to histidine at the amino acid position 1236 of the polypeptide (Q1236H). An additional nt change was identified in the exon 13 (C2254T), a silent change at leucine position 752, which was not found in public databases (Entrez SNP, http://www.ncbi.nlm.nih.gov/). A467T and R627Q were not found in 380 Finnish control chromosomes (<0.2% frequency), but in Belgian controls, A467T has been previously reported to occur with a frequency of 0.6% (7). Q1236H has been reported to occur in Caucasian populations at a frequency of 3.7% (Entrez SNP, http://www.ncbi.nlm.nih.gov/), but our analysis of Finnish control samples showed that in Finns the frequency is higher, 14.8%. A467 is invariant in species, and both R627 and Q1236 are highly conserved (Fig. 4).
Figure 1. shows the detected sequence variants and their locations in the polypeptide. In addition to the index patient, her father II/9, her daughter IV/1 and son IV/3 had the R627Q substitution. The index patient's mother, two maternal aunts and her son IV/2 had the A467T substitution. The Q1236H variant was allelic with R627Q, and therefore, all the subjects with R627Q also carried Q1236H.
Southern blot analysis of total muscle DNA did not reveal multiple mtDNA deletions in the index patient even in the long exposure of the phosphorimaging plate. Furthermore, quantitation of mtDNA versus genomic 18S rRNA gene revealed normal levels of total mtDNA when compared with controls (Fig. 5A and B). PCR analysis designed to amplify preferentially deleted mtDNA species revealed that the index patient's muscle had distinct deleted mtDNA species, clearly more than the age-matched controls. The fraction of aberrant molecules was, however, lower than in the muscle of PEO patients with recessive POLG1 mutations (15). No additional amplifying products were seen under these carefully titrated amplification conditions in the age-matched control samples (Fig. 5C and D).
The A467T mutation had previously been reported exclusively as a recessive mutation (7,17–23). In the present family, it seemed to have a mild dominant phenotype, causing late-onset ptosis. To investigate whether unequal transcript levels, haploinsufficiency, of the mutant and wild-type alleles underlay the dominant effect of A467T, we quantified the POLG1 mRNA levels from lymphoblast total RNA of the subject II/16. Our mini-sequencing analysis showed that the expression level of the mutant allele was slightly lower than that of the wild-type (∼40 and 60%, respectively, data not shown), which excluded expression bias as an explanation for clinical manifestation in A467T carriers.
Functional analyses of recombinant POLGα mutants
First, the variable disease manifestations in the family with three different POLG variants prompted us to study whether the biochemical characteristics of the variants would explain their clinical consequences. Secondly, we wanted to address the question whether common amino acid variants can modify the properties of a mutant enzyme, if occurring in the same polypeptide. This ‘pathological haplotype’ concept has been previously introduced (18) because amino acid variants are common in POLGα. Finally, we wanted to clarify whether the catalytic properties of the mutant R627Q would explain why it was dominant in our pedigree, because a mutation R627W has previously been reported to be recessive (17).
Overexpression and purification of POLGα mutants
We first expressed and purified recombinant proteins carrying different POLGα variants (αA467T, αR627Q, αQ1236H, wild-type), utilizing the baculovirus expression system. We produced the R627Q alone and in combination with Q1236H, mimicking the patients' protein, to examine whether the latter common variant would modify the performance of the R627Q mutant. Furthermore, we expressed and purified a recessive mutant R627W to compare its catalytic performance to the dominant R627Q. To facilitate detection of subtle disease-associated changes in the polymerase functionalities, the proteins were modified to eliminate intrinsic exonuclease activity by substitution of two invariant amino acids in the exo I motif, as described previously (24,25).
Baculoviruses carrying the human POLG1 cDNA with the amino acid substitutions were used to infect Sf9 cells, and the recombinant proteins were purified to near homogeneity from the soluble cytoplasmic fraction. The enzymes were purified 55–130-fold, with an average yield of 6%. About 0.15 mg of near homogeneous POLGα protein was obtained per liter of cultured cells. The chromatographic profiles and peaks upon velocity sedimentation of the mutant constructs were similar to that of the control enzyme. Therefore, we found no indication of structural variation or instability in the mutant enzymes. Immunoblot analysis showed that the full-length protein was present in each chromatographic step of each preparation, although modest proteolytic degradation occurred throughout the purification (data not shown). All the final preparations were of similar purity (Fig. 6).
DNA polymerase activity of recombinant POLGα mutants
DNA polymerase specific activities of the recombinant enzymes were measured on a gapped double-stranded DNA substrate, DNase I-activated DNA. This analysis evaluated the synthesis of short DNA strands, such that minimal DNA-binding affinity and processivity were required. The detected activities were as follows (percentage of control activity shown in parentheses): human control POLGα, 16 836 U/mg; αA467T, 3090 U/mg (18%); αR627W, 15 497 U/mg (92%); αR627Q, 13 000 U/mg (77%); αR627Q/αQ1236H, 25 554 U/mg (151%) and αQ1236H, 19 760/mg (117%). Among the PEO mutant enzymes, only the αA467T mutant exhibited substantially reduced activity of DNA synthesis on a gapped substrate, which is 5.5-fold lower than the activity of the POLGα control enzyme. The higher specific activity observed with the αR627Q/Q1236H enzyme fell outside of the range of ∼8000–18 000 U/mg found in multiple previous purifications of the control POLGα.
DNA-binding affinity of recombinant POLGα mutants
Our recent studies documented that the POLGα spacer region of Drosophila melanogaster is involved in template–primer binding (13). Here, we performed gel mobility shift assays to examine DNA binding of the human POLGα mutants using an oligonucleotide template primer (Fig. 7). DNA binding was measured in the presence or absence of the accessory subunit POLGβ at 100 mm KCl or 30 mm KCl, respectively. The αA467T mutant showed weak DNA-binding affinity, ∼14% of the control, whereas binding by other mutants was not decreased. Addition of the β-subunit enhanced the DNA binding of the αA467T core by 18-fold, increasing it to 70% of the level of the control holoenzyme. DNA binding by the other mutant enzymes was stimulated an average of 2.2-fold, showing somewhat higher activity overall than the control, albeit within the likely range of the control enzyme among various preparations. The DNA-binding data argue that subunit interactions in the holoenzyme are not affected negatively by the A467T mutation; rather, the intrinsic binding affinity of the core to DNA is diminished, and this defect is compensated largely in the holoenzyme form by the accessory β-subunit.
Processivity of recombinant POLGα mutants
To investigate possible defects in the mechanism of DNA strand synthesis by the POLGα mutants, we performed processivity analyses on singly primed M13 DNA (ssDNA) that mimics the substrate for lagging DNA strand synthesis in mtDNA replication, in which long product DNA strands are synthesized from one or a few primers. Assays were performed in the presence or absence of the accessory β-subunit, at 100 mm KCl or 30 mm KCl, respectively. All of the mutant catalytic subunits showed processivities similar to the POLGα control enzyme in the absence of the β-subunit (Fig. 8). This would suggest that the reduced DNA polymerase and DNA-binding activities of αA467T did not result from an intrinsic alteration in the mechanism of DNA strand synthesis but in its ability to bind and initiate nt incorporation at a primer terminus. Once the mutant enzyme has bound the primer terminus, the average product DNA strand length is the same as that for the wild-type core, although the rate of elongation or the rate of recycling upon dissociation, or both, must be reduced in αA467T. Interestingly, addition of the β-subunit enhanced processivity of each of the mutant cores, although to a significantly lower extent for αA467T; processivity increased only 2-fold for αA467T, whereas for the control and other mutants, the increase was ∼7-fold (Fig. 9). The αR627Q core reproducibly produced somewhat longer product DNA strands than the other mutants, suggesting increased binding affinity. Interestingly, in combination with the Q1236H mutation, it was similar to the control. This suggested that common polymorphisms may indeed modify the effect of mutant enzymes, in this case making the peptide more like the control.
Differential stimulation of recombinant POLGα mutants by POLGβ
To investigate the effect of the accessory β-subunit on the DNA polymerase activity of the POLGα mutants, we measured nt incorporation on the singly primed M13 DNA substrate used in the processivity analysis, in the presence and absence of the accessory β-subunit, at 100 mm KCl. In the absence of the β-subunit, the αA467T mutant exhibited only ∼20% of the DNA polymerase activity of the wild-type POLGα, similar to the 5.5-fold decrease in its activity on the gapped DNA substrate. The other mutants exhibited activities similar to the wild-type enzyme, and in this assay, the slightly higher activity of R627Q/Q1236H that was found on the gapped DNA substrate was not observed (Fig. 10). This would argue that 2-fold variations among enzymes in the various assays were possibly not significant. Notably, however, the accessory β-subunit stimulated ∼20-fold the activity of αA467T when compared with ∼6-fold for other mutants and the control POLGα, to yield a holoenzyme activity of ∼60% of the control holoenzyme. These data as well as the DNA-binding and processivity analyses show that interaction with POLGβ mitigates partially the functional defects of the A467T mutation.
DISCUSSION
We and others have recently discovered that mutations affecting the spacer region of the catalytic subunit of mitochondrial POLG commonly underlie severe neurological diseases from spinocerebellar ataxia to Alpers hepato-encephalopathy (18–23). Furthermore, we have recently shown that the spacer region in the fruit fly enzyme affects the enzyme activity, processivity, DNA-binding affinity and the balance of the polymerase and exonuclease activities (13). Here we report an Austrian family whose members carried different combinations of three amino acid changes in POLGα, two of which affected the spacer region. We show here that this family shows phenotype–genotype correlation and that the effect of the mutations is additive in the index patient carrying all three changes. Furthermore, we show evidence that common polymorphisms can modify the performance of mutant enzymes, if occurring in the same polypeptide. This is especially interesting because amino acid polymorphisms are common in POLGα. We also present the first biochemical characterization of human spacer mutations and show that the common αA467T affects severely the catalytic properties of the enzyme. The properties of our other spacer mutants suggest that pathological mechanisms of POLGα must involve mechanisms that are not directly involved in DNA synthesis because the basic catalytic activities were not severely affected.
Two mutant POLG1 alleles segregated in the present family. These alleles carried mutations predicting spacer region amino acid substitutions A467T in one and R627Q in cis, with polymerase domain Q1236H polymorphism in the other polypeptide. The phenotype resulting from the heterozygous A467T mutation was late-onset ptosis, requiring surgical correction, but without ophthalmoplegia. Patients with R627Q/Q1236H had early-onset ptosis and gait disturbance. The index patient, who was the most severely affected, carried all three changes A467T/R627Q/Q1236H. She displayed several CNS manifestations, namely, ataxia, dysarthria, cognitive decline and nystagmus. She had also early-onset PEO and sensory neuropathy. Her symptomatic restless legs syndrome was responsive to treatment with L-dopa, which is of interest, because we have recently described the association of POLG1 mutations and parkinsonism (15). Both of the two spacer mutations in this pedigree resulted in dominant manifestations, unlike previously reported spacer mutations, which have exclusively been recessive (7,18–21).
POLG1 mutations have been found to be the most frequent cause of autosomal dominant and recessive forms of PEO and SANDO (sensory ataxic neuropathy, dysarthria and ophthalmoplegia) (17). Recently, we and others have shown that POLG1 spacer and polymerase region mutations are also associated with new phenotypes such as adult-onset recessive spinocerebellar ataxia, parkinsonism, premature menopause and Alpers syndrome (15,18–20). The patients with neurodegeneration and POLG1 spacer mutations may lack all the traditional findings of mitochondrial myopathy, such as ragged-red fibers or mtDNA deletions in Southern blot analysis, and, therefore, the mitochondrial background of their disease has remained undetected in routine histological and DNA analyses (18,21,26). In our index patient's muscle, only sparse ragged-red fibers were seen (<1%), and Southern blot analysis showed normal-sized mtDNA. However, in our carefully designed amplification by long-PCR, we could show the existence of multiple deleted mtDNA molecules, which clearly exceeded the levels seen in age-matched controls. Therefore, two important aspects for mitochondrial disease diagnosis are emphasized: 1) even a few COX-negative fibers may be pathology associated, if the patient is <50 years of age and 2) multiple mtDNA deletion diagnosis should be done by a controlled PCR-based amplification assay. Our PCR assay, based on amplification with a distantly located primer pair, but with long and short extension times, provides a reliable test, when including age-matched positive and negative controls, and titrating the template DNA amount to be low. In our routine analysis, we initially screen the total muscle DNA by PCR for mtDNA deletion, and if the result is positive, the POLG1 gene is sequenced.
Of the POLGα variants of the present pedigree, the R627Q substitution is novel, it is not present in controls and the arginine is highly conserved among divergent species. These aspects support the role of R627Q as a disease mutation. In our pedigree, R627Q showed dominant inheritance. The R627 position is adjacent to a predicted β-strand (amino acids 621–626) within the conserved γ3 region (amino acids 581–631) in the POLGα spacer, thought to be involved in DNA binding and potentially in interactions between POLGα and other replicative proteins such as mtSSB (13). Our recently reported recessive spinocerebellar ataxia-associated mutation, a W748S substitution, was also found to occur within a predicted β-strand, in the conserved γ4 region of the spacer (18). This suggests that spacer-region mutations may affect a POLGα function especially important for the function of cerebellar neurons and, therefore, result in ataxia. Why this specific region of CNS is sensitive to spacer defects remains to be studied.
The R627 mutation has been reported previously as a tryptophan substitution (R627W) in a recessive disease (17). A Belgian patient with healthy relatives was compound heterozygous carrying R627W and A467T, and had sensory neuropathy and ataxic gait, resembling closely the symptoms of our index patient (A467T/R627Q/Q1236H). In the light of the present and previous findings, R627Q would be associated with a dominant and R627W with a recessive phenotype. A similar situation has been found in the case of connexin 26 mutations, with R143Q causing dominant and R143W recessive phenotypes (27,28), suggesting a gain-of-function and loss-of-function mechanisms of action, respectively. To clarify the functional consequences of the POLGα R627Q and W variants, we studied the biochemical characteristics of these purified proteins in vitro. These POLGα variants exhibited specific activities within normal range, but their DNA-binding affinities were modestly increased, as was also processivity in the case of αR627Q. It seems plausible that increased affinity for DNA could affect the dynamics of DNA synthesis, leading to polymerase stalling or incomplete proof reading, and mutation formation. We and collaborators have recently suggested that multiple deletions in POLG1 mutant patients might be generated by stalling of the polymerase at complex structures in mtDNA (29). Interestingly, in D. melanogaster, biochemical analysis of a spacer mutant E595A (E640 in humans, located just outside of the γ3 element near R627) also showed normal specific activity, processivity, and DNA-binding affinity (13). However, this mutation had a detrimental effect, causing mitochondrial and nervous system dysfunction and being lethal to the developing fly in the larval third instar (30). From the present human and previous fly data, it is clear that the pathogenic mechanisms of these spacer mutations do not involve the essential catalytic functions of the replicative enzyme. Furthermore, the recessive or dominant modes of action of R627W and R627Q, respectively, were not explained by differences in these catalytic functions—unless the mechanism involved the modestly increased processivity of R627Q.
The pathogenic role of the A467T substitution is supported by previous publications, associating it to a range of neurological symptoms from ptosis and PEO to sensory neuropathy, myoclonic epilepsy and ataxia (7,17,18,21). In all these cases, A467T has been presented as recessive, compound heterozygous in association with a second recessive POLG1 mutation or as homozygous. However, in the present family, it caused a mild dominant manifestation, late-onset ptosis, which required operative correction in all the three affected subjects. The young subject IV/2 carrying the mutation showed probable mild ptosis. To detect digenic effects (31), we sequenced the genes encoding ANT1 and Twinkle-helicase and did not identify additional mutations. Furthermore, we excluded uneven expression of the two POLG1 alleles by showing that the two allelic transcripts were present in equal proportions in the patient's lymphoblasts. On the basis of these data, we suggest that a single allele with the A467T substitution may manifest in a dominant manner, leading to late-onset ptosis. The pathogenic effect is emphasized by our biochemical evidence, which showed that the αA467T mutant had significantly reduced specific activity, processivity and DNA-binding affinity, leading to a considerable polymerase defect. The high frequency of A467T in the Belgian population (0.6%), its severe biochemical defect and its late-onset dominant manifestation emphasize the important role of A467T underlying neurological disease. If the Belgian frequency reflected other countries, A467T would be the most common genetic cause of a mitochondrial disease known to date.
The human A467T substitution maps within the conserved γ1 element of POLGα spacer, which has been proposed to be involved in the assembly of the POLGα and β-subunits of the Drosophila enzyme (13). The interaction with the accessory β-subunit has been shown to increase the DNA binding of the catalytic core in vitro, leading to stimulation of the exonuclease and polymerase activities and to improved processivity (8). Both DNA binding and processivity were reduced in our αA467T, and therefore, we tested the effect of the β-subunit on this mutant. The polymerase activity of the αA467T was <20% of that of the wild-type catalytic core, but, interestingly, POLGβ stimulated this mutant ∼20-fold, which was three to four times higher than the effect of the accessory subunit on control POLGα. Assembly of the catalytic and accessory subunits is not required for the solubility and basic activities of human POLGα. Increased stimulation by POLGβ suggests that its functional or physical interactions with POLGα may compensate partially the biochemical defect of the A467T mutant. In a Drosophila γ1 mutant, reduced processivity suggested a defective interaction between the two subunits, implicating a role of the conserved γ1 element in this interaction (13). Our present data are consistent with this observation, although, interestingly, subunit interaction mitigates the low DNA-binding activity and specific activity of the A467T mutant more than its processivity. Further kinetic studies will be required to address this issue.
The third amino acid variant found in the present pedigree, Q1236H, is located in the POLGα polymerase domain, and this site is conserved from mammals to puffer fish and fruit fly. The variant is common in the general population, with a frequency of 14.8% in Finns and 3.7% in SNP database, and exists also in homozygous state in Finnish controls. These aspects suggest that the variant is not disease associated, at least, if present alone. However, Q1236H maps in the same polypeptide as R627Q in our patients and could potentially modify the defect caused by R627Q. The POLG1 sequence is commonly polymorphic and the idea of a pathological haplotype has been put forward (18); combinations of amino acid variants within the same polypeptide could be synergistic or compensatory. To address this question, we compared the biochemical characteristics of R627Q/Q1236H in combination with the individual mutant proteins. When compared with the single R627Q mutant, the double-mutant R627Q/Q1236H enzyme showed ∼2-fold higher specific activity in the gapped DNA assay and 1.6-fold in the ssDNA assay. In the ssDNA assay, the accessory subunit stimulated both mutants similarly, such that the difference in their specific activities remained 1.6-fold. Both processivity and DNA binding were closer to the wild-type values in the double mutant than in R627Q alone. These data suggest that ‘neutral’ polymorphisms may modify the biochemical characteristics of POLGα variants and in this case, in a compensatory fashion.
In conclusion, our data emphasize the importance of POLGα spacer mutations as one of the most common genetic causes of mitochondrial disorders. Two spacer mutations in our family prompted us to look closer at the molecular mechanisms of these mutations. All functional data reported previously have been based solely on analysis of dominant mutations affecting the polymerase domain of POLGα (24,25,32), whereas spacer-region mutants have not been studied. We show here that the αA467T mutant has a clear biochemical phenotype indicating reduced polymerase function that most likely results from reduced DNA binding and initiation of DNA strand elongation, and that part of the pathogenic mechanism may be explained by modified contacts with the accessory subunit. The severe biochemical defect may not be completely compensated by the wild-type allele in a heterozygous patient, and a mild dominant manifestation was seen in our pedigree. Other mutations did not show as clear defects in their catalytic properties, albeit lead to severe clinical manifestations. This strongly suggests that pathogenic mechanisms of POLGα extend beyond the basic catalytic functions of the polymerase. We also indicated that common polymorphic variants may modify the functions of mutant enzymes. In composite, our data show that POLGα substitutions affecting different regions of the catalytic subunit have different pathogenic mechanisms, not always affecting the basic catalytic functions of the enzyme.
MATERIALS AND METHODS
Subjects
This study has been approved by the Helsinki University Hospital's Ethical Committee. All the blood and muscle samples were taken according to the Helsinki declaration, with oral or written informed consent. The patients and their relatives were examined by the same neurologists. For the index patient, follow-up and detailed neurological examination were performed, including testing for cranial nerve function; sensation in hands and feet (touch and vibration; vibration with Rydel–Seiffer tuning fork on distal phalanx of index finger and great toe; if abnormal, more proximal testing); tendon reflexes (biceps, brachioradialis, patellar and ankle) and testing for Babinski's sign. The coordination was tested by finger-to-nose and heel-to-shin tests; the coordination of the hands; standing, walking and tandem gait with open and closed eyes. In addition, routine laboratory tests, including complete blood cell count, blood urea nitrogen, liver function tests, plasma electrolytes, serum lactate and creatine kinase, blood sedimentation rate, serum protein and electrophoresis and thyroid hormones were performed on several occasions. Electrophysiological studies were performed using a Nicolet Viking IV (Nicolet Biomedical).
The index patient underwent magnetic resonance imaging and spectroscopy of the brain, as well as muscle biopsy. The biopsy sample was processed for histological analyses, including hematoxylin and eosin, modified Gomori trichrome staining, oil red O and periodic acid Schiff stains, as well as histochemical assays for NADH tetrazolium reductase, succinate dehydrogenase and cytochrome c oxidase. Glutaraldehyde fixed samples were processed for electron microscopy and viewed with a Zeiss electron microscope. Respiratory chain enzyme complex I–IV activities were determined according to the method of Fischer et al. (33).
Primary cell lines
Lymphoblasts were isolated from peripheral venous blood with heparin or EDTA as anticoagulant, using Lymphoprep™ Tubes (Nycomed Dharma As) according to the instructions of the manufacturer and immortalized with the Epstein–Barr virus. The cultures were maintained in RPMI 1451 (Gibco BRL), supplemented with 20% fetal bovine serum (Gibco BRL), 2 mm Glutamax (Gibco BRL) and penicillin–streptomycin, respectively (Gibco BRL), at 37oC incubator with 5% CO2, at a density of 3×106 cells/ml.
Nucleotides and nucleic acids
Unlabeled deoxy- and ribonucleotides were from Pharmacia BioTech; for use at concentrations >30 µm; ATP, GTP, TTP and CTP solutions were adjusted to pH 7.5 with Tris base (Research Organics). [3H]dTTP, [α-32P]dATP and [γ-32P]ATP were purchased from ICN Biochemicals. Recombinant M13 DNAs (10 650 and 6407 nt, respectively) were prepared by standard laboratory methods for use in DNA polymerase and processivity assays. Synthetic oligodeoxynucleotides were synthesized in an Applied Biosystems model 477 oligonucleotide synthesizer. Primers (15 nt) for DNA polymerase and processivity assays were complementary to the M13 DNAs. The sequences of the 21-mer primer and the 45-mer template used in the gel electrophoretic mobility shift assay (EMSA) are 5′-GACCCGATCTGATCCGATTCG-3′ and 3′-AACTGCTGGGCTAGACTAGGCTAAGCTATGCTGCGCTCCAACCTA-5′.
Baculovirus transfer vectors carrying POLGα-exo− and wild-type POLGβ were provided by Dr William Copeland (NIEHS, USA). Linearized baculovirus AcMNPV DNA (BaculoGold) was purchased from PharMingen. Synthetic oligodeoxynucleotides used for spacer-region site-directed amino-acid mutations were synthesized in an Applied Biosystems model 477 oligonucleotide synthesizer.(13)
Enzymes, proteins, antibodies
The origins of the used materials were as follows: restriction enzymes (Life Technologies and New England BioLabs), bovine pancreatic DNase I (Sigma) and T4 polynucleotide kinase (Roche-Boehringer Mannheim) and Anti-6×His monoclonal antibody (Clontech). The rabbit antisera raised against recombinant POLGα and POLGβ expressed in bacteria were prepared as described by Wang and Kaguni (34). Amphotericin, penicillin-G, streptomycin, sodium metabisulfite and phenylmethylsulfonyl fluoride (PMSF) were from Sigma. Leupeptin was purchased from the Peptide Institute (Minoh-Shi, Japan).
DNA and RNA extraction
Total lymphocyte, muscle and lymphoblast DNA were extracted using standard methods. Total RNA from lymphoblasts was isolated by RNAeasy Mini Kit (Qiagen) according to the manufacturer's instructions. Samples were treated with RNase free DNase (Stratagene) to avoid carry over of DNA template into the reverse transcription and PCR amplification.
Sequencing of mitochondrial tRNA genes, ANT1 and Twinkle
Sequencing of all 22 mitochondrial tRNA genes was performed as previously described (35). Direct sequencing of ANT1 was done as described previously (4). C10orf2 encoding for Twinkle was sequenced using intronic primers described by Spelbrink et al.(5). Cycle sequencing was performed on gel-purified fragments using the ABI PRISM Big Dye Termination kit (Perkin Elmer).
Sequencing of POLG1
All exons of index patient III/2 and her mother II/13 were amplified by PCR: 94°C for 10 min; 35 cycles of 94°C for 1 min, 60°C for 30 s and 72°C for 1 min and final extension 72°C for 10 min using 1 U of AmpliTaq Gold-polymerase (Roche). Intronic primers flanking all POLG1 exons were according to Van Goethem et al. (7). The PCR products were checked by agarose gel electrophoresis, purified by QIAEX II Gel Extraction Kit (Qiagen) and sequenced using BigDye terminator Ready Reaction Kit v.3 on a 3100 Genetic Analyzer Automatic Sequencer (Applied Biosystems). The samples from the subjects (II/9, 10, 11, 16, IV/1, 2, 3) were analyzed only for those exons where mutations were found in the index patient (exons 7 and 10).
Reverse transcription and polymerase chain reactions
Titan One Tube RT–PCR System (Roche) was used to amplify (see primers below) the fragment under study. Cycling conditions were as follows: 58°C for 30 min; 94°C for 2 min; followed by 35 cycles of 94°C for 20 s, 58°C for 30 s and 68°C for 1 min and finally 68°C for 10 min. The absence of DNA carry over was controlled by parallel reaction without the initial cDNA synthesis before amplification steps. RT–PCR was used together with mini-sequencing in order to quantitate conceivable allele specific expression bias.
Solid-phase mini-sequencing
Initial PCR amplification was done by AmpliTaq Gold-polymerase (Roche). Solid-phase mini-sequencing was done according to standard reaction conditions (36). The part of exon 7 harboring A467T mutation was amplified using a 5′-biotinylated forward primer (5′-accagaactgggagcgttac-3′) and an unmodified reverse primer (5′-ctacctctctcctgagagca-3′). Detection and quantitation of wild-type or mutated base were done utilizing detection primer (5′-cagctggcaggcatcattgg-3′) and extension with a tritiated deoxy [1′,2′,5-3H] cytidine 5′-triphosphate and deoxy [methyl-1′,2′-3H] thymidine 5′-triphosphate (Amersham Biosciences), respectively. Incorporation of the nucleoside triphosphates was done by a thermostable DNA polymerase from Thermus brockianus (DynaZyme™/II, FinnZymes).
The part of exon 23 harboring the Q1236H variant was amplified with a 5′-biotinylated forward primer (5′-ttgaggtggcatcctaacca-3′) and an unmodified reverse primer (5′-acgggagcaaatacagagcc-3′). Detection of both variants was done by detection primer (5′-cagctggcaggcatcattgg-3′) with tritiated deoxy [1′, 2′, 5-3H] cytidine 5′-triphosphate and deoxy [3H] adenosine 5′-triphosphate (Amersham Biosciences).
mtDNA analysis
Long-PCR analysis was performed using DyNAzyme EXT™ polymerase (Finnzymes) according to suppliers instructions for standard reaction conditions (0–10 kb). Primers used for this assay were located at nt positions 8238–8262 (forward) and 16 496–16 465 (reverse), amplifying a region of mtDNA often harboring the multiple mtDNA deletions. The cycling conditions were as follows: 94°C for 2 min; 35 cycles of 94°C for 15 s, 63°C for 30s, 72°C for 2 or 6 min and final extension at 72°C for 10 min. The optimal template amount was titrated to be 10 ng, as also previously published (18,37). By reducing the template amount, we were able to amplify deletions only from patient samples and never in age-matched controls (muscle samples from healthy PEO-patient siblings). Two separate PCR reactions for each sample were set up, with otherwise similar reaction conditions but different extension times. Six minutes extension was used to visualize the 8.2 kb product from the normal mtDNA molecule, and 2 min was used to favor the amplification of the deleted molecules. If 6 min extension gave a prominent amplification of wild-type fragment and no products were seen in the 2 min amplification, the sample was judged to have no mtDNA deletions. The multiple mtDNA deletions amplified clearly in both conditions.
Total DNA extracted from the muscle biopsy sample from the index patient was used for Southern blot analysis as described in detail (38). The DNA was restriction digested with BamHI or PvuII restriction enzymes (Fermentas). Both linearize mtDNA but from separate sites. The samples were electrophoresed through a 0.8% agarose gel, and complete digestion was confirmed in ethidium bromide staining. The DNA was transferred on a Hybond-N (Amersham Biosciences) nylon membrane according to standard procedures, and the membranes were hybridized with a probe, cloned fragment of mtDNA (nt 1–740), and random-labeled to a specific activity of 1×108 c.p.m./µl, in order to detect deleted mtDNA species. The same membrane was rehybridized with a nuclear probe 18S rRNA, cloned in pBR322, in order to quantitate the mtDNA signal versus genomic DNA, to detect possible mtDNA depletion. The radioactive signal was detected by scanning of PhosphorImager screens (Fuji Photo Film Co.) with Typhoon 9400 Imager (Amersham Biosciences) and quantitated with ImageQuant 5.1 software (Amersham Biosciences).
Construction of recombinant baculoviruses
Baculovirus transfer vectors carrying mutant POLG1 cDNAs described were prepared by QuikChange mutagenesis with Pfu DNA polymerase (Stratagene), according to the manufacturer's recommendations. A typical PCR was carried out in a 50 µl reaction mixture with 50 ng of DNA template (His-pVLGA) and 2 U of Pfu DNA polymerase. A specific primer pair was used for each mutant as follows: POLGα-A467T: 5′-AGTCGTTGATGGAG(t)CTC(g)AC(g)CAATGATGCCTGCCA-3′ and 5′-TGGCAGGCATCATTGG(c)TG(c)AGC(a)TCCATCAACGACT-3′; POLGα-R627W: 5′-CTACTTGGTA(g)CCTGGGCGGT(c)GGGACAACCT-3′ and 5′-AGGTTGTCCCA(g)CCGCCCAGGT(c)ACCAAGTAG-3′; POLGα-R627Q: 5′-CTACTTGGTA(g)CCTGGGCGGCA(g)GGACAACCT-3′ and 5′-AGGTTGTCCT(c)GCCGCCCAGGT(c)ACCAAGTAG-3′; POLGα-Q1236H: 5′-AACGAAGCCAT(g)CCC(t)GGG(a)CCATAGCACT-3′ and 5′-AGTGCTATGGC(t)CCG(a)GGA(c)TGGCTTCGTT-3′. Bold letters indicate nucleotides that replace those in the wild-type POLGα in parentheses with lower case letters. The introduced/removed endonuclease cleavage sites are indicated by underscore.
The DNA template was first denatured at 95°C for 45 s, followed by 20–25 three-step cycles of 95°C for 45 s, 50°C for 1 min and 68°C for 2 min per kb DNA template. The reaction mixture was then digested with 10 U of DpnI for 2 h at 37°C to eliminate the methylated parental DNA template. A 2 µl aliquot was used for transformation of competent Escherichia coli XL-1 Blue cells [recA1, endA1, gyrA96, thi, hsdR17, supE44, relA1, lac, (F′proAB, lacIqZ M15, Tn10 (tetr)] by electroporation, using an E. coli Pulser (BioRad). The transformed colonies were screened by restriction endonuclease digestion to the introduced/removed cleavage sites in each primer pair. The correct sequence of the various plasmid constructs was verified by DNA sequence analysis. Transfer vectors encoding the mutant versions of POLGα were purified and baculoviruses prepared as described by Wang and Kaguni (34).
Production and purification of the recombinant POLGα subunit
The Sf9 (Spodoptera frugiperda) insect cells were the gift of Dr Suzanne Thiem. Sf9 cells were maintained in TC-100 insect cell culture medium (United States Biological) containing 10% fetal bovine serum (Life Technologies, Inc.) at 27°C. For protein analysis of whole cell lysates, the cells were collected by centrifugation (at 3000g) at a density of 3×107 cells/ml and lysed in Laemmli gel loading buffer and stored at −20°C. Sf9 cells (600 ml) were grown to a cell density of 2×106 cells/ml, diluted to a density of 1×106 cells/ml (1200 ml total) and infected with recombinant baculovirus at multiplicity of infection value of five and harvested 48 h postinfection. The cells were pelleted and washed with an equal volume of cold phosphate buffered saline buffer. All the following operations were performed at 0–4°C. The cell pellet (∼1.2×109 cells) was resuspended in 36 ml of homogenization buffer (50 mm Tris–HCl, pH 7.5, 100 mm KCl, 5 mm EDTA, 7 mm mercaptoethanol, 10 mm sodium bisulfite, 1 mm PMSF and 2 µg/ml leupeptin) and lysed by 20 strokes in a Dounce homogenizer. The homogenate was centrifuged at 1000g for 7 min. The resulting pellet was twice resuspended in 12 ml of homogenization buffer and rehomogenized and centrifuged as mentioned previously. The combined supernatant fractions were centrifuged at 8000g for 15 min to pellet mitochondria, and the resulting supernatant was centrifuged at 100 000g for 30 min to obtain the cytoplasmic soluble fraction (fraction I). Purification of recombinant POLGα mutants was performed as described by Wernette and Kaguni (39) with the following alterations. Fraction I (140–240 mg protein) was adjusted to 80 mm potassium phosphate and loaded onto a phosphocellulose column equilibrated with 80 mm potassium phosphate buffer (80 mm potassium phosphate, pH 7.6, 20% glycerol, 7 mm 2-mercaptoethanol, 1 mm PMSF, 10 mm sodium metabisulfite and 2 µg/ml leupeptin) at a packing ratio of 6 mg protein per packed milliliter of resin and at a flow rate of 12 ml/h. The column was washed and fractions were analyzed and processed to fraction IIb as described in Wang and Kaguni (34). Fraction IIb (10–20 mg protein) was dialyzed when necessary in 10 mm potassium phosphate buffer in a collodion bag (molecular mass cut-off 10 000 kDa) until an ionic strength of ∼300–400 mm KCl was reached and then loaded onto Ni-NTA resin at a ratio of ∼7.5 mg protein per milliliter of packed resin at a flow rate of 1–2 CV (column volumes) per hour. The resin was washed with 2 CV of buffer containing 20 mm Tris–HCl, pH 7.5, 500 mm KCl, 8% glycerol, 5 mm imidazole, 0.1% Tx-100, 7 mm 2-mercaptoethanol, 1 mm PMSF, 10 mm sodium metabisulfite and 2 µg/ml leupeptin and successively eluted with the same buffer containing 25 mm (2 CV), 250 mm (2 CV) and 500 mm imidazole (2 CV). Active fractions were pooled (fraction III, 1.6–2.3 ml) and loaded onto two 12–30% glycerol gradients as described by Wernette and Kaguni (39), except that the gradient buffer contained 30 mm Tris–HCl, pH 8.0, 100 mm KCl, 2 mm EDTA, 7 mm 2-mercaptoethanol, 10 mm sodium bisulfite, 1 mm PMSF and 2 µg/ml leupeptin. The active fractions were again pooled (fraction IV, 32–300 µg of protein, 3000–28 000 U/mg for various mutants), stabilized by the addition of glycerol to 45% and stored at −20°C or frozen in liquid nitrogen and stored at −80°C. Equivalent units of fraction IV enzymes were electrophoresed and stained using silver nitrate or electrophoresed and immunoblotted as described by Wang et al. (40) using anti-6xHis monoclonal antibody. Quantitation was performed using Kodak 1D version 3.5 software (Eastman Kodak Company).
Functional assays of the purified POLGα mutants
All the assays were performed at least three times in triplicate. Standard error was generally <20%.
DNA polymerase assay
DNA polymerase activity was assayed on DNase I-activated calf thymus DNA or singly primed M13 DNA as previously described (39,41). Specific modifications are indicated in the figure legends. Assays were performed at least three times, in triplicate. One unit of standard activity is that amount that catalyzes the incorporation of 1 nm of deoxynucleoside triphosphate into acid insoluble material in 60 min at 30°C using DNase I-activated calf thymus DNA as the substrate.
Gel electrophoretic mobility shift assay
DNA binding by wild-type and mutant POLGα was assayed by gel electrophoretic mobility shift assay. Purified POLGα (∼20 ng) was incubated with the [γ-32P]ATP-labeled template–primer substrate (0.22 pmol) for 10 min at 30°C in standard reaction buffer containing 50 mm Tris–HCl (pH 8.0), 4 mm MgCl2, 5 mm dithiothreitol and 30 mm KCl (without POLGβ) or 100 mm KCl (with POLGβ), followed by the addition of bromophenol blue and glycerol to 0.01 and 5%, respectively, and electrophoresis in a 6% native polyacrylamide gel (13×13×0.15 cm) in 45 mm Tris-borate (pH 8.3) and 1 mm EDTA. After electrophoresis, the gel was dried under vacuum and exposed to a PhosphorImager screen (Molecular Dynamics). The data were analyzed using the ImageQuant version 5.2a software.
Analysis of products of processive DNA synthesis by gel electrophoresis
Reaction mixtures (50 µl) contained 50 mm Tris–HCl, pH 8.0, 4 mm MgCl2, 10 mm DTT, 100 mm KCl, 400 µg/ml bovine serum albumin, 30 µm each of dTTP, dCTP, dGTP and 10 µm dATP and [α-32P]dATP (2×104 c.p.m./pmol), 10 µm singly primed M13 DNA and 20 ng of purified enzyme. Incubation was for 10 min at 30°C. Products were made 1% in sodium dodecyl sulphate and 10 mm in EDTA, heated for 4 min at 80°C and precipitated with ethanol in the presence of 0.5 µg tRNA as carrier. The ethanol precipitates were resuspended in 80% formamide and 90 mmM Tris-borate. Aliquots were denatured for 2 min at 100°C and electrophoresed in a 6% polyacrylamide slab gel (13×30×0.15 cm) containing 7 m urea in 90 mm Tris-borate (pH 8.3) and 25 mm EDTA or in a 1.5% alkaline agarose gel (13×18×0.7 cm) containing 30 mm NaCl and 2 mm EDTA in 30 mm NaOH and 2 mm EDTA. Approximately equal amounts of radioactivity were loaded in each lane. Gels were washed in distilled water for 20 min, dried under vacuum and exposed to a phosphor screen (Molecular Dynamics). The data were analyzed by scanning the phosphor screen using the Storm 820 Scanner (Molecular Dynamics), and the volume of each of the distinct bands (nine bands in Fig. 8 and 22 bands in Fig. 9) was determined by computer integration analysis using ImageQuant version 5.2 software (Molecular Dynamics); the volume of the bands was normalized to the nt level to correct for the uniform labeling of the DNA products.
Other methods
Protein concentration was determined by the method of Bradford (42), with bovine serum albumin as the standard. Immunoblotting was performed as described by Wang et al. (40).
ACKNOWLEDGEMENTS
The electron micrograph was kindly provided by Professor H. Budka, Institute of Neurology, Vienna Medical University, Austria. The authors wish to thank the following funding sources: Academy of Finland, Sigrid Juselius Foundation and Helsinki University (for A.S.) and the National Institutes of Health (Grant GM45295 to L.S.K.).
Conflict of Interest statement. None declared.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
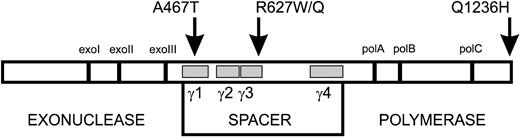
Figure 1. Diagram of POLGα structure indicating the detected patient mutations. POLGα primary sequence of 1239 amino acids is depicted as an open box. Vertical bars represent highly conserved exonuclease and polymerase motifs in the family-A polymerases. The gray boxes indicate the location of the four conserved spacer-region blocks γ1–γ4 (within amino acid boundaries 444–820). The substitutions studied in this report are indicated above the box by arrows.
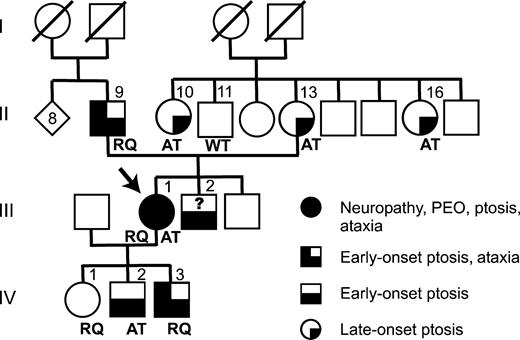
Figure 2. Co-segregation of clinical symptoms and detected POLG1 mutations. Arrow indicates the index patient (proband) of the family. RQ, R627Q mutation with Q1236H polymorphism; AT, A467T mutation of POLGα; WT, wild-type; ?, genotype unknown; slash over a symbol, deceased subject.
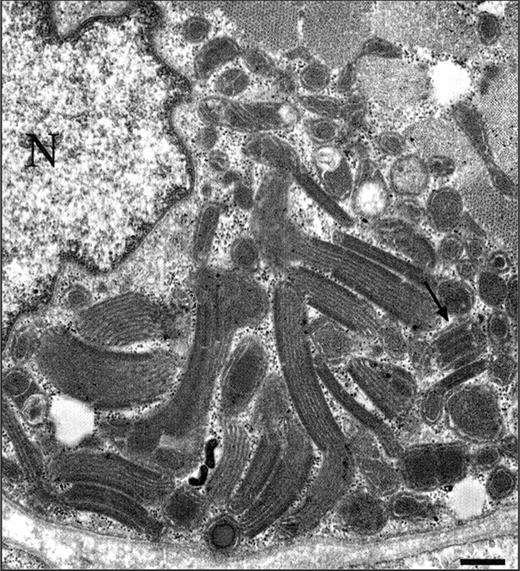
Figure 3. Electron microscopy demonstrating increased amount of structurally abnormal mitochondria with para-crystalline inclusions (arrow) in a muscle fiber (N, nucleus), bar, 1.1 µm.

Figure 4. Alignment shows the POLGα regions in which the amino acid substitutions detected in this study are located. From top to bottom: A467T is invariant among species, R627Q/W is highly conserved and Q1236 is conserved among human, dog, rat, mouse, puffer fish and fruit fly. Asterisks (*) below alignment indicate the invariant amino acids.
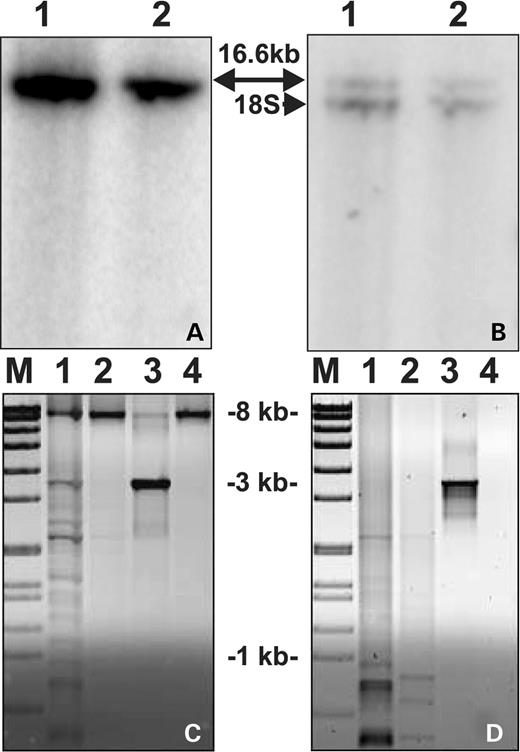
Figure 5. Southern blot analysis of mtDNA/18S rRNA gene and long-PCR of patient's muscle mtDNA. (A)Total muscle DNA was digested with restriction enzyme PvuII and hybridized with a cloned fragment of mtDNA (nt 1–740). Samples: 1, muscle DNA from a healthy control; 2, muscle DNA from the index patient and 16.6 kb indicates the full-size mtDNA. (B) Same membrane as in (A), stripped and reprobed with genomic 18S rRNA clone (lower band). Quantification of the hybridization signals indicated that mtDNA in index patient was 106% of that in control, normalized against the nuclear 18S signal. (C) Long-PCR of muscle mtDNA with 6 min or (D) 2 min extension times. M, molecular weight marker VII (Roche); 1, control patient with multiple mtDNA deletions (15); 2, index patient; 3, control patient with a single mtDNA deletion; 4, healthy control. Only deleted mtDNA is amplified in short 2 min extension conditions, whereas the wild-type mtDNA (8.2 kb) is also amplifying in long 6 min conditions.
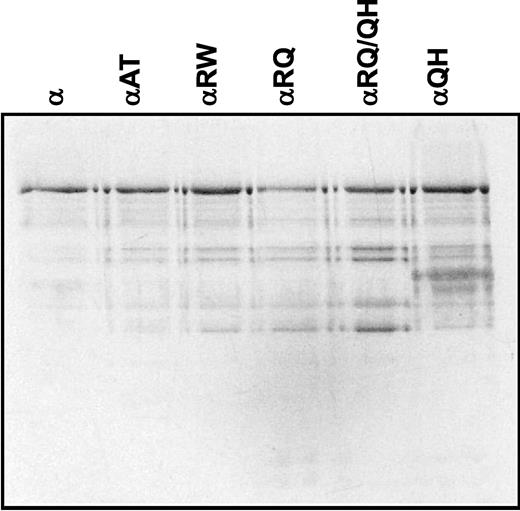
Figure 6. SDS–polyacrylamide gel electrophoresis of mutant human POLGα. Near-homogeneous fractions of baculovirus-expressed and purified-recombinant POLGα (200 ng, as indicated) were denatured and electrophoresed in a 10% SDS–polyacrylamide gel, and the proteins were stained with silver. α, POLGα; αAT, A467T mutant; αRW, R627W mutant; αRQ, R627Q mutant; αRQ/QH, R627Q/Q1236H double mutant; αQH, Q1236H mutant.
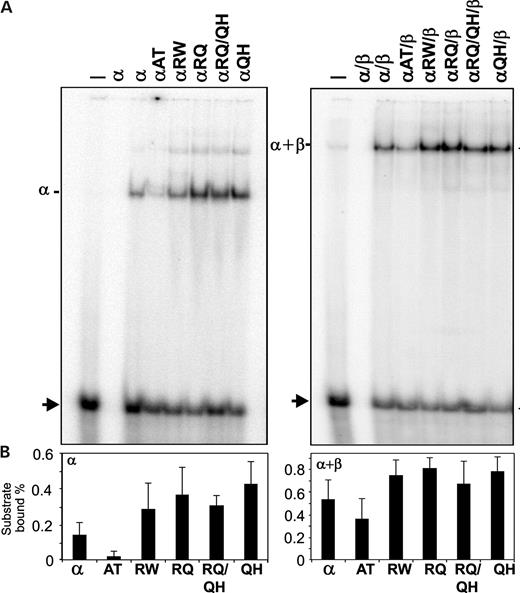
Figure 7. DNA-binding affinity of mutant human POLGα. DNA-binding affinity of mutant POLGα was determined by EMSA using a radiolabeled oligonucleotide template primer. (A) Protein–DNA complexes formed by POLGα in the presence (right) or absence (left) of POLGβ were electrophoresed in a 6% native polyacrylamide gel, and the gel was exposed to a phosphor screen. The first two lanes in each panel represent DNA substrate only and POLGα only controls (−,α), and the adjacent lanes contain both the DNA substrate and the indicated forms of POLGα; mutant codes as in Figure 6. Arrowhead indicates the unbound DNA substrate. (B) Bound and unbound DNA substrate bands were quantitated as described in Materials and Methods. The data in each panel were derived from duplicate assays in three independent experiments.
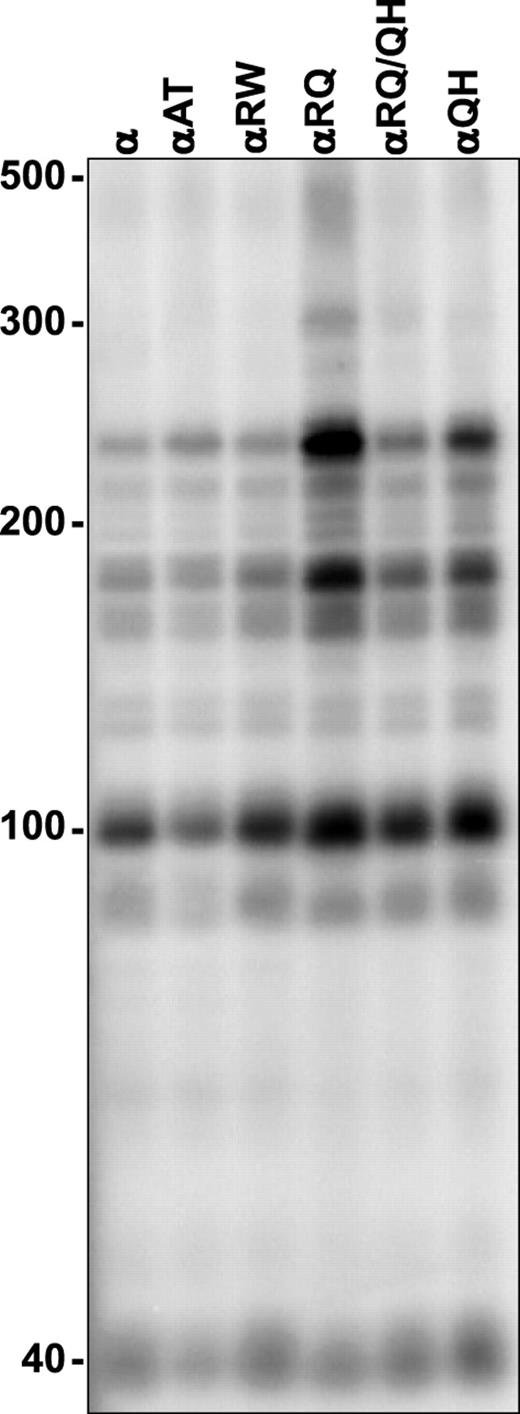
Figure 8. Processivity of mutant human POLGα. DNA synthesis by mutant POLGα was measured at 30 mm KCl on singly primed M13 DNA, as described in Materials and Methods. DNA product strands were isolated, denatured and electrophoresed in a 6% denaturing polyacrylamide gel and the gel was exposed to a phosphor screen. The lengths of the fragments reflect the enzymes' ability to copy the template before dissociation and the intensities of the bands reflect the proportion of the products reaching the specific fragment length. Each of the nine distinct bands derived from two independent experiments were quantitated and yielded the following processivity values (expressed as average processivity units, apu): α, 63 nt; αA467T, 69 nt; αR627W, 70 nt; αR627Q, 108 nt; αR627Q/Q1236H, 77 nt; αQ1236H, 81 nt. The codes for protein variants are as in Figure 5.
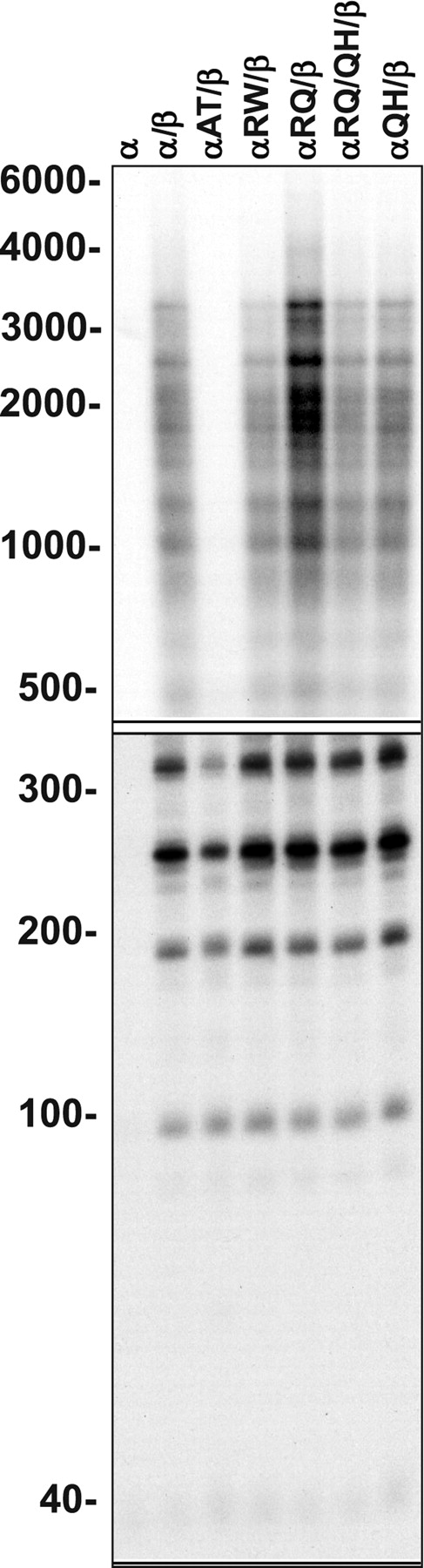
Figure 9. Processivity of reconstituted mutant POLG. Human POLG was reconstituted using a 3-fold molar excess of the accessory POLGβ-subunit over POLGα, and DNA synthesis was measured at 100 mm KCl on singly primed M13 DNA. DNA product strands were isolated, denatured and electrophoresed in denaturing 1.5% agarose (upper panel) and 6% polyacrylamide (lower panel) gels, and the gels were exposed to a phosphor screen. The presence of the accessory POLGβ subunit—also called the processivity factor of POLG—enhances the processivity of the enzyme considerably, and the fragment lengths are, therefore, considerably longer than those with POLGα alone. All the 22 distinct bands derived from two independent experiments were quantitated and yielded the following processivity values (expressed as average processivity units, apu): α, 42 nt; αβ, 437 nt; A467T, 145 nt; R627W, 358 nt; R627Q, 558 nt; R627W/R627Q, 416 nt; Q1236H, 444 nt. The codes for protein variants are as in Figure 6.
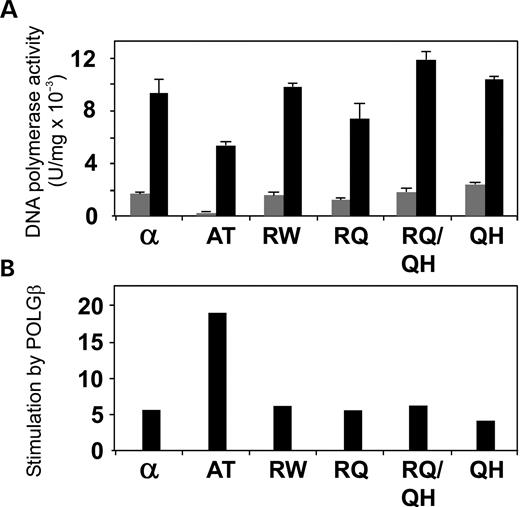
Figure 10. Differential stimulation of mutant human POLGα by POLGβ. (A) DNA synthesis by mutant POLGα was measured at 100 mm KCl on singly primed M13 DNA in the presence (black bars) or absence (gray bars) of POLGβ. (B) Fold stimulation of mutant human POLGα by POLGβ based on the data from (A). The codes for protein variants are as in Figure 6.
References
Zeviani, M., Servidei, S., Gellera, C., Bertini, E., DiMauro, S. and DiDonato, S. (
Suomalainen, A., Majander, A., Haltia, M., Somer, H., Lönnqvist, J., Savontaus, M.L. and Peltonen, L. (
Moslemi, A.R., Melberg, A., Holme, E. and Oldfors, A. (
Kaukonen, J., Juselius, J.K., Tiranti, V., Kyttälä, A., Zeviani, M., Comi, G.P., Keränen, S., Peltonen, L. and Suomalainen, A. (
Spelbrink, J.N., Li, F.Y., Tiranti, V., Nikali, K., Yuan, Q.P., Tariq, M., Wanrooij, S., Garrido, N., Comi, G., Morandi, L. et al. (
Nishino, I., Spinazzola, A. and Hirano A. (
Van Goethem, G., Dermaut, B., Lofgren, A., Martin, J.J. and Van Broeckhoven, C. (
Kaguni, L.S. (
Schmitt, M.E. and Clayton, D.A. (
Bowmaker, M., Yang, M.Y., Yasukawa, T., Reyes, A., Jacobs, H.T., Huberman, J.A. and Holt, I.J. (
Holt, I.J., Lorimer, H.E. and Jacobs, H.T. (
Luo, N. and Kaguni, L.S. (
Lamantea, E., Tiranti, V., Bordoni, A., Toscano, A., Bono, F., Servidei, S., Papadimitriou, A., Spelbrink, H., Silvestri, L., Casari, G. et al. (
Luoma, P., Melberg, A., Rinne, J.O., Kaukonen, J.A., Nupponen, N.N., Chalmers, R.M., Oldfors, A., Rautakorpi, I., Peltonen, L., Majamaa, K. et al. (
Mancuso, M., Filosto, M., Oh, S.J. and DiMauro, S. (
Van Goethem, G., Martin, J.J., Dermaut, B., Lofgren, A., Wibail, A., Ververken, D., Tack, P., Dehaene, I., Van Zandijcke, M., Moonen, M. et al. (
Van Goethem, G., Luoma, P., Rantamäki, M., Al Memar, A., Kaakkola, S., Hackman, P., Krahe, R., Lofgren, A., Martin, J.J., De Jonghe, P. et al. (
Naviaux, R.K. and Nguyen, K.V. (
Ferrari, G., Lamantea, E., Donati, A., Filosto, M., Briem, E., Carrara, F., Parini, R., Simonati, A., Santer, R. and Zeviani, M. (
Van Goethem, G., Mercelis, R., Lofgren, A., Seneca, S., Ceuterick, C., Martin, J.J. and Van Broeckhoven, C. (
Agostino, A., Valletta, L., Chinnery, P.F., Ferrari, G., Carrara, F., Taylor, R.W., Schaefer, A.M., Turnbull, D.M., Tiranti, V. and Zeviani, M. (
Di Fonzo, A., Bordoni, A., Crimi, M., Sara, G., Del Bo, R., Bresolin, N. and Comi, G.P. (
Ponamarev, M.V., Longley, M.J., Nguyen, D., Kunkel, T.A. and Copeland, W.C. (
Longley, M.J., Ropp, P.A., Lim, S.E. and Copeland, W.C. (
Van Goethem, G., Schwartz, M., Lofgren, A., Dermaut, B., Van Broeckhoven, C. and Vissing, J. (
Brobby, G.W., Muller-Myhsok, B. and Horstmann, R.D. (
Loffler, J., Nekahm, D., Hirst-Stadlmann, A., Gunther, B., Menzel, H.J., Utermann, G. and Janecke, A.R. (
Wanrooij, S., Luoma, P., van Goethem, G., van Broeckhoven, C., Suomalainen, A. and Spelbrink, J.N. (
Iyengar, B., Roote, J. and Campos, A.R. (
Van Goethem, G., Lofgren, A., Dermaut, B., Ceuterick, C., Martin, J.J. and Van Broeckhoven, C. (
Graziewicz, M.A., Longley, M.J., Bienstock, R.J., Zeviani, M. and Copeland, W.C. (
Fischer, J.C., Ruitenbeek, W., Gabreels, F.J., Janssen, A.J., Renier, W.O., Sengers, R.C., Stadhouders, A.M., ter Laak, H.J., Trijbels, J.M. and Veerkamp, J.H. (
Wang, Y. and Kaguni, L.S. (
Hofmann, S., Bezold, R., Jaksch, M., Obermaier-Kusser, B., Mertens, S., Kaufhold, P., Rabl, W., Hecker, W. and Gerbitz, K.D. (
Suomalainen, A. and Syvänen, A.C. (
Kajander, O.A., Poulton, J., Spelbrink, J.N., Rovio, A., Karhunen, P.J. and Jacobs, H.T. (
Suomalainen, A., Majander, A., Wallin, M., Setälä, K., Kontula, K., Leinonen, H., Salmi, T., Paetau, A., Haltia, M., Valanne, L. et al. (
Wernette, C.M. and Kaguni, L.S. (
Wang, Y., Farr, C.L. and Kaguni, L.S. (
Farr, C.L., Wang, Y. and Kaguni, L.S. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}