




Abstract
Mutations in the non-lysosomal cysteine protease calpain 3 cause limb-girdle muscular dystrophy type 2A (LGMD2A). Our previous studies of the calpain 3 knockout mouse (C3KO) suggested a role for calpain 3 in sarcomere formation and remodeling. Calpain 3 may mediate remodeling by cleavage and release of myofibrillar proteins, targeting them for ubiquitination and proteasomal degradation. Loss of proper protein turnover may be the basis for this muscle disease. To test this hypothesis in vivo, we used an experimental model of hindlimb unloading and reloading that has been shown to induce sarcomere remodeling. We showed that the rate of atrophy and especially the rate of growth are decreased in C3KO muscles under conditions promoting sarcomere remodeling. In wild-type mice, an elevated level of ubiquitinated proteins was observed during muscle reloading, which is presumably necessary to remove atrophy-specific and damaged proteins. This increase in ubiquitination correlated with an increase in calpain 3 expression. C3KO muscles did not show any increase in ubiquitination at the reloading stage, suggesting that calpain 3 is necessary for ubiquitination and that it acts upstream of the ubiquitination machinery. We found upregulation of heat shock proteins in C3KO muscles following challenge with a physiological condition that requires highly increased protein degradation. Furthermore, old C3KO mice show evidence of insoluble protein aggregate formation in skeletal muscles. These studies suggest that accumulation of aged and damaged proteins can lead to cellular toxicity and a cell stress response in C3KO muscles, and that these characteristics are pathological features of LGMD2A.
INTRODUCTION
Calpain 3 belongs to a family of non-lysosomal, Ca2+-dependent, cysteine proteases (1). Three members of the family are expressed at high levels in skeletal muscles, including the ubiquitous family members calpain 1 (or µ-calpain) and 2 (or m-calpain) and the muscle-specific isoform calpain 3 (CAPN3 or p94). Mutations in CAPN3 cause limb-girdle muscular dystrophy type 2A (LGMD2A), an autosomal recessive muscular dystrophy characterized by progressive atrophy and weakness of the proximal limb muscles (2). Until now, the normal biological function of CAPN3 has not been known nor has it been understood why mutations result in disease.
Several mouse knockout models for calpainopathy have been generated. Studies on mice lacking exons 2 and 3 of CAPN3 showed myonuclear apoptosis due to IkB/NFkB pathway perturbation (3). In our previous studies, CAPN3 knockout mice (C3KO) were generated that completely lack both CAPN3 mRNA and protein in skeletal muscles and it was shown that these mice replicate features of the LGMD2A phenotype (4). These mice are smaller than wild-type (WT) mice and have reduced muscle mass and fiber cross-sectional areas; however, we did not find evidence of myonuclear apoptosis. Histological examination of C3KO muscles revealed small areas of focal necrosis and regeneration similar to those observed in LGMD2A patients with mild or pre-clinical forms of the disease. Studies on primary myoblasts demonstrated that C3KO cells are able to fuse to form myotubes with the same efficiency as that of WT cells; however, sarcomere formation is delayed as was shown by electron microscopy and biochemical analyses. Ultrastructural analysis of adult muscles also showed abnormal sarcomere architecture, namely, misalignment of thick filaments (A-bands), suggesting that CAPN3 is necessary for sarcomere formation and maintenance of sarcomeric structural integrity (4).
CAPN3 may participate in sarcomere formation or maintenance through its interaction with titin, a giant protein that serves as a molecular ruler of the sarcomere, orchestrating alignment of sarcomeric proteins during development and in post-natal muscle (5). An interaction between CAPN3 and titin was first found in yeast two-hybrid assays (6,7) and was later confirmed at the protein level (4).Whether anchorage to titin serves to stabilize CAPN3 or to place CAPN3 in close proximity to its substrates is still unclear; however, it was demonstrated that titin itself can serve as a substrate for CAPN3 in regions adjacent to where it binds, and some pathogenic mutations in CAPN3 reduce the affinity of CAPN3 to titin (4). Taken together, these data have led to the hypothesis that CAPN3 may work together with titin in formation and remodeling of sarcomeres in skeletal muscle.
The observation that CAPN3 plays a role in sarcomere remodeling in vitro has led us to hypothesize that CAPN3 also promotes myofibrillar protein turnover during sarcomere remodeling in post-natal muscle. To test this hypothesis in vivo requires a model system that induces remodeling. Mouse hindlimb suspension provides such a model. In this procedure, animals are suspended by the tails so that the hindlimbs are non-weight bearing. During the suspension stage, non-pathological muscle atrophy is induced. The soleus is the most affected muscle, losing 40–60% of its mass following 2 weeks of suspension (8). Reloading to normal weight bearing following the suspension causes extensive sarcomere formation and muscle growth that restores normal muscle size and strength (9). Thus, hindlimb suspension and remodeling is a good model to test the role of CAPN3 in muscle adaptation.
Numerous studies have shown that the ubiquitin (Ub)–proteasome pathway is the principal mechanism involved in protein breakdown in muscle remodeling and atrophy. The involvement of the Ub–proteasome pathway has been demonstrated in various animal models that includes both pathological (e.g. cancer cachexia, fasting, sepsis, diabetes and denervation), and non-pathological (e.g. muscle disuse) models of muscle wasting (10,11). Although the proteasome is the final degradative mechanism utilized by muscle during wasting, it is clear that there should be additional proteolytic systems operating upstream to disassemble the sarcomere, as proteasomes are not able to degrade intact myofibrils (12). Ca2+-dependent proteolysis may be such a mechanism, as this process also contributes to muscle protein breakdown under catabolic conditions (11,13). On the basis of these observations, it is likely that ubiquitous calpains act upstream of proteasomes to release myofibrillar proteins from these multiprotein complexes (reviewed in 13). The significance of ubiquitous calpains for muscle remodeling during hindlimb unloading has been previously demonstrated in vivo, where it was shown that transgenic animals overexpressing a natural inhibitor of m- and µ-calpains (calpastatin) have a 30% reduction in muscle atrophy during muscle unloading (14). Thus, it is likely that the calpain and proteasomal pathways work together to elicit muscle wasting, although it is unclear whether calpains act upstream or in parallel with the Ub–proteasome system.
The Ub–proteasome pathway also plays a role in muscle recovery and growth following a catabolic condition. It has been demonstrated that during the early growth phase when reloading follows muscle unloading, both muscle protein synthesis and proteasome-mediated breakdown are elevated (15). High-molecular weight Ub–protein conjugates accumulate in reloaded muscles at the early stage of reloading. It is likely that ubiquitination and proteasomal degradation are required to eliminate or replace damaged or atrophy-induced proteins. Ub–proteasome and calpain proteolysis were also shown to be involved in muscle remodeling in response to altered contractile activity (16), injury (17) and in response to eccentric exercise (18). These studies demonstrate that the Ub–proteasome pathway is not only involved in protein breakdown that leads to muscle atrophy, but also involved in processes that lead to muscle regeneration and growth.
Reduced sarcomere formation in C3KO myotubes and the likely involvement of multiple proteolytic systems in sarcomere remodeling led us to test the hypothesis that CAPN3 is involved in muscle remodeling in vivo. In this study, we found that the adaptive remodeling response was reduced in muscles from C3KO mice that had been subjected to hindlimb unloading and reloading. Muscle growth following reloading occurred at a much lower rate when compared with the WT muscles, although the IGF-1/PI3K/Akt pathway was activated normally. We also provide evidence that CAPN3 acts upstream of protein ubiquitination at the muscle reloading stage and that CAPN3 deficient muscle contains evidence of insoluble protein aggregation and cellular stress. Taken together, our data suggest that CAPN3 is involved in sarcomere remodeling in vivo by promoting ubiquitination of muscle proteins. Furthermore, these studies provide insight into mechanisms of the disease process of LGMD2A and suggest that an insufficient rate of protein turnover can lead to muscle disease.
RESULTS
Rates of fiber atrophy and growth are affected by the absence of CAPN3
To determine whether CAPN3 might play a role in muscle remodeling, we used the hindlimb unloading model in which WT and C3KO mice were subjected to hindlimb suspension for 10 days to elicit muscle atrophy. After 10 days of suspension, mice were returned to their standard cage activity for either 2 or 4 days. It has been shown previously that standard cage activity is sufficient to totally reverse muscle atrophy in ∼7–10 days (9). As the soleus is the muscle most affected by unloading and reloading, we measured cross-sectional areas for both fast and slow fibers of solei at each time point of the experiment (Fig. 1A and B, respectively). As we found previously (4), an average soleus fiber cross-sectional area of C3KO mice is ∼25% reduced when compared with the WT. At the end of a 10 day suspension period, both C3KO and WT muscles showed a reduction in fiber cross-sectional areas. However, the rate of change was ∼45% lower in the C3KO when compared with the WT mice (Fig. 1C). At the early stage of reloading (2 days), WT muscles showed a 5.2% increase in the average cross-sectional area when compared with the WT muscles at day 10 of suspension. In contrast, C3KO showed no growth at this early recovery stage. After 4 days of reloading, the average cross-sectional area of WT muscles increased by 43.1%, whereas C3KO muscles showed only 6.1% increase (Fig. 1C and D). Thus, both fiber atrophy and fiber growth occurred more slowly in the C3KO muscles when compared with WT. Interestingly, the rate of recovery of C3KO muscles was more affected than the rate of atrophy.
C3KO muscles do not have an abnormal level of muscle damage at the reloading stage
It is well known that reloading of muscles after a prolonged period of disuse can cause damage to the muscles. One possible explanation for decreased muscle growth in the C3KO mice is that a higher level of muscle damage occurred when compared with the WT. Damaged fibers can be visualized by uptake of mouse FITC-conjugated IgG (19). We assessed fiber damage by this assay and found no difference in the number of damaged fibers between C3KO and WT mice. In both cases, there were zero to three FITC-positive fibers per section (Fig. 2A and B).
To further confirm that the reduced growth of C3KO muscles is not due to more mechanically induced damage, we used another experimental setting that is known to cause muscle damage. C3KO and WT mice were subjected to eccentric running exercise for 4 days followed by injection of Evans blue dye to identify damaged fibers (20). Again, no difference in the number of damaged fibers was found between C3KO and WT mice (Fig. 2C and D). These data suggest that C3KO muscles are not more susceptible to muscle damage when compared with WT and that reduced growth of reloaded C3KO muscles cannot be explained by an abnormally high number of damaged fibers.
C3KO muscles show normal induction of the IGF-1/PI3K/Akt pathway during reloading
The IGF-1/PI3K/Akt pathway was shown to be a critical regulator of muscle mass and fiber size (21). This pathway not only induces anabolic processes (i.e. protein synthesis and muscle growth), but also prevents expression of muscle atrophy-specific genes (22). Activation of the pathway results in phosphorylation of the serine/threonine kinase Akt (or protein kinase B) and increases protein translation. To test whether failure of C3KO mice to recover from muscle unloading is due to inadequate activation of this pathway, we first used primary myoblasts isolated from C3KO and WT mice and induced for differentiation by insulin–transferrin–selenium X. In these differentiating cells, no difference in the concentration of activated (phosphorylated) Akt was found between C3KO and WT myotubes (Fig. 3A).
Muscle extracts from unloaded and reloaded muscles were also probed with a phosphorylated Akt-specific antibody to detect activated Akt. As shown in Figure 3B (upper panel, P-Akt blot), muscle reloading induces activation of Akt (lanes 5 and 6) and this process is not affected by the absence of CAPN3 (compare lanes 11 and 12 with lanes 5 and 6, P-Akt blot). One of the targets of the PI3K/Akt pathway is a protein kinase p70S6K that phosphorylates ribosomal protein S6. This in turn leads to an increase in the rate of translation. We used a phospho-S6 specific antibody to check whether phosphorylation of S6 proteins occurred normally in the C3KO muscles undergoing reloading (Fig. 3B, lower panel). Activation of the IGF-1/PI3K/Akt pathway at the reloading stage caused phosphorylation of S6 protein, and no difference was found between C3KO and WT muscles. Thus, C3KO muscles respond to the reloading by activation of the IGF-1/PI3K/Akt pathway to the same extent as WT muscles; and therefore, failure to grow is not due to failure to properly activate the IGF-1/PI3K/Akt pathway.
Downregulation of proteasomal components occurs normally in C3KO muscles during reloading
The absence of normal muscle growth in C3KO mice undergoing muscle reloading could be due to inadequate downregulation of the protein degradation machinery in response to muscle reloading. To check for proteasomal component concentrations under our experimental conditions, western blots from the muscles at different stages of the experiment were probed with antibodies, which recognize several components of the 20S proteasome (Fig. 4). Reloading of the muscles after 10 days of suspension caused a decrease in the concentration of proteasomal components in both WT and C3KO muscle extracts (compare lanes 5 and 6 with lanes 3 and 4 for WT and lanes 11 and 12 with lanes 9 and 10 for C3KO). This result suggests that C3KO muscles respond normally to muscle reloading by downregulation of proteasomal components and that abnormally high proteasomal proteolysis at the reloading stage is not a reason for the decreased rate of growth of C3KO muscles.
C3KO muscles do not accumulate Ub–protein conjugates during reloading like WT muscles
It was shown previously that Ub–proteasomal proteolysis plays a key role in muscle remodeling that occurs during muscle disuse and recovery (reviewed in 23). However, proteasomes are not able to degrade intact myofibrils (12). Ca2+-dependent proteolysis (ubiquitous calpains 1 and 2) has been previously hypothesized to act upstream of ubiquitination to release proteins from multiprotein complexes to make them available for Ub–proteasomal degradation (reviewed in 13). Because nothing is known about CAPN3 involvement in this process, we used C3KO mice to address the question of whether CAPN3 is necessary for ubiquitination and proteasomal degradation of muscle proteins. Muscle extracts prepared from soleus muscles at different stages of suspension–reloading were probed with two different anti-Ub antibodies to detect accumulation of high-molecular weight Ub–protein conjugates in vivo. Previously, such conjugates were detected in the muscles at the early stage of reloading (15). We also found that WT muscles responded to reloading by accumulation of high-molecular weight Ub–protein conjugates (Fig. 5A, lanes 5 and 6). This increase in ubiquitinated proteins was not detected in C3KO muscles (Fig. 5A, lanes 11 and 12). The same results were observed for all six animals per each group (group 1: ambulatory control; group 2: 10 days suspension and group 3: 10 days suspension followed by 2 days reloading) that were used in this experiment. Two different anti-Ub antibodies were used (left and right panels in Fig. 5A represent four different animals per each group stained with two different antibodies).
The same blots were also probed for CAPN3 to detect changes in CAPN3 expression at different stages of muscle remodeling. In WT mice, there is a decrease in CAPN3 concentration at the suspension stage followed by a significant increase during the reloading period (Fig. 5B). Thus, an increase in CAPN3 concentration in WT muscles undergoing recovery and growth correlates with accumulation of high-molecular weight Ub–protein conjugates, which is not observed in C3KO muscles. These data indicate that CAPN3 is necessary for the ubiquitination of target proteins that occurs during early reloading stage.
To further confirm this observation, ubiquitinated proteins were purified from WT and C3KO muscles after 2 days of reloading. Commercially available S5a beads with high affinity to the Ub–protein conjugates were used in this experiment. As shown in Figure 5C, the concentration of high-molecular weight Ub–protein conjugates was considerably lower in C3KO muscles when compared with WT muscles. The failure of C3KO mice to increase ubiquitination during early reloading suggests that CAPN3 acts upstream of the ubiquitination machinery to promote muscle remodeling.
In the experiment described earlier, muscles at the end of the 10 day unloading period were analyzed. At this stage, the proteasomal degradation system is fully activated and muscles develop a severe atrophy. To see whether changes in CAPN3 expression and Ub–protein conjugate accumulation occur at an earlier stage of muscle atrophy, muscles were unloaded for only 3 days. Neither accumulation of high-molecular weight Ub–protein conjugates nor changes in CAPN3 expression were detected at the early stage of muscle unloading (data not shown). These data suggest that both an increase in CAPN3 concentration and an accumulation of Ub–protein conjugates occur specifically during muscle reloading. One possible explanation for the absence of high-molecular weight Ub–protein conjugates at the unloading stage is that a high level of proteasomal activity and protein degradation are occurring during suspension. The rapid rate of turnover of the Ub-conjugated proteins might prevent their visible accumulation. During reloading, there is a significant decrease in proteolytic activity (15) and proteasomal component concentration (Fig. 4) which might allow for visible accumulation of Ub–protein conjugates in the reloaded muscles.
Heat shock proteins are upregulated in C3KO muscles at the end of a 10 day suspension period and in the insoluble fraction in older mice
Involvement of Ca-dependent proteolysis mediated by ubiquitous calpains (calpains 1 and 2) is well documented for cases of pathological (24) and non-pathological atrophies induced by muscle disuse (11,14). However, there has not previously been specific investigation about CAPN3 involvement in this latter process. Our data show that rates of both atrophy and growth are affected by the absence of CAPN3 (Fig. 1) and that CAPN3 is necessary for protein ubiquitination at least at the early reloading stage (Fig. 5). As ubiquitination tags proteins for degradation, decreased ubiquitination may lead to excessive accumulation of the proteins that should otherwise be degraded. This in turn could trigger a cell stress response, one manifestation of which is upregulation of heat shock proteins.
To determine whether C3KO muscles are experiencing cellular stress, we compared concentrations of several heat shock proteins in C3KO and WT muscles that were subjected to changing loading conditions which are accompanied by increases in protein degradation. No change in expression of Hsp60 or Hsp70 was detected in the muscles undergoing unloading for 3 days (data not shown). However, after 10 days of unloading, hsp70 and especially hsp60 were upregulated in the C3KO muscles but not in the WT muscles (Fig. 6A, compare lanes 8 and 9 with lanes 3 and 4). Thus, heat shock proteins accumulate in C3KO muscles when muscles are challenged with a physiological condition that requires highly increased protein degradation. Protein degradation is the ultimate mechanism in cells to protect against misfolded or damaged proteins (reviewed in 25). When these proteins accumulate, they form insoluble inclusions called aggresomes (26). Aggresomes recruit some cytosolic component, such as heat shock proteins, of the Hsp70 and small Hsp families (reviewed in 27). If abnormal protein turnover is a feature of C3KO muscles, it could lead to a similar response in muscles under normal physiological conditions, especially in older animals. To check this hypothesis, we compared the concentration of several Hsps in soluble and insoluble fractions of tibialis anterior in 1-year-old C3KO and WT mice. Figure 6B and C shows that Hsp70 and Hsp25 concentrations in the insoluble fraction of C3KO muscles are significantly higher than in WT muscles, whereas the soluble fractions contain approximately equal amounts of these proteins in both C3KO and WT muscles. This data support the hypothesis that insoluble protein aggregates may form in muscles lacking CAPN3 due to a reduced capacity for normal protein turnover.
DISCUSSION
Previous studies have reported an important and predominant contribution of the Ub–proteasome pathway to many types of muscle wasting that range from atrophy induced by sepsis to cancer cachexia. Although the role of the Ub–proteasome system in these processes is undisputed, it is clear that other upstream proteolytic systems must be operating to enable myofibrillar protein degradation. As proteasomes are not able to degrade properly folded intact myofibrils, other proteases must be initiating myofibrillar protein release prior to ubiquitination and proteasomal degradation (12). One candidate proteolytic system that could function in this manner is the calpain family. Indeed, in some studies, Ca2+-dependent proteolysis was shown to contribute to total muscle proteolysis under several catabolic conditions (reviewed in 13). Studies examining the relative contributions of different proteolytic systems have relied on the use of inhibitors in vitro or have examined the concentration of mRNAs for various protease family members. In some cases, controversial results were obtained when different methods were used to evaluate a role for Ca2+-dependent proteolysis in muscle wasting. For example, during sepsis-related atrophy, both increased (28) and unchanged (29) calpain activities were reported. The difference in these findings was likely due to the use of two different substrates in these respective studies (30). For our studies, we have relied on the use of genetically modified animals, because this approach is the only way to analyze the contribution of individual calpain family members in vivo. Taking this approach, an involvement of calpain-1 and calpain-2 in muscle wasting during hindlimb disuse was previously demonstrated (14). In this case, transgenic animals overexpressing an endogenous inhibitor of the ubiquitous calpains, called calpastatin, were used. These studies do not provide information pertaining to the role of CAPN3 in muscle wasting; because CAPN3 is not inhibited by calpastatin. Furthermore, CAPN3 is not inhibited by leupeptin or E64, inhibitors used in prior studies, and it is also not clear whether Ca2+ is required for CAPN3 activation as autolysis can occur in the presence of EDTA (31). Thus, inhibitor studies or investigations examining Ca2+-dependent proteolysis do not address the role of CAPN3 in the process of the muscle adaptive response. The current study is the first to specifically examine the role of CAPN3 in muscle protein breakdown and growth.
In this study, we used mice lacking CAPN3 to address the question of CAPN3 involvement in muscle remodeling during hindlimb suspension and reloading. Our data suggest that CAPN3 is involved in muscle protein breakdown during the catabolic stage (muscle unloading) as the rate of muscle mass loss was reduced in C3KO mice. This result is not surprising because all proteolytic systems are activated during catabolic conditions in muscle to facilitate massive protein breakdown. More interestingly, CAPN3 appears to be necessary for muscle growth during the anabolic stage (muscle reloading), and in fact, the rate of muscle growth was more affected by the absence of CAPN3 than the rate of muscle mass loss (Fig. 1). This indicates a unique role for CAPN3 as a protease that is involved in new sarcomere formation in vivo. Data presented here are in good agreement with our previous results, which demonstrated that in primary muscle cells in vitro, CAPN3 is necessary for proper myotube differentiation. Although initial steps of myoblast fusion to form myotubes appeared to be normal, consequent steps of sarcomere formation were severely affected by the absence of CAPN3 (4). Thus, CAPN3 may play a role in promoting myofibrillogenesis during muscle cell differentiation in vitro and muscle growth in vivo.
Although extensive research has been done on the signaling pathways that control myogenic differentiation, very little is known about molecular mechanisms of muscle adaptation in post-natal muscle, particularly, how muscles translate increases in mechanical loading into adaptive hypertrophic responses. The IGF/PI3K/Akt pathway was shown to be a principal mechanism that induces myogenic differentiation and is also important for muscle adaptation (32). Ectopic expression of IGF-1 inhibits disuse-mediated atrophy (33). It was shown that IGF-1 is expressed in the muscles in response to stretch or increased mechanical activity and functional overload (reviewed in 34). Our results show that activation of this pathway, as judged by accumulation of activated Akt and phosphorylation of the ribosomal protein S6, also occurs in muscles that are reloaded after a prolonged period of disuse (Fig. 3). CAPN3 is not necessary for this activation because the IGF/PI3K/Akt pathway is activated normally in C3KO muscle.
Very little information is available on the molecular and cellular mechanisms of muscle recovery following catabolism. Activation of the IGF/PI3K/Akt pathway leads to an increase in protein synthesis that is necessary for muscle growth. In contrast, this pathway also inhibits expression of atrophy-specific genes, namely, atrophy-related Ub ligases MAFbx and MuRF1 (22) leading to downregulation of the Ub–proteasome pathway. This question has also been addressed by investigating the protein turnover rate at different times of muscle reloading following muscle suspension (15). In these studies, it was found that at the early recovery stage, both muscle protein synthesis and breakdown were elevated by 65 and 22%, respectively, when compared with the ambulatory control. However, protein breakdown was significantly lower compared with the catabolic condition of muscle suspension (66% increase compared with ambulatory control) (11). While during muscle suspension, all three main muscle proteolytic systems (Ub-proteasomal, lysosomal and Ca2+-dependent) were coordinately activated, only the Ub–proteasome system remained activated during muscle reloading. Accumulation of high-molecular weight Ub–protein conjugates was also detected during early recovery (15). These findings suggest that elevated Ub–proteasomal proteolysis is required to eliminate atrophy-related or damaged proteins at the early reloading stage. We similarly observed accumulation of high-molecular weight Ub–protein conjugates in WT muscles during recovery which was absent in C3KO muscles and found a 2.5–3-fold increase in CAPN3 concentration at this stage. Taken together, these data suggest that CAPN3 is necessary for ubiquitination of muscle proteins and that it acts upstream of this process. It is still unclear whether CAPN3 directly cleaves proteins to make them available for ubiquitination or whether the effect of CAPN3 is indirect (i.e. through regulation of other proteins involved in ubiquitination).
It was proposed earlier that calpains act upstream of the Ub–proteasome pathway to release proteins and make them available for proteasomal degradation (reviewed in 13). Although inhibitor studies provided information on the relative contribution of different proteolytic systems, including calpains, it was not clear whether they were operating in parallel or in series. In this study, using a genetic approach, we showed in vivo evidence to support the hypothesis that CAPN3 and the Ub–proteasome system act in series. The absence of CAPN3, thus, may lead to decreased protein turnover and accumulation of damaged or misfolded proteins. Protein degradation is important for normal cellular homeostasis as a way to protect cells against misfolded or damaged proteins (25,35). Numerous examples exist in which a loss or decrease of proteolytic capability via the Ub–proteasome pathway leads to disease. Alzheimer's disease, juvenile recessive Parkinson's disease, Angleman's syndrome and several forms of cancer can result from mutations in proteins associated with the Ub–proteasome pathway (35). Accumulation of damaged or misfolded proteins can trigger a cell stress response, upregulation of heat shock proteins, aggregation of these proteins and consequent formation of large inclusions often called ‘aggresomes’ (27). Many cytosolic proteins such as Ub–proteasome pathway components and heat shock proteins are directed to aggregated proteins presumably in the attempt to renature or to degrade these proteins (27). In this study, we found an accumulation of heat shock proteins, Hsp25 and Hsp70, in insoluble but not in soluble fractions of C3KO mice that may be an indication of an aggregation process in C3KO muscles. Although further studies are necessary to confirm protein aggregate formation in C3KO muscles, taken together our findings allow us to hypothesize that accumulation of aged and damaged proteins may lead to cellular toxicity and cell stress in C3KO muscles leading to muscle pathology. Involvement of CAPN3 in generalized proteolysis does not exclude the possibility that CAPN3 may also play a role in more specific cellular pathways controlling sarcomere formation. We are currently attempting to identify CAPN3 substrates using several proteomic approaches to further elucidate a role for CAPN3 in muscle and to gain insight into pathological mechanisms of LGMD2A.
MATERIALS AND METHODS
Animal procedures and muscle analysis
Male mice (4–5 months of age, six in each group) of C3KO or WT genotype were subjected to hindlimb unloading for 10 days, as described previously (14). Briefly, the tail of each mouse was placed in a harness, which is used to elevate the pelvis so that the feet of the hindlimbs did not contact the cage floor. Other animals were subjected to 10 days of hindlimb suspension followed by reloading for 2 or 4 days by allowing them to return to their normal cage activity. For each group, there were age- and sex-matched ambulatory controls. At the end of experimental treatments, soleus muscles were dissected from each animal; one soleus was frozen for sectioning and second one was placed in liquid nitrogen and used for western analysis. Cross-sectional areas of slow and fast fibers were measured using a digital imaging system (Bioquant, Nashville, TN, USA). All experimental protocols and use of animals were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the UCLA Institutional Animal Care and Use Committee.
Downhill running exercise
Mice were placed on treadmill to run with a downhill inclination of 15°, and the speed was gradually increased from 5 to 15 m/min for 30 min. The running sessions were repeated daily for 4 days. After the last session, mice were injected intraperitoneally with Evans blue [Sigma, 1% solution in phosphate-buffered saline (PBS), 0.1 ml/10 g body weight]. The mice were sacrificed 24 h after injection. Cross-sections of muscles were examined under a fluorescence microscope (Zeiss).
Immunohistochemistry
Muscles frozen in isopentane were cross-sectioned mid-belly and sections were stained with monoclonal antibodies to myosin fast type or slow type heavy chains (1:50, Novacastra) using M.O.M. kit (Vector Laboratories). To visualize all the fibers, sections were stained with anti-distrophin antibody (kindly provided by Dr L. Anderson). Staining for IgG to visualize damaged fibers was performed as described (19). Briefly, slides were blocked in 1% gelatin for 15 min and incubated with FITC-conjugated anti-mouse IgG (1:150, Vector Laboratories) for 1 h at room temperature. After washing in PBS, slides were mounted in Gel/Mount (Biomeda Corp.) and examined under a fluorescence microscope (Zeiss).
Muscle extract preparation and western blot analysis
For western blot analysis, muscles were homogenized in a Dounce homogenizer in reducing sample buffer [80 mm Tris, pH 6.8, 0.1 m dithiothreitol (DTT), 2% sodium dodecyl sulfate (SDS) and 10% glycerol with protease inhibitors cocktail (Sigma)]. For analysis of insoluble fractions, muscles were homogenized in 80 mm HEPES, pH 7.0 with protease inhibitors cocktail (as described in 36). The homogenates were sonicated and centrifuged at 125 000g for 20 min at 4°C. The supernatants were used as soluble fractions; the pellets were resuspended in reducing sample buffer with 2% SDS and used as insoluble fractions.
Antibody
The following antibodies were used for western blot analysis: anti-Akt total and anti-Thr308 phosphorylated Akt, rabbit 1:500 (Signal Transduction); anti-S6 ribosomal protein and anti-phospho-S6(Ser240/244), rabbit 1:1000 (Cell Signalling); anti-20S proteasome αβ subunits, rabbit 1:1000 (BioMol International); anti-β-tubulin, mouse 1:500 (BD Biosciences); anti-Ub, mouse 1:500 (Zymed) and mouse 1:500 (Transduction Laboratories), rabbit 1:1000 (Calbiochem); anti-calpain 3, mouse 12A2 1:100 (Novocastra); anti-hsp60, mouse 1:500 (Transduction Laboratories); anti-hsp70, mouse 1:1000 (Transduction Laboratories) and anti-hsp25, rabbit 1:2000 (Stressgen).
Isolation of ubiquitinated proteins
The soleus muscle was homogenized in a sample buffer containing 9 m urea, 2% CHAPS and 1% DTT. After centrifugation at 15 000g for 10 min at 4°C, the supernatant was collected and diluted at a 1:1 ratio. After measuring protein concentration, the supernatant was allowed to bind commercially available S5a beads (BioMol) for 4 h at room temperature. Following that, the beads were washed three times and the proteins were eluted in a solution containing 4.5 m urea, 1% CHAPS and 0.4 m KCl. The eluted samples were desalted and concentrated and 5 µg of each sample was loaded on to a 9% gel. Rabbit anti-Ub antibody (Calbiochem) was used for immunostaining of western blots.
ACKNOWLEDGEMENTS
The authors acknowledge the excellent technical support of Ms Jane Wen and Mr Benjamin Wu. Authors would like to thank Drs J.G. Tidball and H.X. Nguyen for their help and advice on hindlimb suspension experiments. This work was supported by the NIH (NIAMS, AR48177 to M.J.S.) and the Muscular Dystrophy Association (to M.J.S.).
Conflict of Interest statement. None declared.
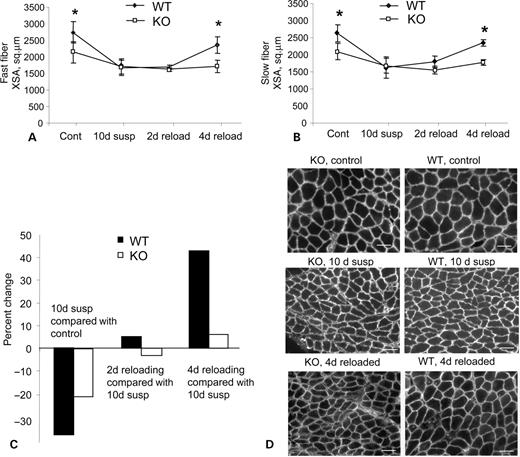
Figure 1. Rates of fiber atrophy and growth are affected by the absence of CAPN 3. Mid-belly cross-sectional areas of soleus muscle stained with anti-slow or anti-fast myosin heavy chain antibodies were measured separately for the fast (A) and slow (B) type fibers. Four groups of mice were analyzed: (1) ambulatory control (cont); (2) mice subjected to 10 days of suspension (10d susp); (3) mice subjected to 10 days of suspension followed by 2 days (2d reload) or (4) 4 days (4d reload) of reloading. (C) Comparison of fiber cross-sectional area changes in WT and C3KO mice indicated as percentage change, relative to ambulatory controls (for mice subjected to 10 days of suspension) or to mice after 10 days of suspension (for groups that were subjected to 2 or 4 days of reloading). (D) Representative sections of WT and C3KO soleus muscles stained with anti-dystrophin antibody to show fiber size at different stage of the experiment. Bars, 50 µm.
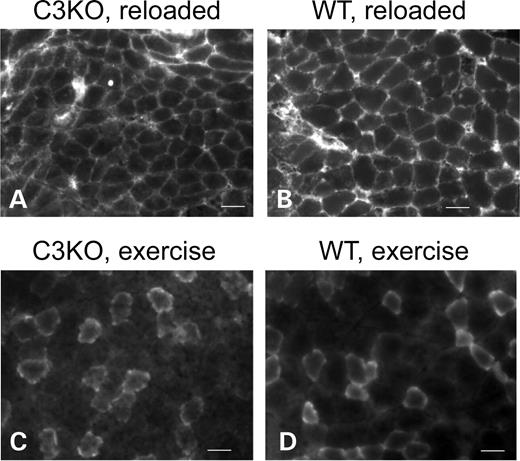
Figure 2. C3KO muscles are not more susceptible to muscle damaging conditions. Representative sections of C3KO (A) and WT (B) soleus muscles from mice subjected to 10 days of suspension followed by 2 days of reloading stained with FITC-conjugated anti-mouse IgG to visualize damaged fibers (19). (C and D) Representative sections of C3KO and WT gastrocnemius muscles after 4 days of treadmill running exercise and Evans blue injection to visualize damaged fibers. In both cases, staining is due to unspecific uptake of the labeled molecules by fibers with damaged membrane. Bars, 50 µm.
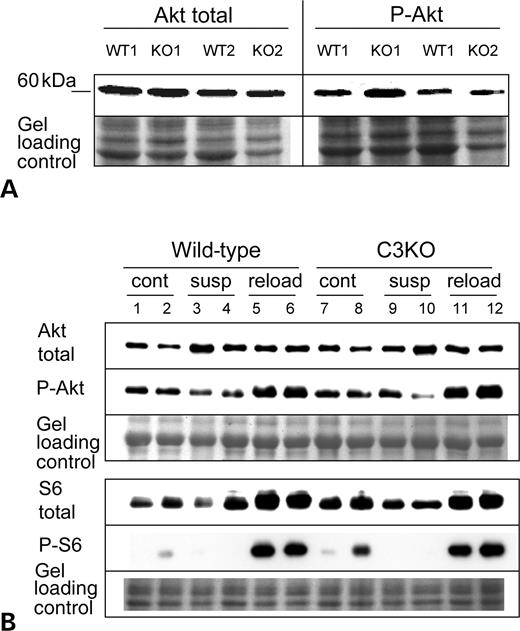
Figure 3. Activation of the IGF-1/PI3K/Akt pathway is not affected by the absence of CAPN3. (A) Western blots of extracts of C3KO and WT myotubes undergoing differentiation during 2.5 days in the presence of insulin–transferrin–selenium X were stained with antibodies that recognize all forms of Akt (total Akt) or Akt activated by phosphorylataion (P-Akt). No difference was found between C3KO and WT myotubes. (B) Representative western blot of muscle extracts from ambulatory control (cont), suspended for 10 days (susp) or reloaded for 2 days after suspension (reload) mice were stained with total Akt or activated Akt (P-Akt)-specific antibodies (upper panel) and with total ribosomal protein S6 and phospho-S6 antibodies (lower panel).
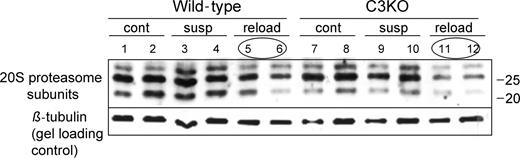
Figure 4. Downregulation of proteasomal components occurs normally in C3KO muscles during reloading. Western blot of muscle extracts from ambulatory control (cont), suspended for 10 days (susp) or reloaded for 2 days after suspension (reload) mice was probed with anti-20S proteasome αβ subunits rabbit polyclonal antibody (BioMol International) that recognizes several components of 20S proteasome. Note the decrease in concentration of all three components in the reloaded muscles in both C3KO and WT. The same blot was also probed with anti-β tubulin antibody to show gel loading.
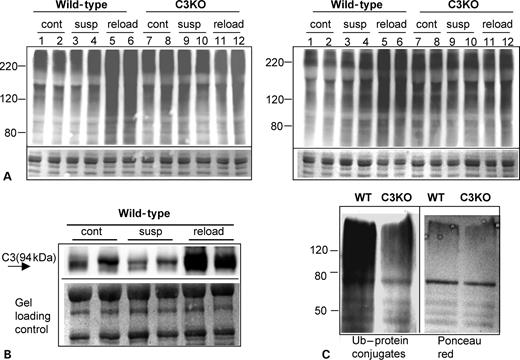
Figure 5. Accumulation of high-molecular weight Ub–protein conjugates is absent in C3KO muscles. (A) A representative western blot of muscle extracts from ambulatory control (cont), suspended for 10 days (susp) or reloaded for 2 days after suspension (reload) mice stained for Ub demonstrates an accumulation of high-molecular weight Ub–protein conjugates in WT reloaded muscles. Comparison of lanes 5 and 6 (WT) with lanes 11 and 12 (C3KO) shows that no accumulation of high-molecular weight Ub–protein conjugates occurs in C3KO muscles. Left and right panels represent muscle extracts from four different mice per each group stained with two different antibodies. (B) CAPN3 expression as revealed by staining with anti-CAPN3 antibody is upregulated in WT muscles during reloading. (C) Ub–protein conjugates were purified from soleus muscles reloaded for 2 days using Ub-binding S5a beads. Immunostaining with anti-Ub antibody shows decreased amount of Ub–protein conjugates in C3KO muscle when compared with WT (left panel). Right panel shows Ponceau red staining of the same blot.
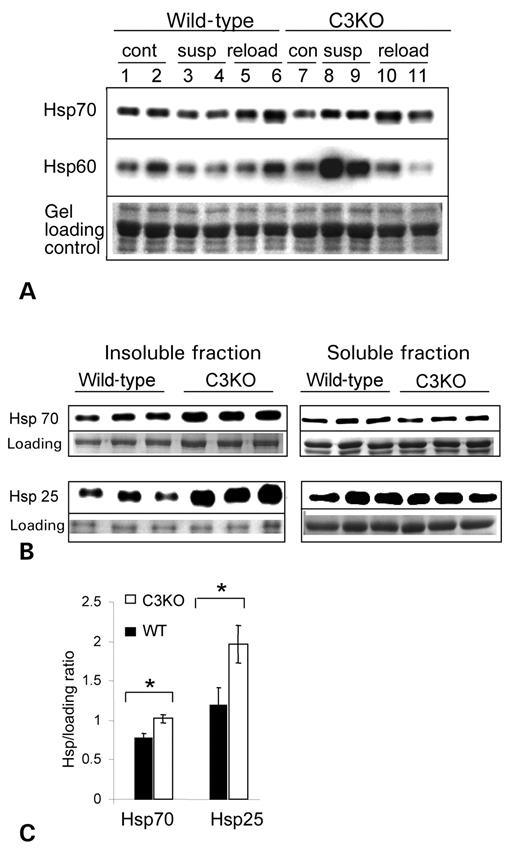
Figure 6. Heat shock proteins expression in C3KO and WT muscles. (A) Western blots of muscle extracts from ambulatory control (cont), suspended for 10 days (susp) or reloaded for 2 days after suspension (reload) mice were stained with antibodies against several heat shock proteins. Concentrations of hsp60 and hsp70 were elevated in C3KO muscles after 10 days of suspension (compare lanes 3 and 4 with lanes 8 and 9). (B) Soluble and insoluble fractions were prepared from tibialis anterior muscle of 12–13 months old mice and probed with antibodies against several heat shock proteins. Accumulation of Hsp70 and Hsp25 was found in insoluble fractions of C3KO muscles. Quantitative analysis of these blots is presented in (C).
References
Goll, D.E., Thompson, V.F., Li, H., Wei, W. and Cong, J. (
Richard, I., Broux, O., Allamand, V., Fougerousse, F., Chiannilkulchai, N., Bourg, N., Brenguier, L., Devaud, C., Pasturaud, P., Roudaut, C. et al. (
Richard, I., Roudaut, C., Marchand, S., Baghdiguian, S., Herasse, M., Stockholm, D., Ono, Y., Suel, L., Bourg, N., Sorimachi, H. et al. (
Kramerova, I., Kudryashova, E., Tidball, J.G. and Spencer, M.J. (
Gregorio, C.C., Granzier, H., Sorimachi, H. and Labeit, S. (
Kinbara, K., Sorimachi, H., Ishiura, S. and Suzuki, K. (
Sorimachi, H., Kinbara, K., Kimura, S., Takahashi, M., Ishiura, S., Sasagawa, N., Sorimachi, N., Shimada, H., Tagawa, K., Maruyama, K. et al. (
Thomason, D.B. and Booth, F.W. (
Krippendorf, B.B. and Riley, D.A. (
Jagoe, R.T. and Goldberg, A.L. (
Taillandier, D., Aurousseau, E., Meynial-Denis, D., Bechet, D., Ferrara, M., Cottin, P., Ducastaing, A., Bigard, X., Guezennec, C.Y., Schmid, H.P. et al. (
Solomon, V. and Goldberg, A.L. (
Hasselgren, P.O. and Fischer, J.E. (
Tidball, J.G. and Spencer, M.J. (
Taillandier, D., Aurousseau, E., Combaret, L., Guezennec, C.Y. and Attaix, D. (
Ordway, G.A., Neufer, P.D., Chin, E.R. and DeMartino, G.N. (
Duguez, S., Bihan, M.C., Gouttefangeas, D., Feasson, L. and Freyssenet, D. (
Feasson, L., Stockholm, D., Freyssenet, D., Richard, I., Duguez, S., Beckmann, J.S. and Denis, C. (
Hainsey, T.A., Senapati, S., Kuhn, D.E. and Rafael, J.A. (
Straub, V., Rafael, J.A., Chamberlain, J.S. and Campbell, K.P. (
Glass, D.J. (
Stitt, T.N., Drujan, D., Clarke, B.A., Panaro, F., Timofeyva, Y., Kline, W.O., Gonzalez, M., Yancopoulos, G.D. and Glass, D.J. (
Taillandier, D., Combaret, L., Pouch, M.N., Samuels, S.E., Bechet, D. and Attaix, D. (
Spencer, M.J. and Mellgren, R.L. (
Goldberg, A.L. (
Johnston, J.A., Ward, C.L. and Kopito, R.R. (
Garcia-Mata, R., Gao, Y.S. and Sztul, E. (
Bhattacharyya, J., Thompson, K. and Sayeed, M.M. (
Fischer, D.R., Sun, X., Williams, A.B., Gang, G., Pritts, T.A., James, J.H., Molloy, M., Fischer, J.E., Paul, R.J. and Hasselgren, P.O. (
Wei, W., Fareed, M.U., Evenson, A., Menconi, M.J., Yang, H., Petkova, V. and Hasselgren, P.O. (
Kinbara, K., Ishiura, S., Tomioka, S., Sorimachi, H., Jeong, S.Y., Amano, S., Kawasaki, H., Kolmerer, B., Kimura, S., Labeit, S. et al. (
Hornberger, T.A. and Esser, K.A. (
Alzghoul, M.B., Gerrard, D., Watkins, B.A. and Hannon, K. (
Baldwin, K.M. and Haddad, F. (
Glickman, M.H. and Ciechanover, A. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}