




Abstract
Despite the marked advances in drug therapy, some patients do not respond favorably or suffer severe adverse drug effects. Pharmacogenetic studies have shown that polymorphisms of drug metabolizing enzymes, transporters and receptors contribute to variable drug response. Owing to the complexity of drug actions, a broader genomics approach aims at finding new drug targets and optimizing therapy for the individual patient. However, pharmacogenomics has made only a few inroads into clinical practice to date. This review evaluates obstacles that need to be overcome. These include the complexity of mechanisms underlying drug response, given singly or in combination, uncertainty about the genetic underpinnings of complex diseases, such as cancer, diabetes, cardiovascular and mental disorders and a lack of quantitative understanding of the scope of genetic variations, even for well-studied genes. By resolving these hurdles, pharmacogenomics will yield significant, but incremental, therapeutic advances paving the way towards personalized health care.
INTRODUCTION
Pharmacogenetic studies of several decades have documented pervasive effects of genetic polymorphisms on drug response (1–9). Availability of high-density genomic SNP maps, and a rapidly expanding slate of known functional polymorphisms, have generated high expectations for applying pharmacogenetics to the optimization of therapies for individual patients. Because of increasing use of genomics and other -omics technologies, the term pharmacogenomics has emerged to reflect this broadening approach in drug discovery, development and therapy. Pharmacogenomics is a harbinger of personalized medicine, a paradigm shift from the mindset of ‘one-drug-fits-all’ to ‘the right drug for the right patient at the right dose and time.’ This does not mean that each patient will be treated differently from every other patient, an economically untenable proposition. Rather, patients are divided into groups by genetic and other markers that predict disease progression and treatment outcome. For drug therapy, one needs to avoid lack of response or toxicity. If the frequency of an adverse event can be reduced from 5 to 2%, by excluding 10% of the targeted population, a drug gains a more favorable risk/benefit ratio and could advance to first-choice treatment, thereby gaining market share. One can expect a growing trend to link the launch of new drugs with diagnostic markers, often genetic, to improve treatment outcome for individual patients—the hallmark of personalized medicine.
Numerous factors contribute to variable drug response, including age, sex, body weight, nutrition, organ function, infections, comedications and genetic factors. Pharmacogenomics is one of many approaches in personalized medicine, with medical informatics providing integration of relevant data. However, disease processes and drug therapies are complex systems, the behavior of which cannot be precisely predicted, limiting predictive power of pharmacogenomics. This review summarizes the current status of pharmacogenomics and then delves into challenges that we need to overcome for successful personalized medicine.
GENETIC CAUSES OF INTERINDIVIDUAL VARIABILITY IN DRUG RESPONSE
Highly variable drug response and toxicity preclude clinical use of a drug. Even with some of the most advanced current drugs, favorable response occurs in only 30–70% of patients, with a significant portion showing adverse effects, yielding a poor risk/benefit ratio for a diverse patient population. Pharmacokinetics (PK) and pharmacodynamics (PD) provide quantitative measures of drug exposure and effect, and therefore, are critical disciplines for understanding variability. PK focuses on absorption, distribution, metabolism and excretion (ADME), whereas PD concerns drug targets (receptors and enzymes), downstream signaling events and pharmacological response. Polymorphic genes relevant to PK–PD are listed in Table 1. Because ADME governs drug exposure, drug level monitoring yields phenotypic markers useful for individualizing therapy. Large interindividual variability is often associated with frequent mutations in cytochrome P450s and in conjugating enzymes such as glucuronyl transferases (Table 1). In a meta-analysis, we had addressed the question whether severe adverse drug effects, a leading cause of death in the US (10), are linked to polymorphic drug metabolizing enzymes. Called ‘unavoidable’ because adverse drug effects occur upon proper drug treatment excluding medication errors, we nevertheless found a high level of association between polymorphic CYP450 genes and the lead drugs causing adverse reactions (11). This suggests a potential approach for reducing the incidence of adverse effects with the use of genetic information.
Drug transporters are encoded by several hundred genes, playing a pervasive role in ADME and drug targeting (12). Numerous functional polymorphisms appear to affect drug response (http://www.pharmgkb.org/), but their impact is poorly understood. Similarly, the effect of polymorphisms in genes encoding drug receptors is difficult to assess. Activating mutations are a possible exception, particularly those of tyrosine kinases involved in cancer progression. For example, constitutive activity of the fusion protein BCR/ABL (generated through chromosomal translocation in leukemia) conveys high sensitivity to imatinib (Gleevec), whereas activating mutations of EGFR appear to correlate with responsiveness to gefitinib (Iressa) (13,14). Over-expression of ErbB2 is a requisite for successful herceptin treatment of breast cancer (15).
PROSPECTIVE GENOTYPING AS PART OF DRUG THERAPY
If a genetic factor is substantial and frequent, and failure to achieve optimal drug therapy has dire consequences, prospective genotyping may be indicated (Table 1). However, in many cases, it may suffice to understand the genetic components affecting variable drug response to minimize the potential for severe adverse effects. Currently, under discussion for obligatory prospective genotyping is the potentially life-threatening toxicity of thiopurines in the treatment of childhood leukemias, in patients lacking functional thiopurine methyltransferase (TPMT) (2,16–19). In homozygous carriers of TPMT null mutations, the dose of thiopurines has to be reduced 10-fold or more to avoid myelosuppression. However, only one in 300 individuals is homozygous, so that genotyping of 300 patients is needed for detecting a null carrier. The logistics of genotyping in the clinical setting, and legal and economical considerations, have led to different opinions on prospective genotyping in this case. Alternatively, careful monitoring of white blood cell count could suffice to avoid serious toxicity. Clearly, medical, legal, ethical and economic issues all play a role in the implementation of prospective genotyping at the bedside.
DRUG RESPONSE IS MULTIFACTORIAL
Interindividual variability in drug response may be governed by mutations in a single gene, such as CYP2D6, with 5–10% poor metabolizers in a Caucasian population (carriers of two null alleles). However, most drugs interact with several CYP isozymes, conjugating enzymes such as glucuronyl transferase, UGT1A1, and transporters, each adding to genetic variability. The topoisomerase inhibitor irinotecan (camptothecin) serves to illustrate this point.
A case analysis: irinotecan
Irinotecan has become a first-line defense against colon carcinomas, showing enhanced efficacy over the use of 5-FU alone. However, leukopenia and diarrhea may become limiting and can be severe. Because UGT1A1 appears to play a major role in detoxifying the active metabolite of irinotecan, prospective genotyping has been suggested to avoid undue toxicity in carriers of defective UGT1A1. However, irinotecan interacts with multiple polymorphic drug metabolizing enzymes and transporters (20–28), being inactivated by CYP3A4 to APC, and requiring conversion by carboxyesterases to the active metabolite SN38. The latter in turn is inactivated by UGT1A1 glucuronidation as the main degradation pathway. In addition, irinotecan and its metabolites serve as substrates for transporters, including the ABC transporters (ATP-drive extrusion pumps) MDR1, MRP2 and BCRP. Each of these factors displays interindividual variability, with functional polymorphisms potentially contributing to variable irinotecan response.
UGT1A1 is responsible for bilirubin glucuronidation (29). Null mutations of UGT1A1 lead to Criggler–Najjar syndrome, whereas less complete defects are associated with Gilbert's syndrome. The most common functional polymorphism is a A(TA)6TAA repeat in the promoter region of 1A1. The most common form carries six (TA) repeats (wt), whereas UGT1A1*28 carries seven repeats, associated with lower UGT activity. However, additional polymorphisms (for example in the phenobarbital responsive enhancer module PBREM) appear to contribute to variable UGT expression. Haplotype analysis has provided additional insight into the regulation of gene transcription (20,24), but a quantitative assessment of all factors is lacking. As a result, use of TA repeat polymorphisms in predicting in vivo UGT activity and SN38 exposure after irinotecan administration has been only partially successful. Hence, the value of prospective genotyping for UGT1A1 in irinotecan therapy has to be determined empirically, in the intended target populations.
A NETWORK OF INTERACTIONS IN COMBINATION DRUG THERAPY
Treatment with single drugs targeting a specific receptor is no longer considered optimal in the treatment of complex diseases, such as cancer and HIV/AIDS. With the administration of multiple drug simultaneously, however, possible drug–drug interactions multiply, leading to unexpected adverse effects that may be difficult to trace. For example, if drug A, metabolized by both CYP2D6 and CYP2C9, is administered together with a second drug acting as an inhibitor of CYP2C9, metabolism of drug A is sharply reduced in CYP2D6 poor metabolizers. For anti-HIV therapy, up to three antiviral drugs are given concomitantly, with ritonavir serving as an antiviral ‘boosting’ agent (30). Ritonavir is a potent mechanism-based inhibitors of CYP3A4 and an inhibitor of membrane transporters such as Pgp (MDR1) (31). This permits the reduction of the dose of other antivirals that are also metabolized by CYP3A4 and transported by Pgp; however, dose-titration becomes erratic. Moreover, most patients are comedicated with antibiotics, antidepressants and statins to overcome lipodystrophic side effects of the antivirals. This leads to a high rate of severe adverse effects, with polymorphisms in ADME-related genes likely determining frequency and severity. A causal relationship may be difficult to ascertain with a one gene-one drug approach, because effects distribute over a network of interactions. Rather, a systems approach is needed to integrate overall adverse effects with functional polymorphisms in multiple genes. We suggest a medical informatics approach involving large patient populations and assessment of all side effects in associations with the most prevalent pharmacogenetic markers.
INCOMPLETE KNOWLEDGE OF THE GENETIC CONTRIBUTION TO PHENOTYPIC VARIABILITY
Even for well-studied genes, the overall genetic variability remains uncertain. Once a functional polymorphism has been experimentally validated, this marker is applied to clinical studies, often without assessing its relative contribution to overall genetic variability. Most genes harbor multiple functional polymorphisms. For example, the serotonin transporter gene, SERT (SLC6A4), has been implicated in multiple mood and cognitive disorders. A polymorphism in the promoter region (LPR, long and short form) has been extensively analyzed in association studies and shown to affect SERT mRNA levels in lymphocytes and using a reporter gene assay (32,33). Yet, SERT is mainly transcribed in neurons located in the pontine area of brain stem, but there is ambiguous evidence that SERT levels differ between LPR genotypes in the CNS. Whereas non-synonymous polymorphisms in the SERT coding region are rare, further regulatory polymorphisms may contribute to disease susceptibility or treatment response. A quantitative assessment is needed to evaluate the penetrance of SERT polymorphisms in mental disorders or in treatment with specific serotonin reuptake inhibitors, widely used antidepressant agents with efficacy reported in 60–70% of patients. Use of haplotypes has the potential to integrate the effects of multiple functional SNPs in phase, including epistasis, but haplotypes also do not yield complete genetic information in various patient populations. Our goal must be to identify all functional polymorphisms occurring at a sufficient frequency in the target population.
The highly variable expression of CYP3A4 provides another example of unresolved genetic factors in interindividual variability. CYP3A4 is involved in the metabolism of ∼40% of drugs, showing 30-fold variability in hepatic enzyme activity between individuals. Polymorphisms in the coding region are exceedingly rare (<3% for the most abundant synonymous SNP) and cannot account for variability. In contrast, enzyme induction via transcription factors such as PXR and CAR has been well documented with CYP3A4 (34,35), but it also can account for only a portion of variability. A recent analysis of allelic expression imbalance of CYP3A4 mRNA (measured in unspliced hnRNA) in human liver has revealed 4-fold variations in the ratio of one allele over the other, providing strong evidence for the presence of cis-acting regulatory factors (36). Therefore, genetic factors appear to contribute substantially to interindividual variability, but the functional polymorphisms remain to be determined. These examples demonstrate the need for quantitative analysis of genetic variability, for optimal interpretation of clinical studies.
GENETIC CAUSES OF PHENOTYPIC VARIABILITY
DNA sequence variations can affect protein structure and function, regulation of gene expression and mRNA processing and stability (Fig. 1). Large-scale surveys of genetic variation suggest that cis-regulatory polymorphisms appear to be considerably more abundant than those affecting primary protein structure and function (37–40). Practically, all genes are likely to harbor polymorphism(s) at one or more cis-regulatory sites that can locate anywhere in the extended gene locus (41), but most remain unknown. Genetic variations can further affect mRNA processing, such as alternative splicing and mRNA stability. Current estimates indicate that 35–59% of human genes undergo alternative splicing (42). Although numerous polymorphisms have already been discovered to affect splicing, including mutations of CYP2D6 (43–47), few have been shown to affect mRNA stability, such as a synonymous SNP in the dopamine DRD2 receptor (48). However, computational analysis of mRNA folding suggests that a majority of SNPs have the potential to affect mRNA folding, and hence, mRNA stability and processing or translation.
Our analysis indicates that cis-acting polymorphisms affecting mRNA functions may account for much phenotypic variability. The common outcome is a difference in mRNA generated from one allele versus the other (allelic expression imbalance, as discussed for CYP3A4 above). Allelic expression imbalance can be quantitated by a method involving PCR amplification of genomic DNA and mRNA (as cDNA) of a transcribed region of the gene containing a frequent marker SNP. This is followed by detection of the allelic ratios in DNA and mRNA as shown in Figure 2 (49). Each allele functions as the internal control for the other, thereby canceling out trans-acting factors. We can select several marker SNPs to test whether splicing events vary polymorphically. The assay must be performed in relevant target tissues, as regulation of transcription and mRNA processing is tissue specific. Where mRNA levels are genetically variable via trans-acting mechanisms (transcription factors), we must find the underlying cis-acting polymorphisms upstream in the signaling cascades.
Even though analysis of allelic expression imbalance is potentially powerful in revealing cis-acting factors, it has been used sparingly, mainly because of poor reproducibility. We have optimized the procedure, yielding low error rates, using the peptide transporter hPepT2 as an example (49). Applying this assay to multiple genes, we find that more than half show significant and frequent allelic expression imbalance, consistent with other such surveys (40). Bray et al. (50) have demonstrated allelic expression imbalance in brain autopsy tissues for catechol-O-methyltransferase (COMT), implicated as a susceptibility gene for schizophrenia. The COMT allele showing lower expression was more frequently represented in schizophrenic patients (50). [A non-synonymous SNP, Val158Met, additionally affects COMT protein function, and clinical response to a pain stressor (51).] Allelic expression imbalance of several key genes could suffice for manifestation of disease or variation in treatment outcome.
It is noted that allelic mRNA imbalance can also result from epigenetic effects. Measuring allelic DNA versus mRNA ratios is sensitive to all these events, yielding quantitative phenotypes that can serve to identify the cis-acting factors responsible for interindividual variability in gene expression and mRNA processing.
EPIGENETIC EFFECTS AND REGULATION OF GENE EXPRESSION AT THE mRNA AND PROTEIN LEVEL
Altered gene expression can also be transmitted from generation to generation, or across somatic cell divisions, involving imprinting or chromatin remodeling in the absence of genomic DNA polymorphisms (Fig. 1). Methylation of CpG islands and histone modifications by acetylation and methylation represent main mechanisms underlying these transmissible traits (52). Directed by the Xist transcript, X chromosome inactivation also relies on global methylation, to compensate for gene dosage in females versus males, but X-inactivation is often biased, leading to allelic expression imbalance (53,54). Recent results have revealed pervasive roles for epigenetic modulation in disease and possibly therapy. The long lasting phases of bipolar disorder with consecutive manic and depressive phases may result from epigenetic alteration of gene regulation (55), which is metastable and can be reversed (56). Silencing of tumor suppressor genes by epigenetic mechanisms also appears to play a role in cancer (57), leading to the use of decitabine and HDAC inhibitors (e.g. depsipeptide) to reverse CpG methylations and increase histone acetylation, in an attempt to force expression of suppressor genes (58,59). In contrast, methylation of the promoter of MGMT, a DNA repair enzyme, enhances the response to anticancer therapy with cisplatin and BCNU (60). Although epigenetic changes clearly affect disease and treatment outcomes, our knowledge is still insufficient to use this information prospectively in personalized medicine.
An astounding complexity of gene regulation and translation has been revealed with recent studies on small regulatory RNAs (61), including antisense transcripts from the opposite DNA strand of a number of genes (62), siRNA mechanisms (63) and the emerging world of microRNAs (64) (Fig. 1). With potentially up to 1000 microRNAs present in the human genome that target multiple genes each, one can anticipate that microRNAs play a significant role in disease and treatment outcome. Specifically, microRNAs could be related to chemoresistance or -sensitivity in cancer chemotherapy. Future studies will address the genetics of small regulatory RNAs in disease progression and treatment outcomes.
CONCLUSIONS
Although pharmacogenomics continues to improve understanding of drug response, progress is gradual, with clinical implementation lagging far behind. Several obstacles need to be overcome for successful application of pharmacogenomics to drug therapy.
Multiple processes contribute to the response to drugs and drug combinations, with drug–drug interactions leading to unexpected outcomes linked to polymorphic genes. Genetic analysis of overall drug response requires a systems analysis using medial informatics for integration of all relevant information.
It is essential to understand the contribution of genetic factors to the target phenotype in quantitative terms. In addition to molecular genetic analysis of polymorphisms affecting protein primary structure, we propose the systematic use of allelic expression imbalance, for quantitative assessment of cis-acting factors in transcription and mRNA processing.
We must assess the role of epigenetic factors, and of small regulatory RNAs, in determining interindividual variability.
A regulatory framework is needed to assure that pharmacogenomic data are incorporated into drug development and post-approval surveillance. Because the impact of genetic and genomic data is still poorly understood, the FDA has implemented a ‘safe haven policy’ by which pharmaceutical companies are encouraged to include genomic data for the New Drug Approval process without risking delays or other regulatory actions. In time, our knowledge will progress to a point where such data will become a cornerstone of the drug approval process. The inclusion of pharmacogenetic data (e.g. on CYP polymorphisms) on drug package inserts has already been implemented, providing genetic information accessible to patients and physicians alike.
To summarize, pharmacogenomics serves as an increasingly powerful tool in understanding interindividual variability in drug response and toxicity. Yet, decisive advances in drug therapy require an integrative systems approach, using medical informatics for optimizing personalized medicine.
ACKNOWLEDGEMENTS
Part of the work cited in this review was supported by NIH research grants DA018744 and GM61390, and a grant from the Ohio Biomedical Research and Technology Transfer Commission.
Conflict of Interest statement. None declared.
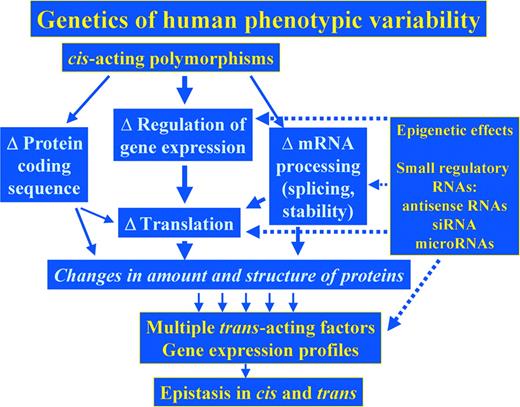
Figure 1. Main paths of genetic and epigenetic factors in cis and trans, determining activity of encoded proteins. The thickness of the arrows provides a rough estimate of the relative contribution of each path to overall variability. For broken arrows, relative impact remains unknown.
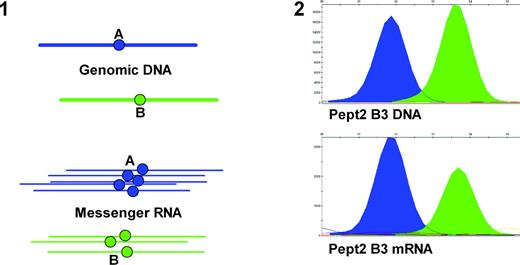
Figure 2. Schematics of allelic expression imbalance (panel 1) and quantitative analysis of genomic DNA and mRNA alleles of the hPept2 gene measured in a single brain autopsy sample (B3) (panel 2). DNA and cDNA are amplified by PCR, and the alleles detected by primer extension using fluorescent dideoxynucleotides. The labeled extension primers are then measured on an ABI3730 capillary electrophoresis apparatus. The two allele peaks (blue and green) provide information on relative allele frequency (49).
Survey of polymorphic genes relevant to drug response
| Gene symbol | Description | Polymorphisms | Clinical phenotype |
|---|---|---|---|
| Phase I | |||
| CYP2D6a | Cytochrome P450 2D6 | Numerous and frequent functional polymorphisms (non-synonymous SNPs, splice variants, regulatory, gene amplification) (http://www.imm.ki.se/cypalleles/) | Variable metabolism of CYP substrates depending on enzyme specificity. Allele frequencies vary among ethnic groups |
| CYP2C9a | Cytochrome P450 2C9 | ||
| CYP2C19 | Cytochrome P450 2C19 | ||
| CYP3A5 | Cytochrome P450 3A5 | ||
| CYP2B6a | Cytochrome P450 2B6 | ||
| CYP2E1 | Cytochrome P450 2E1 | ||
| DPYD | Dihydropyrimidine dehydrogenase | Multiple polymorphims including splice variant IVS14+1G>A (DYPD*2A) | *2A increases 5FU neurotoxicity |
| Phase II | |||
| TPMTa | Thiopurine methyltransferase | Multiple polymorphisms | Null genotype associated with hematopoietic thiopurine toxicity, homozygous frequency 1/300 |
| COMT | Catechol-O-methyltransferase | Val158Met, haplotypes | Met158 associated with higher daily neuroleptic dosage and poor response; haplotye affects mRNA levels |
| NAT2 | N-acetyltransferase 2 | Arg197Gln, Ile114The, Lys268Arg, G191GA, G857A | Affect acetylation rates for drugs (e.g. isoniazid) |
| GSTM1 | Glutathione-S-transferase M1 | Deletion causing null genotype | Decreased glutathione conjugation |
| GSTT1 | Glutathione-S-transferase T1 | Deletion causing null genotype | |
| UGT1A1a | UDP-glycosyltransferase 1A1 | Multiple polymorphisms in the promoter and coding regions | UGT1A1*28 variants associated with increased irinotecan toxicity |
| Drug transport | |||
| ABCB1a | Multiple-drug resistance gene 1 | Several polymorphisms including C3435T | C3435 associated with higher drug transport activity |
| BCRP (ABCG2) | Breast cancer related protein | G34A, C421A and 944–949 deletion | Minor alleles with lower BRCP expression, enhanced drug sensitivity |
| Drug targets/pathway protein | |||
| MTHFR | 5, 10-Methylenetetrahydrofolate reductase | C(677)T and A(1298)C | T677 increases toxicity to methotrexate, drug resistance |
| TYMS | Thymidylate synthase | Two to nine 28 bp repeats in the 5′ enhancer | TSER*3 associated with drug resistance |
| VKORC1a | Vitamin K epoxide reductase complex 1 | Many, VKORC1 haplotypes | Variable anticoagulant effect of warfarin |
| ADRB2 | β2-adrenergic receptor | Arg16Gly, Gln27Glu and Thr164Ile | 16Gly decreases response to altuterol and salbutamol |
| ADRB1 | β1-adrenergic receptor | Arg389Gly | Gly389 with decreased cardiovascular response to β1-antagonists |
| ALOX5 | Arachidonate 5-lipoxygenase | Variant promoter SP1 binding site | Decreased drug response with the mutant genotype |
| SLC6A3 | Dopamine transporter | 3′-VNTR 10 repeat allele | 10/10 repeat genotype associated with poor response to methylphenidate |
| SLC6A4 | Serotonin transporter (5-HTT) | Long/short promoter polymorphism | May affect antidepressant drug response efficacy |
| HTR2A | Serotonin receptor 2A | His452Tyr | Tyr452 decreases response to the antipsychotic clozapine |
| ACE | Angiotensin-1 converting enzyme | Ins/del in intron 16 | Del/Del decreases efficacy of the reduction of proteinuria by ACE inhibitors |
| CETP | Cholesteryl ester transfer protein | TaqIB polymorphism B1/B2 | B1/B1associated with better response to statins |
| t(9,22) translocationa | t(9,22) BCR-ABL translocation | Translocation | Gleevec effective against chronic myeloid leukemia with translocation only |
| ERBB2a | ERBB2, HER/Neu | Overexpression of protein | Herceptin for breast cancer with ERBB2 overexpression |
| EGFRa | Epidermal growth factor receptor | Exon 18–21 mutations | Human lung cancers with mutations response better for Iressa |
| DNA repair | |||
| XRCC1 | X-ray repair cross-complementing gene 1 | Arg194Trp, Arg280His and Arg399Gln | Gln399 associated with oxaliplatin/5-FU resistance |
| XPD | Xeroderma Pigmentosum, group D | Several SNPs including Lys 751Gln | Lys751 associated with improved oxaliplatin/5-FU treatment outcome |
| Others | |||
| HLA-Ba | Major histocompatibility complex, class I B | HLA-B*5701 | HLA-B*5701 associated with hypersensitivity to abacavir |
| Gene symbol | Description | Polymorphisms | Clinical phenotype |
|---|---|---|---|
| Phase I | |||
| CYP2D6a | Cytochrome P450 2D6 | Numerous and frequent functional polymorphisms (non-synonymous SNPs, splice variants, regulatory, gene amplification) (http://www.imm.ki.se/cypalleles/) | Variable metabolism of CYP substrates depending on enzyme specificity. Allele frequencies vary among ethnic groups |
| CYP2C9a | Cytochrome P450 2C9 | ||
| CYP2C19 | Cytochrome P450 2C19 | ||
| CYP3A5 | Cytochrome P450 3A5 | ||
| CYP2B6a | Cytochrome P450 2B6 | ||
| CYP2E1 | Cytochrome P450 2E1 | ||
| DPYD | Dihydropyrimidine dehydrogenase | Multiple polymorphims including splice variant IVS14+1G>A (DYPD*2A) | *2A increases 5FU neurotoxicity |
| Phase II | |||
| TPMTa | Thiopurine methyltransferase | Multiple polymorphisms | Null genotype associated with hematopoietic thiopurine toxicity, homozygous frequency 1/300 |
| COMT | Catechol-O-methyltransferase | Val158Met, haplotypes | Met158 associated with higher daily neuroleptic dosage and poor response; haplotye affects mRNA levels |
| NAT2 | N-acetyltransferase 2 | Arg197Gln, Ile114The, Lys268Arg, G191GA, G857A | Affect acetylation rates for drugs (e.g. isoniazid) |
| GSTM1 | Glutathione-S-transferase M1 | Deletion causing null genotype | Decreased glutathione conjugation |
| GSTT1 | Glutathione-S-transferase T1 | Deletion causing null genotype | |
| UGT1A1a | UDP-glycosyltransferase 1A1 | Multiple polymorphisms in the promoter and coding regions | UGT1A1*28 variants associated with increased irinotecan toxicity |
| Drug transport | |||
| ABCB1a | Multiple-drug resistance gene 1 | Several polymorphisms including C3435T | C3435 associated with higher drug transport activity |
| BCRP (ABCG2) | Breast cancer related protein | G34A, C421A and 944–949 deletion | Minor alleles with lower BRCP expression, enhanced drug sensitivity |
| Drug targets/pathway protein | |||
| MTHFR | 5, 10-Methylenetetrahydrofolate reductase | C(677)T and A(1298)C | T677 increases toxicity to methotrexate, drug resistance |
| TYMS | Thymidylate synthase | Two to nine 28 bp repeats in the 5′ enhancer | TSER*3 associated with drug resistance |
| VKORC1a | Vitamin K epoxide reductase complex 1 | Many, VKORC1 haplotypes | Variable anticoagulant effect of warfarin |
| ADRB2 | β2-adrenergic receptor | Arg16Gly, Gln27Glu and Thr164Ile | 16Gly decreases response to altuterol and salbutamol |
| ADRB1 | β1-adrenergic receptor | Arg389Gly | Gly389 with decreased cardiovascular response to β1-antagonists |
| ALOX5 | Arachidonate 5-lipoxygenase | Variant promoter SP1 binding site | Decreased drug response with the mutant genotype |
| SLC6A3 | Dopamine transporter | 3′-VNTR 10 repeat allele | 10/10 repeat genotype associated with poor response to methylphenidate |
| SLC6A4 | Serotonin transporter (5-HTT) | Long/short promoter polymorphism | May affect antidepressant drug response efficacy |
| HTR2A | Serotonin receptor 2A | His452Tyr | Tyr452 decreases response to the antipsychotic clozapine |
| ACE | Angiotensin-1 converting enzyme | Ins/del in intron 16 | Del/Del decreases efficacy of the reduction of proteinuria by ACE inhibitors |
| CETP | Cholesteryl ester transfer protein | TaqIB polymorphism B1/B2 | B1/B1associated with better response to statins |
| t(9,22) translocationa | t(9,22) BCR-ABL translocation | Translocation | Gleevec effective against chronic myeloid leukemia with translocation only |
| ERBB2a | ERBB2, HER/Neu | Overexpression of protein | Herceptin for breast cancer with ERBB2 overexpression |
| EGFRa | Epidermal growth factor receptor | Exon 18–21 mutations | Human lung cancers with mutations response better for Iressa |
| DNA repair | |||
| XRCC1 | X-ray repair cross-complementing gene 1 | Arg194Trp, Arg280His and Arg399Gln | Gln399 associated with oxaliplatin/5-FU resistance |
| XPD | Xeroderma Pigmentosum, group D | Several SNPs including Lys 751Gln | Lys751 associated with improved oxaliplatin/5-FU treatment outcome |
| Others | |||
| HLA-Ba | Major histocompatibility complex, class I B | HLA-B*5701 | HLA-B*5701 associated with hypersensitivity to abacavir |
This list is not meant to be comprehensive.
aGenes or proteins for prospective genotyping tests are in clinical use or under consideration.
Survey of polymorphic genes relevant to drug response
| Gene symbol | Description | Polymorphisms | Clinical phenotype |
|---|---|---|---|
| Phase I | |||
| CYP2D6a | Cytochrome P450 2D6 | Numerous and frequent functional polymorphisms (non-synonymous SNPs, splice variants, regulatory, gene amplification) (http://www.imm.ki.se/cypalleles/) | Variable metabolism of CYP substrates depending on enzyme specificity. Allele frequencies vary among ethnic groups |
| CYP2C9a | Cytochrome P450 2C9 | ||
| CYP2C19 | Cytochrome P450 2C19 | ||
| CYP3A5 | Cytochrome P450 3A5 | ||
| CYP2B6a | Cytochrome P450 2B6 | ||
| CYP2E1 | Cytochrome P450 2E1 | ||
| DPYD | Dihydropyrimidine dehydrogenase | Multiple polymorphims including splice variant IVS14+1G>A (DYPD*2A) | *2A increases 5FU neurotoxicity |
| Phase II | |||
| TPMTa | Thiopurine methyltransferase | Multiple polymorphisms | Null genotype associated with hematopoietic thiopurine toxicity, homozygous frequency 1/300 |
| COMT | Catechol-O-methyltransferase | Val158Met, haplotypes | Met158 associated with higher daily neuroleptic dosage and poor response; haplotye affects mRNA levels |
| NAT2 | N-acetyltransferase 2 | Arg197Gln, Ile114The, Lys268Arg, G191GA, G857A | Affect acetylation rates for drugs (e.g. isoniazid) |
| GSTM1 | Glutathione-S-transferase M1 | Deletion causing null genotype | Decreased glutathione conjugation |
| GSTT1 | Glutathione-S-transferase T1 | Deletion causing null genotype | |
| UGT1A1a | UDP-glycosyltransferase 1A1 | Multiple polymorphisms in the promoter and coding regions | UGT1A1*28 variants associated with increased irinotecan toxicity |
| Drug transport | |||
| ABCB1a | Multiple-drug resistance gene 1 | Several polymorphisms including C3435T | C3435 associated with higher drug transport activity |
| BCRP (ABCG2) | Breast cancer related protein | G34A, C421A and 944–949 deletion | Minor alleles with lower BRCP expression, enhanced drug sensitivity |
| Drug targets/pathway protein | |||
| MTHFR | 5, 10-Methylenetetrahydrofolate reductase | C(677)T and A(1298)C | T677 increases toxicity to methotrexate, drug resistance |
| TYMS | Thymidylate synthase | Two to nine 28 bp repeats in the 5′ enhancer | TSER*3 associated with drug resistance |
| VKORC1a | Vitamin K epoxide reductase complex 1 | Many, VKORC1 haplotypes | Variable anticoagulant effect of warfarin |
| ADRB2 | β2-adrenergic receptor | Arg16Gly, Gln27Glu and Thr164Ile | 16Gly decreases response to altuterol and salbutamol |
| ADRB1 | β1-adrenergic receptor | Arg389Gly | Gly389 with decreased cardiovascular response to β1-antagonists |
| ALOX5 | Arachidonate 5-lipoxygenase | Variant promoter SP1 binding site | Decreased drug response with the mutant genotype |
| SLC6A3 | Dopamine transporter | 3′-VNTR 10 repeat allele | 10/10 repeat genotype associated with poor response to methylphenidate |
| SLC6A4 | Serotonin transporter (5-HTT) | Long/short promoter polymorphism | May affect antidepressant drug response efficacy |
| HTR2A | Serotonin receptor 2A | His452Tyr | Tyr452 decreases response to the antipsychotic clozapine |
| ACE | Angiotensin-1 converting enzyme | Ins/del in intron 16 | Del/Del decreases efficacy of the reduction of proteinuria by ACE inhibitors |
| CETP | Cholesteryl ester transfer protein | TaqIB polymorphism B1/B2 | B1/B1associated with better response to statins |
| t(9,22) translocationa | t(9,22) BCR-ABL translocation | Translocation | Gleevec effective against chronic myeloid leukemia with translocation only |
| ERBB2a | ERBB2, HER/Neu | Overexpression of protein | Herceptin for breast cancer with ERBB2 overexpression |
| EGFRa | Epidermal growth factor receptor | Exon 18–21 mutations | Human lung cancers with mutations response better for Iressa |
| DNA repair | |||
| XRCC1 | X-ray repair cross-complementing gene 1 | Arg194Trp, Arg280His and Arg399Gln | Gln399 associated with oxaliplatin/5-FU resistance |
| XPD | Xeroderma Pigmentosum, group D | Several SNPs including Lys 751Gln | Lys751 associated with improved oxaliplatin/5-FU treatment outcome |
| Others | |||
| HLA-Ba | Major histocompatibility complex, class I B | HLA-B*5701 | HLA-B*5701 associated with hypersensitivity to abacavir |
| Gene symbol | Description | Polymorphisms | Clinical phenotype |
|---|---|---|---|
| Phase I | |||
| CYP2D6a | Cytochrome P450 2D6 | Numerous and frequent functional polymorphisms (non-synonymous SNPs, splice variants, regulatory, gene amplification) (http://www.imm.ki.se/cypalleles/) | Variable metabolism of CYP substrates depending on enzyme specificity. Allele frequencies vary among ethnic groups |
| CYP2C9a | Cytochrome P450 2C9 | ||
| CYP2C19 | Cytochrome P450 2C19 | ||
| CYP3A5 | Cytochrome P450 3A5 | ||
| CYP2B6a | Cytochrome P450 2B6 | ||
| CYP2E1 | Cytochrome P450 2E1 | ||
| DPYD | Dihydropyrimidine dehydrogenase | Multiple polymorphims including splice variant IVS14+1G>A (DYPD*2A) | *2A increases 5FU neurotoxicity |
| Phase II | |||
| TPMTa | Thiopurine methyltransferase | Multiple polymorphisms | Null genotype associated with hematopoietic thiopurine toxicity, homozygous frequency 1/300 |
| COMT | Catechol-O-methyltransferase | Val158Met, haplotypes | Met158 associated with higher daily neuroleptic dosage and poor response; haplotye affects mRNA levels |
| NAT2 | N-acetyltransferase 2 | Arg197Gln, Ile114The, Lys268Arg, G191GA, G857A | Affect acetylation rates for drugs (e.g. isoniazid) |
| GSTM1 | Glutathione-S-transferase M1 | Deletion causing null genotype | Decreased glutathione conjugation |
| GSTT1 | Glutathione-S-transferase T1 | Deletion causing null genotype | |
| UGT1A1a | UDP-glycosyltransferase 1A1 | Multiple polymorphisms in the promoter and coding regions | UGT1A1*28 variants associated with increased irinotecan toxicity |
| Drug transport | |||
| ABCB1a | Multiple-drug resistance gene 1 | Several polymorphisms including C3435T | C3435 associated with higher drug transport activity |
| BCRP (ABCG2) | Breast cancer related protein | G34A, C421A and 944–949 deletion | Minor alleles with lower BRCP expression, enhanced drug sensitivity |
| Drug targets/pathway protein | |||
| MTHFR | 5, 10-Methylenetetrahydrofolate reductase | C(677)T and A(1298)C | T677 increases toxicity to methotrexate, drug resistance |
| TYMS | Thymidylate synthase | Two to nine 28 bp repeats in the 5′ enhancer | TSER*3 associated with drug resistance |
| VKORC1a | Vitamin K epoxide reductase complex 1 | Many, VKORC1 haplotypes | Variable anticoagulant effect of warfarin |
| ADRB2 | β2-adrenergic receptor | Arg16Gly, Gln27Glu and Thr164Ile | 16Gly decreases response to altuterol and salbutamol |
| ADRB1 | β1-adrenergic receptor | Arg389Gly | Gly389 with decreased cardiovascular response to β1-antagonists |
| ALOX5 | Arachidonate 5-lipoxygenase | Variant promoter SP1 binding site | Decreased drug response with the mutant genotype |
| SLC6A3 | Dopamine transporter | 3′-VNTR 10 repeat allele | 10/10 repeat genotype associated with poor response to methylphenidate |
| SLC6A4 | Serotonin transporter (5-HTT) | Long/short promoter polymorphism | May affect antidepressant drug response efficacy |
| HTR2A | Serotonin receptor 2A | His452Tyr | Tyr452 decreases response to the antipsychotic clozapine |
| ACE | Angiotensin-1 converting enzyme | Ins/del in intron 16 | Del/Del decreases efficacy of the reduction of proteinuria by ACE inhibitors |
| CETP | Cholesteryl ester transfer protein | TaqIB polymorphism B1/B2 | B1/B1associated with better response to statins |
| t(9,22) translocationa | t(9,22) BCR-ABL translocation | Translocation | Gleevec effective against chronic myeloid leukemia with translocation only |
| ERBB2a | ERBB2, HER/Neu | Overexpression of protein | Herceptin for breast cancer with ERBB2 overexpression |
| EGFRa | Epidermal growth factor receptor | Exon 18–21 mutations | Human lung cancers with mutations response better for Iressa |
| DNA repair | |||
| XRCC1 | X-ray repair cross-complementing gene 1 | Arg194Trp, Arg280His and Arg399Gln | Gln399 associated with oxaliplatin/5-FU resistance |
| XPD | Xeroderma Pigmentosum, group D | Several SNPs including Lys 751Gln | Lys751 associated with improved oxaliplatin/5-FU treatment outcome |
| Others | |||
| HLA-Ba | Major histocompatibility complex, class I B | HLA-B*5701 | HLA-B*5701 associated with hypersensitivity to abacavir |
This list is not meant to be comprehensive.
aGenes or proteins for prospective genotyping tests are in clinical use or under consideration.
References
Evans, W.E. and Relling, M.V. (
Weinshilboum, R. and Wang, L. (
Wilkinson, G.R. (
Pinsonneault, J. and Sadee, W. (
Meyer, U.A. (
Roses, A.D. (
Lesko, L.J. and Woodcock, J. (
Efferth, T. and Volm, M. (
Goldstein, D.B., Tate, S.K. and Sisodiya, S.M. (
Lazarou, J., Pomeranz, B.H. and Corey, P.N. (
Phillips, K.A., Veenstra, D.L., Oren, E., Lee, J.K. and Sadee, W. (
Huang, Y., Anderle, P., Bussey, K.J., Barbacioru, C., Shankavaram, U., Dai, Z., Reinhold, W.C., Papp, A., Weinstein, J.N. and Sadee, W. (
Druker, B.J., Talpaz, M., Resta, D.J., Peng, B., Buchdunger, E., Ford, J.M., Lydon, N.B., Kantarjian, H., Capdeville, R., Ohno-Jones, S. et al. (
Lynch, T.J., Bell, D.W., Sordella, R., Gurubhagavatula, S., Okimoto, R.A., Brannigan, B.W., Harris, P.L., Haserlat, S.M., Supko, J.G., Haluska, F.G. et al. (
Slamon, D.J., Leyland-Jones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A., Fleming, T., Eiermann, W., Wolter, J., Pegram, M. et al. (
Wang, L., Sullivan, W., Toft, D. and Weinshilboum, R. (
Krynetskiy, E.Y. and Evans, W.E. (
Relling, M.V., Hancock, M.L., Boyett, J.M., Pui, C.H. and Evans, W.E. (
Kaniwa, N., Kurose, K., Jinno, H., Tanaka-Kagawa, T., Saito, Y., Saeki, M., Sawada, J., Tohkin, M. and Hasegawa, R. (
Ando, Y., Saka, H., Ando, M., Sawa, T., Muro, K., Ueoka, H., Yokoyama, A., Saitoh, S., Shimokata, K. and Hasegawa, Y. (
Innocenti, F., Undevia, S.D., Iyer, L., Chen, P.X., Das, S., Kocherginsky, M., Karrison, T., Janisch, L., Ramirez, J., Rudin, C.M. et al. (
Iyer, L., Das, S., Janisch, L., Wen, M., Ramirez, J., Karrison, T., Fleming, G.F., Vokes, E.E., Schilsky, R.L. and Ratain, M.J. (
Innocenti, F., Grimsley, C., Das, S., Ramirez, J., Cheng, C., Kuttab-Boulos, H., Ratain, M.J. and Di Rienzo, A. (
Mathijssen, R.H., Marsh, S., Karlsson, M.O., Xie, R., Baker, S.D., Verweij, J., Sparreboom, A. and McLeod, H.L. (
Rouits, E., Boisdron-Celle, M., Dumont, A., Guerin, O., Morel, A. and Gamelin, E. (
Sai, K., Saeki, M., Saito, Y., Ozawa, S., Katori, N., Jinno, H., Hasegawa, R., Kaniwa, N., Sawada, J., Komamura, K. et al. (
Strassburg, C.P., Kneip, S., Topp, J., Obermayer-Straub, P., Barut, A., Tukey, R.H. and Manns, M.P. (
Zeldin, R.K. and Petruschke, R.A. (
Zhou, S., Chan, E., Lim, L.Y., Boelsterli, U.A., Li, S.C., Wang, J., Zhang, Q., Huang, M. and Xu, A. (
Heils, A., Teufel, A., Petri, S., Stober, G., Riederer, P., Bengel, D. and Lesch, K.P. (
Lesch, K.P., Bengel, D., Heils, A., Sabol, S.Z., Greenberg, B.D., Petri, S., Benjamin, J., Muller, C.R., Hamer, D.H. and Murphy, D.L. (
Goodwin, B., Hodgson, E., D'Costa, D.J., Robertson, G.R. and Liddle, C. (
Bombail, V., Taylor, K., Gibson, G.G. and Plant, N. (
Hirota, T., Ieiri, I., Takane, H., Maegawa, S., Hosokawa, M., Kobayashi, K., Chiba, K., Nanba, E., Oshimura, M., Sato, T. et al. (
Cheung, V.G., Conlin, L.K., Weber, T.M., Arcaro, M., Jen, K.Y., Morley, M. and Spielman, R.S. (
Johnson, A.D., Wang, D. and Sadee, W. (
Rockman, M.V. and Wray, G.A. (
Yan, H., Yuan, W., Velculescu, V.E., Vogelstein, B. and Kinzler, K.W. (
West, A.G. and Fraser, P. (
Rogan, P.K., Svojanovsky, S. and Leeder, J.S. (
Attaie, A., Kim, E., Wilcox, E.R. and Lalwani, A.K. (
Maillet, P., Dalla Venezia, N., Lorenzo, F., Moriniere, M., Bozon, M., Noel, B., Delaunay, J. and Baklouti, F. (
Bodzioch, M., Lapicka, K., Aslanidis, C., Kacinski, M. and Schmitz, G. (
Howe, D. and Lynas, C. (
Duan, J., Wainwright, M.S., Comeron, J.M., Saitou, N., Sanders, A.R., Gelernter, J. and Gejman, P.V. (
Pinsonneault, J., Nielsen, C.U. and Sadee, W. (
Bray, N.J., Buckland, P.R., Williams, N.M., Williams, H.J., Norton, N., Owen, M.J. and O'Donovan, M.C. (
Zubieta, J.K., Heitzeg, M.M., Smith, Y.R., Bueller, J.A., Xu, K., Xu, Y., Koeppe, R.A., Stohler, C.S. and Goldman, D. (
Murrell, A., Rakyan, V.K. and Beck, S. (
Carrel, L. and Willard, H.F. (
Sado, T. and Ferguson-Smith, A.C. (
Petronis, A. (
Rakyan, V.K., Blewitt, M.E., Druker, R., Preis, J.I. and Whitelaw, E. (
Byrd, J.C., Marcucci, G., Parthun, M.R., Xiao, J.J., Klisovic, R.B., Moran, M., Lin, T.S., Liu, S., Sklenar, A.R., Davis, M.E. et al. (
Dowell, J.E. and Minna, J.D. (
Esteller, M., Garcia-Foncillas, J., Andion, E., Goodman, S.N., Hidalgo, O.F., Vanaclocha, V., Baylin, S.B. and Herman, J.G. (
Mattick, J.S. and Makunin, I.V. (
Chen, J., Sun, M., Hurst, L.D., Carmichael, G.G. and Rowley, J.D. (
Cogoni, C. and Macino, G. (

{kind=link}
{kind=link}