



Abstract
Hutchinson–Gilford progeria syndrome (HGPS) is typically caused by mutations in codon 608 (G608G) of the LMNA gene, which activates a cryptic splice site resulting in the in-frame loss of 150 nucleotides from the lamin A message. The deleted region includes a protein cleavage site that normally removes 15 amino acids, including a CAAX box farnesylation site, from the lamin A protein. We investigated the processing of the C-terminus of the mutant protein, ‘progerin’, and found that it does not undergo cleavage and, indeed, remains farnesylated. The retention of the farnesyl group may have numerous consequences, as farnesyl groups increase lipophilicity and are involved in membrane association and in protein interactions, and is likely to be an important factor in the HGPS phenotype. To further investigate this, we studied the effects of farnesylation inhibition on nuclear phenotypes in cells expressing normal and mutant lamin A. Expression of a GFP–progerin fusion protein in normal fibroblasts caused a high incidence of nuclear abnormalities, as was also seen in HGPS fibroblasts, and resulted in abnormal nuclear localization of GFP–progerin in comparison with the localization pattern of GFP–lamin A. Expression of a GFP–lamin A fusion containing a mutation preventing the final cleavage step, causing the protein to remain farnesylated, displayed identical localization patterns and nuclear abnormalities as in HGPS cells and in cells expressing GFP–progerin. Exposure to a farnesyltransferase inhibitor (FTI), PD169541, caused a significant improvement in the nuclear morphology of cells expressing GFP–progerin and in HGPS cells. These results implicate the abnormal farnesylation of progerin in the cellular phenotype in HGPS cells and suggest that FTIs may represent a therapeutic option for patients with HGPS.
INTRODUCTION
Hutchinson–Gilford progeria Syndrome (HGPS) is a rare disease that affects children in the first decade of life and causes a remarkable phenotype resembling many aspects of aging. Affected children have an extremely aged appearance, a lack of subcutaneous fat, growth retardation and severe atherosclerosis. The average life span of HGPS patients is 13 years, with most dying from complications associated with atherosclerosis. Recently, the gene responsible for HGPS was identified, and HGPS joined a group of syndromes—the laminopathies—all of which have an underlying defect in the lamin A/C gene (LMNA) (1).
The lamins are a component of the nuclear lamina, a fibrous matrix located at the interior of the nuclear membrane, responsible for nuclear integrity and organization. In addition, lamins are present in the nucleoplasm and may be involved in more complex spatial organization of the nucleus. They play a role in a wide array of nuclear processes, including transcription, replication and cell cycle functions (2). In humans and other mammals, there are three lamin genes. The LMNA gene encodes the A-type lamins, A, AΔ10, C and C2. The B-type lamins are encoded by two genes, LMNB1, which encodes lamin B1, and LMNB2, which encodes lamin B2 and B3. All vertebrate cells express at least one B-type lamin, which is essential for cell viability (3). The A-type lamins are developmentally regulated and expressed primarily in differentiated cells (4). Lamins C2 and B3 are expressed only in germ cells.
Mature lamin A is formed by post-translational processing of a pre-lamin A precursor. The pre-lamin A protein has a CAAX box at the C-terminal, which signals isoprenylation, the addition of a farnesyl group to the cysteine by the enzyme farnesyltransferase (FTase). The CAAX box is a cysteine followed by two aliphatic amino acids with the X denoting methionine, serine, alanine or leucine. The final amino acid defines the specificity for the addition of the isoprenyl group with methionine, serine, or alanine signaling farnesylation and leucine signaling the addition of a geranylgeranyl group by the enzyme geranylgeranyl transferase-I (GGTase-I) (5). Following farnesylation, the three terminal amino acids are removed, and the cysteine is methylated. Although both the B-type lamins and lamin A are farnesylated and carboxymethylated, unique to lamin A is the removal of an additional 15 C-terminal amino acids from the mature protein, including the farnesylated cysteine, by the protein Zmpste24 (6–8). Farnesylation of lamin A is an essential step in processing the protein. Addition of the farnesyl moiety allows lamin A to localize to the nuclear periphery where the final cleavage step occurs (9).
The most common mutation found in HGPS patients is a silent mutation (G608G) creating an abnormal splice donor site in exon 11 of the LMNA gene (1). The result of the mis-splicing is an in-frame deletion of 150 nucleotides from the message resulting in a protein missing 50 amino acids near the C-terminus. The deleted region contains two potential cyclin-dependent kinase target serines (652 and 657) that may be involved in signaling the disassembly of the lamina during cell division (10). The deleted region also includes the protein cleavage site that is targeted by Zmpste24 for the final cleavage event removing the C-terminal 15 amino acids, including the farnesylated cysteine.
Why the specific mutation in the LMNA gene causes the premature aging phenotype found in HGPS is unknown. The progerin deletion could have a number of consequences that might give rise to the abnormal phenotype, including abnormal protein processing and deletion of critical protein binding domains. The retention of the farnesyl group may change important properties of the protein, making it more lipophilic and altering interactions with other proteins. Incomplete processing of lamin A caused by mutations in Zmpste24 are found in patients with a severe form of mandibuloacral dysplasia (MAD), which is phenotypically similar to HGPS (11). In addition, Zmpste24 knockout mice are phenotypically similar to MAD and HGPS with defects in hair, bone and fat and muscle (12,13). We therefore hypothesized that retention of the farnesyl group on progerin may be a significant factor in the development of the HGPS phenotype. Although the conserved cleavage site is missing in progerin, it remained possible that the protein was still cleaved, as there are other potential sites in the progerin protein that may have substituted for the missing cleavage sequence. Using fluorography, we have demonstrated that progerin is incompletely processed and does retain the farnesyl group. We have also shown that expression of an N-terminal GFP–progerin fusion in normal fibroblasts caused abnormal nuclear morphology similar to that seen on HGPS fibroblasts and displayed abnormal nuclear localization patterns. Furthermore, a GFP-tagged cleavage-minus mutant lamin A (L647R) gives rise to cellular phenotypes very similar to a GFP–progerin construct when expressed in normal fibroblasts, causing an abnormal localization pattern and a significant increase in abnormal nuclear morphology. Exposure to the farnesyltransferase inhibitor (FTI) significantly reduced nuclear morphology abnormalities of both normal fibroblasts expressing GFP–progerin and HGPS fibroblasts. These studies indicate that abnormal farnesylation of progerin plays a major role in the abnormal cellular phenotype and suggest that FTI treatment may be a possible therapeutic option for HGPS.
RESULTS
Farnesylation of progerin
In order to determine if the progerin protein retains a farnesyl group, we labeled normal and HGPS fibroblasts with 3H-mevalonate and performed an immunoprecipitation using anti-lamin A/C antibodies. This step was necessary because many proteins are modified by the addition of farnesyl or geranylgeranyl groups. Progerin was efficiently detected as a protein of approximately 74 kDa that runs between lamin C (70 kDa) and lamin A (78.5 kDa) on an immunoblot, a size consistent with the loss of 50 amino acids from lamin A (Fig. 1A). The presence of a 3H-labeled farnesyl group on the progerin protein was detected by fluorography of the immunoprecipitated proteins, demonstrating that progerin is farnesylated (Fig. 1B). The addition of 2 µm FTI prevented the farnesylation of progerin. Normal fibroblasts expressing the cleavage-minus mutant construct, GFP–LA (L647R), were included as a positive control and retained a farnesyl group as expected.
Transient expression studies
When GFP-tagged normal lamin A was expressed in normal fibroblasts, the nuclei had an oval shape with fluorescence evenly distributed at the periphery of the nucleus with a few intranuclear structures (Figs 2A and E). In contrast, transient expression of the GFP-tagged progerin in normal fibroblasts caused abnormal nuclear morphology, similar to that seen in HGPS fibroblasts, including nuclear blebbing (Fig. 2B). In addition, altered distribution of the GFP-tagged progerin into nuclear foci was observed in many cells. The most commonly seen localization pattern for transiently expressed GFP–progerin was thick regions in the nuclear lamina, as visualized by standard florescent (Fig. 2B) and confocal microscopy (Fig. 2G). These were also detected by ICC using antibodies specific for lamin A in HGPS fibroblasts (Fig. 2D) but with less resolution, most likely owing to a lack of availability of the epitope (14) within these aggregates. Quantitation of abnormal nuclei revealed that expression of progerin in normal fibroblasts for 72 h caused a substantial increase in the percent abnormal nuclei to levels typically observed in HGPS cells (Fig. 3A). However, expression of normal lamin A in HGPS fibroblasts for 72 h did not correct the nuclear blebbing phenotype seen in these cells (Fig. 3A). In addition, the normal GFP–lamin A was often mislocalized in these cells, indicating that the presence of the endogenous mutant can cause mislocalization of the normal GFP-tagged lamin A (data not shown).
The effects of several concentrations of the specific FTI, PD169451, on nuclear morphology and localization of transiently expressed lamin A and progerin were examined. Exposure of 100 nm FTI for 72 h had a significant effect on the localization of transiently expressed GFP–lamin A and GFP–progerin (Figs 2F and H, respectively). Transiently expressed GFP–lamin A and GFP–progerin were localized in very similar fashion to intranuclear filaments when exposed to the FTI rather than localizing to the nuclear periphery. Confocal microscopy revealed that the localization pattern for GFP–progerin was different when the cells were exposed to 100 nm FTI for 72 h than from the foci seen with GFP–progerin without the FTI (compare Figs 2G with H). In addition, short-term exposure to the FTI had a significant effect on the nuclear morphology of fibroblasts transiently expressing GFP–progerin. These fibroblasts had a 33% decrease in the percentage of cells with abnormal nuclear morphology when exposed to 100 nm FTI (P=0.01) (Fig. 3B). To verify this result in HGPS fibroblasts, the AG11498 line was exposed to various FTI concentrations for 3 days. Treating these HGPS cells for 72 h with the FTI resulted in as much as a 40% drop in the percent of cells with abnormal nuclear morphology (P=0.008) (Fig. 3C). However, the FTI appears to have a negative effect on the nuclear morphology of normal fibroblasts in a dose-dependent manner (Figs 3B and C). High concentrations of the FTI for a short duration had significant effects on the localization of GFP–lamin A and GFP–progerin and caused a significant improvement in the nuclear morphology of the normal cells expressing GFP–progerin and on HGPS fibroblasts.
Stable expression studies
To confirm and extend these results and to control protein expression levels, GFP–lamin A and GFP–progerin were expressed from Tet-off retroviral constructs that allowed for sustained expression and modulation of expression to address the possibility of results due to overexpression of the fusion proteins when the proteins were transiently expressed. In addition, a cleavage-minus mutant, GFP–LA (L647R), was included to examine the effect of retaining the farnesyl-group without the 50 amino acid deletion found in progerin. When no tetracycline was present, the GFP fusions were expressed at relatively high levels as measured by western blot (Fig. 4D). Addition of 0.05 µg/ml tetracycline allowed for reduced expression of the GFP fusions such that near-endogenous levels were obtained, whereas 8 µg/ml tetracycline reduced, but did not altogether prevent, expression from these constructs (Fig. 4D). When GFP–progerin was stably expressed in normal fibroblasts (0.05 µg/ml tetracycline) at levels similar to progerin levels in HGPS fibroblasts, 10 nm FTI substantially reduced the percent abnormal nuclei by 71% (P<0.0001) (Fig. 4B). When GFP-tagged L647R mutant was expressed at intermediate levels (0.05 µg/ml tetracycline), the increase in abnormal nuclear morphology was reduced 67% by exposure to 10 nm FTI (P<0.0001) (Fig. 4B). The improvement in abnormal nuclear morphology with intermediate expression levels of GFP–progerin and GFP–LA (L647R) was actually less profound at the higher concentrations of the FTI (100 nm) in comparison with the decrease observed for 10 nm (Fig. 4B). Treatment with 10 nm FTI could not overcome the effects of very high expression levels of the GFP–progerin or GFP–LA (L647R) that were obtained when no tetracycline was present to repress expression (Fig. 4A). However, the cleavage-minus mutant may be more tractable to FTI treatment at the highest expression level, especially when exposed to 100 nm FTI, perhaps owing to slightly lower expression levels. Some leaky expression was observed even at 8 µg/ml tetracycline, and these low levels did affect nuclear morphology, causing a moderate increase in the percent abnormal nuclei in the GFP–progerin and GFP–LA (L647R) cells (Fig. 4C). Thus, it is unlikely that the effects seen with the progerin constructs are due solely to protein overexpression.
As with transient expression, stable expression of GFP–lamin A did not have a significant effect on nuclear morphology, and the GFP signal was evenly distributed at the nuclear periphery (Fig. 5A). When GFP–progerin was expressed in normal fibroblasts at levels near what is observed in HGPS patients, nuclear aggregates at the periphery of the nucleus and intranuclear structures were observed, along with a substantial increase in abnormal nuclear morphology including folding and blebbing of the nuclear membrane (Fig. 5D). In addition, chromatin staining by DAPI was altered with a loss of staining often observed at the nuclear periphery and in regions adjacent to the progerin aggregates. Expression of intermediate levels of the cleavage-minus mutant, GFP–LA (L647R), had very similar effects on localization of the GFP signal and on nuclear morphology as the GFP–progerin (Fig. 5G). Also, as observed in the transient expression studies, the FTI caused the GFP–lamin A and GFP–progerin to localize to intranuclear filaments when expressed in cells exposed to the FTI at 100 nm concentrations. Localization of GFP-tagged L647R mutant was also redistributed to intranuclear filaments when exposed to 100 nm FTI (Fig. 5I). However, 10 nm FTI did not lead to this drastic reorganization of GFP signal for any of the fusion proteins. Rather, alterations in GFP localization at this FTI concentration were less noticeable (Figs 5B, E and H).
Effects of FTI on HGPS fibroblasts
A longer duration analysis on the effects of the FTI on the nuclear morphology of two normal and four HGPS fibroblast lines revealed that a sustained low dose of 10 nm, substantially reduced the percentage of HGPS cells displaying abnormal nuclear morphology in three of the four HGPS lines (Fig. 6). Following 14 days of exposure to 10 nm FTI, each of the three HGPS lines had over a 70% reduction (P<0.0001) in the relative number of abnormal nuclei, reducing the occurrence of abnormal nuclei to that of normal controls. The HGPS line (HGADFN004) that did not show a significant decrease in abnormal nuclear morphology after 14 days of exposure did have an initial improvement at the two-day time point. However, it must be noted that all cell lines in the experiment were at a passage six except for the HGADFN004 line, which was at passage 12 at the beginning of the experiment. Extended exposure to 200 nm or higher concentration of the FTI caused cells to stop dividing after a week to 10 days (data not shown) and all experiments displayed a dose-dependent effect of the FTI, causing an increase in abnormal nuclear morphology in normal fibroblasts. However, 10 nm FTI did not significantly alter the nuclear morphology of normal cells and substantially lowered the percentage of abnormal nuclei in HGPS fibroblasts (Fig. 6). As in previously discussed experiments, the higher concentration of FTI, 100 nm, was less effective at reducing the abnormal nuclear morphology phenotype in the HGPS cells. In addition, there was a significant increase in abnormal nuclei in normal cells at 100 nm concentrations at the 6-day and 14-day time points (Fig. 6). This effect was observed in all normal controls at concentrations higher than 100 nm and involved some unusual nuclear structures, including binucleated cells and donut-shaped nuclei (Fig. 5C). Although doses of the FTI, PD169541, at 100 nm or higher had some adverse affects on nuclear morphology of normal cells, the 10 nm concentration had a dramatic effect, lowering the percentage of nuclei with abnormal morphology in fibroblasts derived from HGPS patients without a significant adverse effect on normal nuclear morphology.
DISCUSSION
To date, there have been 11 diseases associated with mutations in the LMNA gene. The disorders include muscular dystrophy, lipodystrophy, neuropathy and premature aging disorders, and the tissues most affected in these disorders are striated muscle, adipose tissue and bone (reviewed in 15). The remarkable heterogeneity and phenotypic differences of the laminopathies are somewhat reflected in the distribution of mutations in LMNA. Mutations causing EDMD2 are distributed throughout the gene, while those resulting in CMD1A are mainly clustered within the coiled-coil domain, suggesting that the latter may act by disrupting dimerization or subsequent polymerization of lamin A/C. In contrast, the mutations found in HGPS, FPLD and MAD patients are often found within exons encoding the C-terminal globular domain of lamin A/C and, more specifically, are typically restricted to just a few codons in exons 8, 9 and 11 (1,16–18).
We and others have observed the mis-splicing caused by the activation of a cryptic splice site due to the codon 608 mutation in HGPS. This mis-splicing results in the loss of 150 nucleotides from the lamin A message, between the cryptic splice site and the end of exon 11. The frame of the transcript is retained so that the final 9 codons from exon 12 are transcribed, including a CAAX box farnesylation site. The deletion of 50 amino acids may have a number of effects, including effects on protein interactions and on protein processing. We investigated the processing of the C-terminus to determine if the processing of progerin is incomplete. Although the conserved cleavage site is missing in progerin, it was possible that the protein was either not farnesylated or was aberrantly cleaved, as there are potential sites in the progerin protein that may have substituted for the missing cleavage sequence. Here, we have directly determined that progerin indeed retains the farnesyl group.
The retention of the farnesyl group by progerin could have deleterious consequences for several reasons. Farnesyl groups increase lipophilicity and are involved in membrane association (5) and may, therefore, alter protein localization. Also, farnesyl groups are involved in protein interactions (19). For example, the nuclear pre-lamin A recognition factor, NARF, has been shown to interact only with the farnesylated form of lamin A (20). We have shown that retention of the farnesyl group on a GFP–progerin protein and on a GFP-labeled cleavage-minus mutant alters the distribution of these proteins in the nucleus in comparison with the normal GFP–lamin A in a manner comparable to that of progerin in HGPS cells. The expression of normal lamin A in HGPS fibroblasts did not correct the abnormal nuclear morphology and also often displayed an abnormal localization pattern in the nucleus, suggesting a dominant-negative effect as the endogenous mutant protein could lead to mis-localization of GFP-labeled lamin A. This abnormal distribution of the mutant proteins and lamin A may contribute to the loss of integrity of the lamina and, thus, result in the abnormal nuclear phenotype observed in HGPS cells and in normal cells expressing the mutant progerin protein.
Presently, there are few well-characterized cellular phenotypes that can be used in the analysis of HGPS cells. The abnormal nuclear morphology observed in cells isolated from HGPS, and in normal cells expressing progerin, is consistent with the direct role of lamins in maintaining nuclear shape in interphase nuclei. Disruption in lamin organization may be related to the HGPS phenotype in several ways. The lamin matrix acts as a ‘shock absorber’ (21), and disruption of the lamin structure may cause cell types to become particularly affected owing to mechanical stresses. This effect may be observed in other laminopathies and their affected tissues, such as striated muscle in limb-girdle muscular dystrophy. Similarly, the severe atherosclerosis in HGPS may be at least partially because of an increased fragility of nuclei in vessel walls. These stresses may be exacerbated by altered transcription in affected cells (22,23). However, it is difficult to imagine that mechanical stress plays a role in all the affected tissues in HGPS such as fat, bone and hair.
Altered localization of progerin, and its disruption of the lamin structure, not only leads to increased nuclear abnormalities, but also appears to effect heterochromatin localization and may have effects on transcription. Lamin A interacts with pRB (24) and the transcription factor, SREBP1 (25). In addition, the expression of a dominant-negative form of lamin A with an N-terminal deletion disrupts lamin organization in the nucleus and decreases the synthesis of RNA polymerase II-dependent transcripts (26). Further, in a study examining the expression of 33 000 genes in HGPS and normal cell lines, transcription factors were the largest functional category with altered expression in HGPS cells (27). The LMNA gene is primarily expressed in differentiated tissues in the fetus and adult (4) and may be important in maintaining the differentiated state, perhaps by influencing heterochromatin organization (28,29). In support of this hypothesis, cells from HGPS patients have altered heterochromatin localization (10), and we have observed altered chromatin staining in cells expressing GFP–progerin.
Incomplete processing of lamin A appears to be an important factor not only for nuclear phenotypes in culture but also in the development of the disease phenotype. In support of this hypothesis, mutations in Zmpste24, the protease that performs the final cleavage step removing the farnesyl group from lamin A, are found in patients with a severe form of MAD, which is often misdiagnosed as HGPS. It may be that retention of the farnesyl group by incompletely processed lamin A, or by progerin, creates a protein that is toxic to the cell. Reduced lamin A/C expression in mice heterozygous for an LMNA knockout eliminates the Zmpste24 KO phenotype and reduces the percent of cells with mis-shapen nuclei (30). In this case, the mice heterozygous for lamin A are protected from the effects of the Zmpste24 KO owing to lower levels of incompletely processed lamin A. Similarly, FTIs may improve the nuclear phenotype of HGPS fibroblasts and normal fibroblasts expressing progerin by simply reducing the levels of farnesylated progerin. The FTI may also improve the phenotype by sequestering the mutant protein into intranuclear filaments and preventing it from localizing to the nuclear membrane, as exposure to the FTI does not appear to reduce progerin levels (Fig. 1A) (data not shown). The lowest concentration of the FTI used in these experiments (10 nm) caused a substantial improvement in the nuclear morphology of HGPS cells and normal cells expressing GFP–progerin. However, this concentration did not cause the drastic redistribution of GFP–lamin A and GFP–progerin to intranuclear filaments that were observed at higher FTI concentrations. Therefore, it seems likely that reduced levels of farnesylated progerin, below a threshold level, may be responsible for the improvement in nuclear phenotype observed at the 10 nm FTI concentration.
Our results suggest that there may be a therapeutic window for effective concentrations of the FTI, and a window of opportunity for use of the drug at least in alleviation of the nuclear morphology phenotype in HGPS cells in culture. At 10 nm, a significant reduction in abnormal nuclei in HGPS cells was observed with little effect on normal nuclear morphology. At FTI concentrations of 100 nm, the nuclear morphology of normal cells was affected with some interesting nuclear shapes occurring, notably ‘donut-shaped’ nuclei. This shape was never observed in normal cells without the FTI. The increase in abnormal nuclear morphology at the higher FTI concentrations may be directly related to the drug's effect on lamin structure, as the higher concentrations of the FTI cause a substantial reorganization of GFP-labeled lamin A and progerin distribution in the nucleus. Further, overexpression of lamin A appears to have a protective effect when cells are exposed to 100 nm FTI (Fig. 4A). In addition to a window for effective concentrations, there may be a window for timeliness of treatment. The HGPS cell line that did not respond to 14 days exposure to 10 nm FTI was of a higher passage number than the three HGPS lines that showed substantial reduction in abnormal nuclear morphology under the same conditions.
Therapeutic interest in FTIs arose as a means of preventing oncogenic Ras signaling. Approximately 30% of human cancers have a mutation in a ras proto-oncogene leading to constitutively active Ras signaling. However, Ras must localize to the inner plasma membrane in order to propagate signaling. To accomplish this, Ras is post-translationally modified by addition of a farnesyl group by FTase. The compound used in these studies, PD169451, is a peptidic farnesyl diphosphate (FPP) competitor, but is not a FPP analog, and is 4500-fold more selective for FTase inhibition than for inhibition of the related GGTase-I (31). These experiments serve as a proof of principle that FTIs can improve the phenotype of HGPS cells. Different FTIs, or combination therapies with drugs like the statins, may prove to be even more effective in treatment of the disease. Other FTIs have been used in clinical studies and are well tolerated by patients (32). The improvement in the morphology of the HGPS cells when exposed to low doses of the FTI provides a positive sign that these agents may be useful in treating the disease. Examination of these drugs in animal models for HGPS to determine if there is improvement in the overall phenotype of the animals is an obvious next step.
MATERIALS AND METHODS
Expression constructs
Normal and mutant lamin A alleles were cloned by isolating RNA from the cell lines AG11513a and NF, respectively. An oligo-dT primer was used for first-strand synthesis using the Superscript II kit (Invitrogen). The first-strand cDNA was used as a template to amplify normal and mutant products using the Platinum Taq high fidelity kit from Invitrogen with the primers lamin AF1 (AGCAGTCTCTGTCCTTCGACCC) and lamin AR1 (CTTCCACCTCCCACCTCATTCC). Owing to the GC-rich nature of the template, DMSO and betaine were added to final concentrations of 4% and 500 mm, respectively (33). The reaction was run for 20 cycles with an annealing temperature of 62°C, and the resulting product was used for TOPO-TA cloning (Invitrogen). These cDNA constructs were verified by sequencing and used as templates for subcloning into the pEGFP-C2 and pRevTRE vectors (Clontech).
The L647R mutation described by Hennekes and Nigg (34) was introduced into the pRevTRE-GFP–Lamin A construct using a PCR-based site-directed mutagenesis. Two overlapping PCR products were generated using the following primer pairs: PCR product 1 using the primers LMNA-1320-For (GTGGAGGAGGTGGATGAGGAGGGCAAG) and LMNA-L647R-Rev (TGGAGTTGCCCAGGCGGTAGGAGCGGGTGA), and PCR product 2 using the primers LMNA-L647R-For (TCACCCGCTCCTACCGCCTGGGCAACTCCA) and pRevTRE-3700-Rev (CTGAGGGCTGGACCGCATCTGGGGAC). Note that the reverse primer for product 1 is the complement sequence to the forward primer for product 2 and that both of these primers contain a single mismatch with the target sequence that will generate the L647R mutation. Both products generated separately using the pRev-TRE-lamin A construct as a template. The two products were gel-purified and used together as the template for a third PCR reaction using the primers LMNA-1390-For (CATGGGCAATTGGCAGATCAAG) and pRevTRE-3440-Rev (CATGCCTTGCAAAATGGCGTTAC). Although the template is two overlapping fragments, a single PCR product results, which was gel-purified and cloned using the TOPO-TA kit from Invitrogen. The resulting insert, containing the L647R mutation, was subcloned into the pRevTRE-GFP–Lamin A construct using Mfe I and Cla I restriction endonucleases. The resulting construct, pRevTRE-GFP–LA (L647R) was sequenced to verify the presence of the introduced mutation without any random base changes owing to the use of PCR to generate the construct.
Cell culture
Cell lines used were normal fibroblast lines, GM08398, HGMDFN090 and AG06278, and HGPS fibroblast lines: P01 (HGADFN003), HGADFN004, HGADFN136, AG03513d, AG06297C, AG10750 and AG11498. Cell lines with AG or GM designations were purchased from Coriell Repositories. Cell lines beginning with HG were obtained from the Progeria Research Foundation. Fibroblasts were cultured in minimal essential media (Gibco, no. 10370-021) supplemented with 15% FBS (HiClone), 2 mml-glutamine, penicillin (50 U/ml) and streptomycin (50 mg/ml). The Amphotropic Phoenix retroviral packaging line (Peter Nolan, Stamford University) was maintained in DMEM supplemented with 10% FBS, 2 mml-glutamine, penicillin (50 U/ml) and streptomycin (50 mg/ml). The FTI used in these experiments, PD169451, was a gift from Pfizer. The FTI was dissolved in DMSO and stored as a stock solution of 10 mm. When added to media for experiments, the amount of DMSO was equal in all samples.
Retroviral constructs
The pRevTetOff vector and pRevTRE constructs were transfected using FuGENE 6 (Roche) into the Amphotropic Phoenix retroviral packaging line. Forty-eight hours after transfection, the media containing retrovirus was filtered through a 0.22 µm filter and used immediately or stored at −70°C. Retroviral infection was performed by exposing target cells to a 1:1 mix of retroviral supernatant and growth media with 4 µg/ml polybrene.
GFP localization
Normal and HGPS fibroblasts were plated on coverslips in six-well plates or in chambered microscope slides (BD Biosciences) 24 h prior to transfection. Fibroblasts were transiently transfected with the GFP–lamin A or GFP–progerin expression constructs using FuGENE 6 (Roche). Seventy-two hours later, the cells were washed two times with PBS (pH 7.2) and fixed for 20 min at 4°C with 4% paraformaldehyde in PBS containing 62.5 µg/ml LPC palmitoyl and 1 U/ml rhodamine phalloidin (Molecular Probes) to stain the cytoskeleton. Following three washes with PBS, 10 µl of ProLong Gold mounting medium with DAPI (Molecular Probes) was applied, and the cells were overlaid with a coverslip.
To examine stable expression from the Tet-off constructs, the cell lines were maintained in growth medium containing 8 µg/ml tetracycline. To induce expression, cells were rinsed three times with Hanks balanced saline solution (HBSS), released from the flask with 0.05% trypsin, pelleted at 800g, washed once with HBSS and resuspended in growth medium. About 10 000 cells were plated per chamber in chambered microscope slides containing the appropriate tetracycline and FTI concentrations. To observe nuclear morphology and GFP localization, cells were fixed as described earlier.
Immunocytochemistry
Cells were cultured on coverslips in six-well plates for 72 h. Coverslips were washed twice with PBS and fixed with 4% paraformaldehyde in PBS with 0.18% Triton X-100 for 20 min at 4°C. Coverslips were washed three times with PBS and incubated for 30 min at 37°C in blocking buffer (1% BSA in PBS). The cells were probed with 1:10 dilution of the lamin A specific antibody (US Biological) in blocking buffer for 1 h at room temperature followed by washing three times with PBS. Secondary antibody (Goat Anti-Mouse IgG, FITC) was diluted 1:1000 in blocking buffer and applied to the coverslips for 1 h at room temperature. After washing three times for 5 min with PBS, the coverslips were inverted and placed on slides with ProLong Gold mounting medium containing DAPI (Molecular Probes).
Immunoprecipitation
Cells from nearly confluent flasks were harvested and lysed in RIPA buffer (150 mm NaCl, 1.0% NP-40, 0.5% Na-deoxycholate, 50 mm Tris, pH 8.0) containing a protease inhibitor cocktail (Roche). Cell pellets were vortexed and sonicated briefly to ensure thorough lysing. A total cellular protein of 300 µg in a final volume of 500 µl from each sample was immunoprecipitated as follows. Cell lysate was pre-cleared by incubating for 1 h with protein G–Sepharose at 4°C on a tube rotator. The protein G–Sepharose was removed by a brief spin, 100 µl anti-lamin A/C antibody (Abcam ab8984) was added to the supernatant and incubated overnight at 4°C. Fifty microliters of protein G–Sepharose was added to each sample and incubated for 3 h at 4°C on a tube rotator. After washing four times with RIPA buffer, 50 µl of 2× NuPAGE sample buffer with reducing agent (Invitrogen) was added to the protein G pellet followed by heating to 80°C for 10 min and loading onto a 4–12% Bis–Tris NuPAGE gel (Invitrogen).
Immunoblotting
Proteins were transferred to Immobilon-P membrane (Millipore) using a Trans-Blot semi-dry electrophoretic transfer cell (BioRad) blocked with 5% dry milk in TBST and probed with the lamin A/C specific antibodies, MAB3211 (Chemicon), according to standard protocols.
Fluorography
To determine whether progerin retains the farnesyl group, normal and HGPS fibroblasts were plated into T25 flasks and labeled for 24 h with 125 µCi/ml 3H-mevalonate (Amersham), as previously described (35). Twenty-five micromoles mevinolin (Sigma) was added to the media to inhibit endogenous mevalonate synthesis. Immunoprecipitation was performed as described earlier. Following electrophoresis on a 4–12% Bis–Tris NuPAGE gel (Invitrogen), the gel was fixed in a solution of 10% glacial acetic acid, 30% methanol for 1 h. The gel was impregnated with Enhance (Perkin-Elmer) according to manufacturer's instructions, dried and exposed to film for 4 weeks.
ACKNOWLEDGEMENTS
This work was supported by a grant from the Progeria Research Foundation. We are very grateful to Charles Omer for valuable advice and discussion, and thank Pfizer Inc., for their generous gift of the FTI, PD169541.
Conflict of Interest statement. None declared.
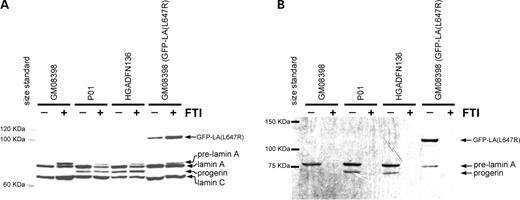
Figure 1. Normal (GM08398) and HGPS (P01 and HGADFN136) fibroblasts were plated in T25 flasks. One flask of each line was labeled with 3H-mevalonate (125 µCi/ml) and mevinolin (25 µm). Cells were lysed in the flask with RIPA and 300 µg of cellular protein was immunoprecipitated with lamin A/C antibodies. Protein levels prior to immunoprecipitation are displayed in (A). Fluorography (B) of the immunoprecipitated proteins shows a unique band, progerin, in the HGPS lines demonstrating that progerin is farnesylated. Addition of 2 µm FTI (+) causes accumulation of pre-lamin A (A) and completely prevents incorporation of the 3H-labeled farnesyl precursor (B). Normal fibroblasts expressing an uncleavable form of lamin A (GFP–LA (L647R)) that remains farnesylated were included as a positive control.
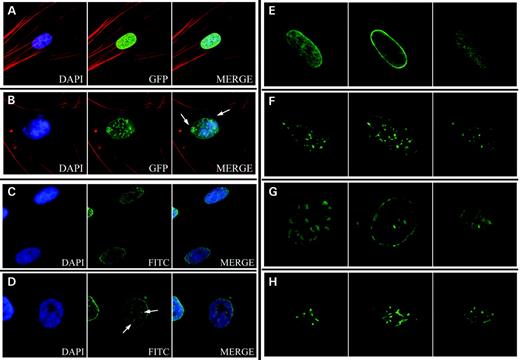
Figure 2. Localization of transiently-expressed GFP–lamin A and GFP–progerin. GFP–lamin A was evenly distributed around the nuclear periphery (A), whereas expression of GFP–progerin formed aggregates at the nuclear periphery and caused blebbing (arrows) (B). Immunocytochemistry using a lamin A-specific antibody to detect localization of endogenous lamin A is shown for normal (C) and HGPS (D) fibroblasts. Lamin A aggregates were observed in HGPS fibroblasts (arrows) (D). Images in (A) through (D) show focal planes at the near side of the nucleus. Signals were obtained with a Zeiss Axiophot Fluorescent microscope with a Sensys CCD camera and Applied Imaging digital imaging capture software. Localization of GFP–lamin A (E and F) and progerin (G and H) was analyzed using a Zeiss LSM 510 confocal microscope mounted on a Zeiss Axiovert 100M inverted microscope. Three focal planes are shown: top, middle and bottom (from left to right). These images show the location of the GFP–progerin aggregates as thicker regions at the nuclear periphery (G). Fibroblasts expressing normal GFP–lamin A have GFP signal around the nuclear periphery (E). Exposure to 100 nm FTI (PD169451) for 72 h caused a redistribution of the GFP–lamin A signal (F) and GFP–progerin (H) to intranuclear structures.
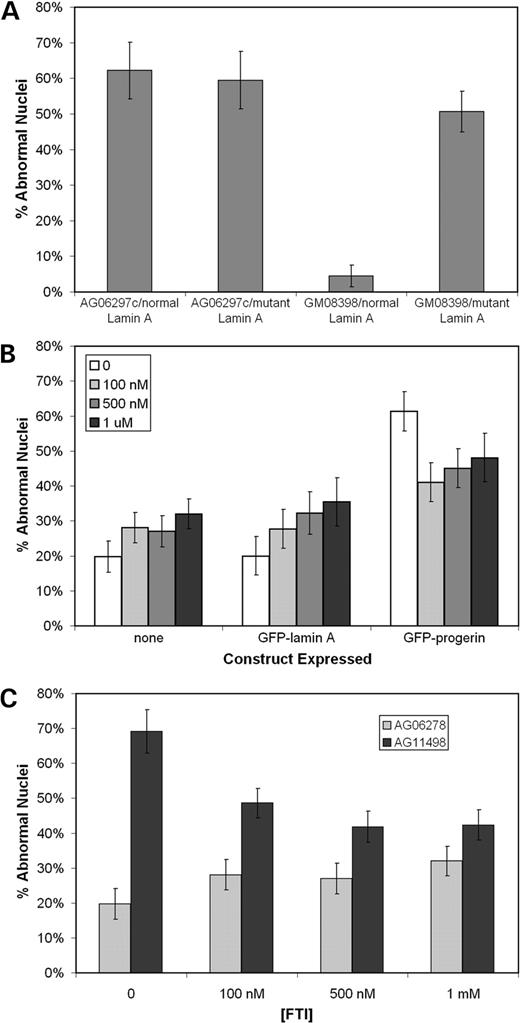
Figure 3. Quantitation of the effects on nuclear morphology of transiently expressing the GFP–lamin A and GFP–progerin constructs in HGPS (AG06297c) and normal (GM08398) fibroblasts (A). These constructs were transiently expressed in normal cells and exposed to various concentrations of FTI for 72 h (B). A significant drop in the percentage of abnormal nuclei was observed in cells expressing GFP–progerin when exposed to FTI for 72 h. However, increasing FTI concentrations caused increased percentage of cells with abnormal nuclear morphology in mock-transfected cells (none) and cells expressing GFP–lamin A. The same concentrations of FTI were applied to normal (AG06278) and HGPS (AG11498) fibroblasts for 72 h (C) and caused a significant drop in the percentage of abnormal nuclei in this HGPS cell line. Analysis of nuclear morphology were performed on blinded samples, independently by two individuals with no significant differences between individuals. At least 50 cells were analyzed per sample.
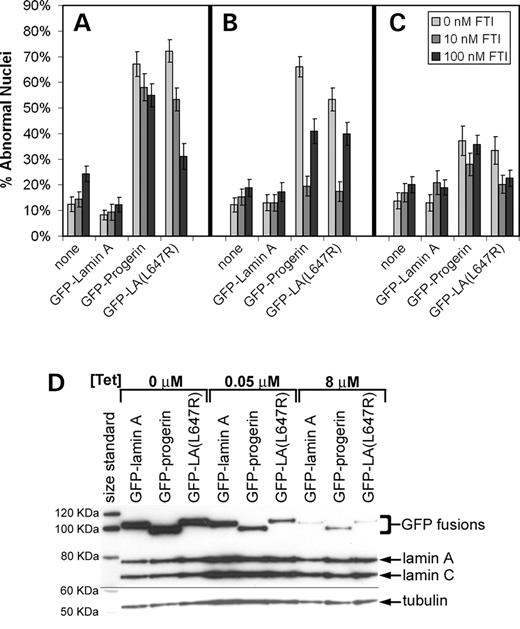
Figure 4. The effect of the FTI, PD169451, on the nuclear morphology of normal fibroblasts (GM08398) stably expressing GFP–lamin A, GFP–progerin and GFP–LA (L647R) using a retroviral Tet-off system (Clontech). Fibroblasts were maintained in medium with 0, 10 or 100 nm FTI for six days. Cultures were plated in chambered microscope slides 24 h prior to fixing with 4% paraformaldehyde for nuclear morphology studies. Cells were maintained in three predetermined tetracycline concentrations, 0 µm for full expression (A), 0.05 µm for intermediate expression levels (B), and 8 µm for very low expression levels (C). Using a Tet-off system allowed for modulation of expression levels with tetracycline concentration as demonstrated by immunoblot of cells from the previous experiment (D). When GFP–progerin and GFP–LA (L647R) mutant was stably expressed in normal fibroblasts at intermediate levels (0.05 µg/ml tetracycline), 10 nm FTI substantially reduced the percent abnormal nuclei by 71% and 67%, respectively (P<0.0001) (B). Analysis of nuclear morphology was performed in a double-blinded fashion with at least 70 cells analyzed per sample.
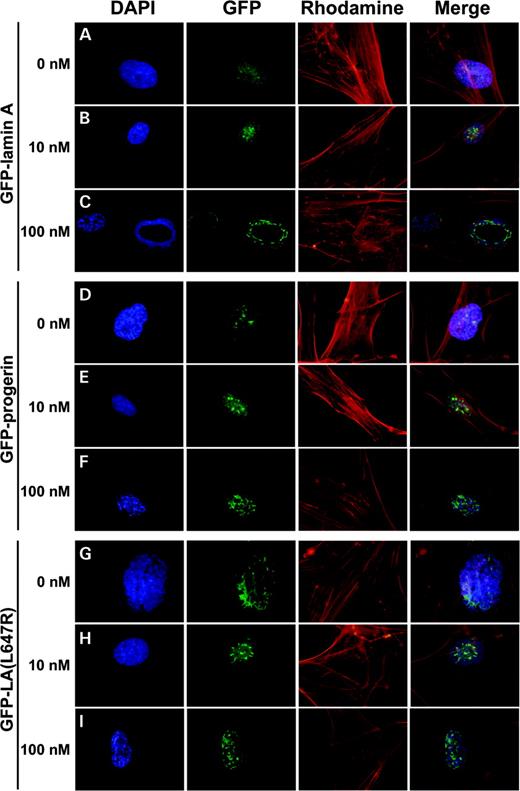
Figure 5. The effect of FTI on the distribution of GFP signal in normal fibroblasts (GM08398) expressing GFP–lamin A (A, B and C), GFP–progerin (D, E and F) and GFP–LA (L647R) (G, H and I) using a retroviral Tet-off system with various FTI concentrations. Fibroblasts were maintained in medium with 0 nm (A, D and G), 10 nm (B, E and H) or 100 nm (C, F and I) FTI for six days. Expression levels of the GFP fusions were maintained at levels similar to endogenous levels of lamin A by the addition of 0.05 mg/ml tetracycline. Signals were visualized with a Zeiss Axiophot Fluorescent microscope and imaging was performed using the Olympus DP70 Digital Camera System.
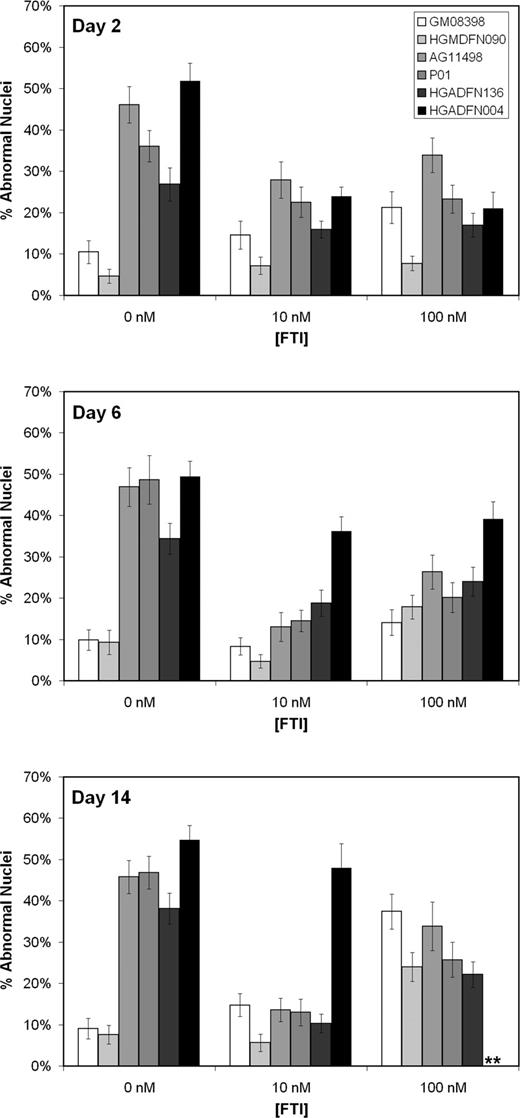
Figure 6. The effect of long-term exposure to low doses of FTI (PD169451) on the nuclear morphology of normal (GM08398 and HGMDFN090) and HGPS (AG11498, P01, HGADFN136 and HGADFN004) fibroblasts. Fibroblasts were maintained in medium with 0, 10 or 100 nm FTI. Cultures were plated in chambered microscope slides 24 h prior to fixing with 4% paraformaldehyde for nuclear morphology studies. After 14 days of exposure to 10 nm FTI, three of the HGPS lines had over a 70% reduction (P<0.0001) in the relative number of abnormal nuclei, reducing the occurrence of abnormal nuclei to that of normal controls. **Too few cells to count. The HGADFN004 line failed to grow in 100 nm FTI to the 14-day time point. Analysis of nuclear morphology was performed in a double-blinded fashion with over 100 cells analyzed per sample.
References
Eriksson, M., Brown, W.T., Gordon, L.B., Glynn, M.W., Singer, J., Scott, L., Erdos, M.R., Robbins, C.M., Moses, T.Y., Berglund, P. et al. (
Gruenbaum, Y., Wilson, K.L., Harel, A., Goldberg, M. and Cohen, M. (
Harborth, J., Elbashir, S.M., Bechert, K., Tuschl, T. and Weber, K. (
Rober, R.A., Weber, K. and Osborn, M. (
Zhang, F.L. and Casey, P.J. (
Weber, K., Plessmann, U. and Traub, P. (
Kilic, F., Dalton, M.B., Burrell, S.K., Mayer, J.P., Patterson, S.D. and Sinensky, M. (
Corrigan, D.P., Kuszczak, D., Rusinol, A.E., Thewke, D.P., Hrycyna, C.A., Michaelis, S. and Sinensky, M.S. (
Lutz, R.J., Trujillo, M.A., Denham, K.S., Wenger, L. and Sinensky, M. (
Goldman, R.D., Shumaker, D.K., Erdos, M.R., Eriksson, M., Goldman, A.E., Gordon, L.B., Gruenbaum, Y., Khuon, S., Mendez, M., Varga, R. et al. (
Agarwal, A.K., Fryns, J.P., Auchus, R.J. and Garg, A. (
Pendas, A.M., Zhou, Z., Cadinanos, J., Freije, J.M., Wang, J., Hultenby, K., Astudillo, A., Wernerson, A., Rodriguez, F., Tryggvason, K. et al. (
Bergo, M.O., Gavino, B., Ross, J., Schmidt, W.K., Hong, C., Kendall, L.V., Mohr, A., Meta, M., Genant, H., Jiang, Y. et al. (
Dyer, J.A., Kill, I.R., Pugh, G., Quinlan, R.A., Lane, E.B. and Hutchison, C.J. (
Burke, B. and Stewart, C.L. (
Shackleton, S., Lloyd, D.J., Jackson, S.N., Evans, R., Niermeijer, M.F., Singh, B.M., Schmidt, H., Brabant, G., Kumar, S., Durrington, P.N. et al. (
Speckman, R.A., Garg, A., Du, F., Bennett, L., Veile, R., Arioglu, E., Taylor, S.I., Lovett, M. and Bowcock, A.M. (
Novelli, G., Muchir, A., Sangiuolo, F., Helbling-Leclerc, A., D'Apice, M.R., Massart, C., Capon, F., Sbraccia, P., Federici, M., Lauro, R. et al. (
Marshall, C.J. (
Barton, R.M. and Worman, H.J. (
Dahl, K.N., Kahn, S.M., Wilson, K.L. and Discher, D.E. (
Ly, D.H., Lockhart, D.J., Lerner, R.A. and Schultz, P.G. (
Park, W.Y., Hwang, C.I., Kang, M.J., Seo, J.Y., Chung, J.H., Kim, Y.S., Lee, J.H., Kim, H., Kim, K.A., Yoo, H.J. et al. (
Ozaki, T., Saijo, M., Murakami, K., Enomoto, H., Taya, Y. and Sakiyama, S. (
Lloyd, D.J., Trembath, R.C. and Shackleton, S. (
Spann, T.P., Goldman, A.E., Wang, C., Huang, S. and Goldman, R.D. (
Csoka, A.B., English, S.B., Simkevich, C.P., Ginzinger, D.G., Butte, A.J., Schatten, G.P., Rothman, F.G. and Sedivy, J.M. (
Peter, M. and Nigg, E.A. (
Muller, P.R., Meier, R., Hirt, A., Bodmer, J.J., Janic, D., Leibundgut, K., Luthy, A.R. and Wagner, H.P. (
Fong, L.G., Ng, J.K., Meta, M., Cote, N., Yang, S.H., Stewart, C.L., Sullivan, T., Burghardt, A., Majumdar, S., Reue, K. et al. (
Sebolt-Leopold, J.S., Leonard, D.M. and Leopold, W.R. (
Caraglia, M., Budillon, A., Tagliaferri, P., Marra, M., Abbruzzese, A. and Caponigro, F. (
Baskaran, N., Kandpal, R.P., Bhargava, A.K., Glynn, M.W., Bale, A. and Weissman, S.M. (
Hennekes, H. and Nigg, E.A. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}