



Abstract
The XRCC3 variant T241M, but not D213N, has been reported to be associated with an increased risk of some cancers. XRCC3 is one out of five RAD51 paralogues and is involved in homologous recombination, as are the BRCA1 and BRCA2 proteins. However, in contrast to mutations in BRCA1 and BRCA2, the XRCC3T241M protein is proficient in homologous recombination and reverts sensitivity to mitomycin C found in XRCC3-deficient cells, whereas XRCC3D213N is defective in homologous recombination. Here, we report that both the XRCC3 D213N and T241M alleles are associated with an increase in centrosome number and binucleated cells. However, only the D213N allele gives an increase in spontaneous levels of apoptosis. We suggest that the inability of XRCC3 T241M to apoptotically eliminate aberrant cells with mitotic defects could increase cancer susceptibility in individuals carrying this variant. In contrast, cells carrying the XRCC3 D213N variant are able to eliminate aberrant cells by apoptosis, and consistent with this observation, this variant does not seem to be associated with cancer susceptibility.
INTRODUCTION
The individual risk of cancer depends on inherited as well as environmental factors. Mutations in certain high penetrance genes such as BRCA1 and BRCA2 greatly increase the risk of breast and ovarian cancer. It is generally believed that the tumour suppressor function of the BRCA1 and BRCA2 proteins is related to their role in DNA homologous recombination (1). Loss of the error-free homologous recombination repair pathway may result in the use of error prone repair mechanisms (2), resulting in translocations and genetic instability in BRCA1/2 deficient tumours. Independently of their role in recombination repair, BRCA1 and BRCA2 are also involved in maintaining the correct number of centrosomes (3,4). This role is supported by the observations that BRCA1 binds to and causes ubiquitination of γ-tubulin (5,6), and increased cytokinesis failure is observed in cells with BRCA2 defect (7). Loss of the centrosome maintenance function could also contribute to aneuploidy, by causing aberrant segregations at mitosis. Like BRCA1 and BRCA2, the RAD51 paralogue XRCC3 also plays a central role in homologous recombination and is important for maintenance of the correct centrosome number in mammalian cells (8,9).
We previously identified a rare variant of XRCC3 that contains a substitution of asparagine for aspartic acid at amino acid position 213, in the ATP-binding Walker box domain. The D213N variant is defective in recombination repair, shown by its inability to correct the mitomycin C (MMC)-sensitive phenotype found in XRCC3-deficient cells (10). However, in contrast to the situation with BRCA1 and BRCA2 mutations, so far there is no evidence that the D213N variant is related to cancer susceptibility (10). A common variant of XRCC3, comprising a threonine to methionine substitution at amino acid position 241, has been proposed as a low-penetrance cancer allele associated with breast cancer (11), lung cancer (12), acute myeloid leukaemia (13) and a reduced risk of cancer in the upper aerodigestive tract (14). However, there are also reports that do not find a link between XRCC3 T241M and cancer (15,16) (reviewed in 17). The functional basis for any possible association of the XRCC3 variant T241M with an increased risk of cancer has not been identified. The XRCC3T241M protein is proficient in homologous recombination repair of a DNA double-strand-break (18) and complements the sensitivity of XRCC3-deficient cells to MMC as efficiently as the wild-type protein (10).
In this study, we further investigate the phenotypes of the D213N and T241M variants that could relate to cancer susceptibility. We make use of the Chinese hamster ovary (CHO) cell line irs1SF, which does not express any detectable XRCC3 protein. The D213N and T241M cell lines are stable transfectants of irs1SF, which express the D213N or T241M variant forms of the human XRCC3 gene under the control of the human cytomegalovirus gene promoter. We compare centrosome number, the frequency of binucleated cells, the rate of spontaneous apoptosis in the parental cell line AA8 (containing the endogenous wild-type XRCC3 gene), the XRCC3-deficient cells irs1SF and the D213N- and T241M-transfected cells. We find an increase in bi- and multinucleated cells in irs1SF, D213N and T241M. However, apoptosis was increased only in irs1SF and D213N. We speculate that the reason that the D213N variant is not associated with increased cancer risk is explained by the high background apoptotic levels, killing aberrant cells. In contrast, the T241M cells show high levels of binucleated cells without the corresponding increase in apoptosis. This might allow aberrant cells to survive and thus could increase cancer risk.
RESULTS
Increased centrosome numbers in XRCC3-deficient cells
XRCC3 defective cells have an increased number of centrosomes, resulting in missegregation of chromosomes (9), which is an important driving force in the aetiology of cancer (19). Although the XRCC3T241M protein is functional for homologous recombination, it may still be defective in maintaining the correct number of centrosomes. To test this, we compared centrosome number in a series of cell lines derived by transfection from the XRCC3-deficient irs1SF cell line (20), which itself originated from the wild-type AA8 CHO cell line (21). The cell lines comprised irs1SF complemented with the human XRCC3 (WT), XRCC3 T241M or XRCC3 D213N cDNAs, expressed from the pcDNA3.1 vector stably integrated into the genome of irs1SF cells (10). At least two independent transfected clones were examined in each case in order to avoid clonogenic variability. To identify the expressed XRCC3 protein, a western blot was carried out (Fig. 1). The irs1SF cell line shows no detectable XRCC3 protein, whereas all irs1SF clones expressing hXRCC3 variants produce an XRCC3 protein, showing that the human XRCC3 is expressed in every clone.
Centrosomes were identified by immunofluorescence labelling with γ-tubulin, and the number of centrosomes was counted in mitotic and interphase cells. In agreement with the published data, we found that 20% of the irs1SF mitotic cells had more than two centrosomes compared with 5% in AA8 cells (P<0.001) (Fig. 2A) (9). The irs1SF cells complemented with hXRCC3wt (WT) also had significantly higher numbers of centrosomes when compared with AA8 cells (12%, P<0.001), indicating that complementation with the human XRCC3 gene does not fully compensate for the hamster cell defect in this regard. However, significantly more irs1SF cells had more than two centrosomes than WT cells (P<0.01). We also found that 28% of the D213N cells and 16% of the T241M cells had increased numbers of centrosomes, which is more than the 5% AA8 cells with increased numbers of centrosomes (statistically significant P<0.001 in t-test). Although hXRCC3T241M expressing cells have increased numbers of centrosomes when compared with AA8 cells, there was no statistically significant difference when comparing hXRCC3T241M with hXRCC3WT expressing cells (P=0.68 in t-test) (Fig. 2A).
The majority of interphase cells had one centrosome and we found no significant increases in centrosome numbers in irs1SF, T241M or WT interphase cells when compared with AA8 (Fig. 2B). However, significantly higher numbers of D213N cells have more than two centrosomes when compared with AA8 (P < 0.001) (Fig. 2B).
An increased number of centrosomes in mitosis may lead to the formation of multipolar spindles, aberrant chromosome segregation and aneuploidy. Hence, it is important to determine whether the centrosomes are functional in these cells. We found that most centrosomes co-localized with microtubules (Fig. 3), indicating that they are likely to be functional. Also, we observed that the majority of the centrosomes share the same size and shape as the spherical ones observed in the parental AA8 cells (Fig. 3). Interestingly, we found several cells with a high number of centrosomes (>20) (Fig. 3C). This indicates a severe defect in maintaining the correct centrosome numbers in XRCC3-deficient cells.
Increased frequency of binucleated cells in XRCC3 T241M expressing cells
There are several routes for centrosome amplification (see Discussion). One pathway is coupled to cytokinesis because a cleavage failure would yield binucleated cells with twice the normal amount of centrosomes (22). Cells that have gone through endoreduplication also have increased numbers of centrosomes. If these cells enter mitosis, the result would be cells with multipolar spindles and aneuploid cells, if viable. Another possibility is that these cells also face problems in cytokinesis. This would probably also result in multinucleated cells with aberrant centrosome numbers. It has been shown that BRCA2-deficient cells have an impaired cytokinesis (7), which could also be the case with XRCC3-deficient cells because XRCC3, as BRCA2, is implicated in homologous recombination. To test this, we studied the frequency of binucleated cells among the XRCC3 variants. The number of nuclei in the different cell types was scored: two nuclei of equal size, of unequal size and multinucleated cells (Fig. 4A). It was found that the XRCC3 variant cell lines had significantly higher numbers of binucleated cells than control AA8 (3.6%), D213N (8%, P<0.001) and T241M (6%, P<0.05) (Fig. 4B). Also, the level of cells with two nuclei of unequal size and cells with more than two nuclei was higher in D213N (11%, P<0.01) and T241M (9%, P<0.001) when compared with AA8 (4%) or WT (4%, P<0.05 in t-test) (Fig. 4).
The XRCC3 variant T241M suppresses the high level of apoptosis in XRCC3-deficient cells
One role of apoptosis is to destroy aberrant cells to prevent them becoming cancerous. It has been shown that spontaneous levels of apoptosis are increased in XRCC3-deficient irs1SF cells, most likely as a result of unrepaired endogenous DNA damage (23). We compared the levels of apoptosis between the XRCC3 variants D213N and T241M. We used the Annexin-V assay to detect early apoptotic cells in which the membrane phospholipid phosphatidylserine has been translocated from the inner to the outer leaflet of the plasma membrane in cells entering apoptosis (24). In agreement with the previous results, we found that the level of apoptosis was higher in XRCC3-deficient irs1SF and D213N cells when compared with wild-type AA8 cells (P<0.05) (Fig. 5A) (23). Expression of the wild-type XRCC3 in WT cells resulted in reduced apoptosis to levels not statistically different to those found in wild-type AA8 cells (P=0.29). Expression of the hXRCC3T241M variant reduced the apoptotic level to the same extent as when expressing hXRCC3WT and again to a level of apoptosis that is not statistically different when compared with control AA8 cells (P=0.14). Both the wild-type and the T241M expressing cells reduced the level of apoptosis when compared with D213N expressing cells (P<0.05).
As a second assay for apoptosis, we scored apoptotic bodies in the immunofluorescently labelled cells (Fig. 5B). In agreement with the Annexin-V results, we found that the D213N variant has a significantly higher frequency of apoptosis when compared with T241M and the parental AA8 cells (P<0.05) (Fig. 5C). In contrast, there was no significant difference in the level of apoptosis between XRCC3 T241M and WT expressing cells.
The amount of apoptosis was also scored in a third assay, investigating the percent of cells in the sub-G1 fraction. We found that the D213N and irs1SF cells contained a more pronounced sub-G1 peak when compared with the wild-type control or T241M expressing cells (Fig. 6). This indicates more apoptosis in irs1SF or D213N expressing cells, in agreement with the two other apoptosis assays.
DISCUSSION
Some reports have associated the XRCC3 variant T241M with cancer, but the evidence so far indicates that D213N is not a cancer susceptibility allele (10,18). This is not easily explained because the T241M is functional for homologous recombination (18), whereas the D213N is likely to be deficient in homologous recombination (10). Here, we assessed XRCC3 function in respect of retaining the correct centrosome number and genetic stability. We used the XRCC3-deficient cell line irs1SF and complemented the defect by expression of either hXRCC3 T241M, hXRCC3 D213N or the wild-type hXRCC3. We found that all the XRCC3-deficient and complemented cells had significantly more cells with more than two centrosomes in mitosis than AA8 cells. Consistent with previous reports, the human XRCC3 gene does not fully complement the hamster protein (20,23,25), and thus hXRCC3WT expressing cells also have raised numbers of centrosomes when compared with AA8. There was no increase in centrosome number in interphase cells except in the hXRCC3 D213N expressing cells. This increase may be caused by the overexpression of the non-functional hXRCC3 D213N influencing the accessibility of other proteins that might normally compensate for the XRCC3 defect in interphase cells, producing a dominant negative phenotype. This would not be a problem in the irs1SF cells, as no XRCC3 protein is detected (Fig. 1). One caveat to our conclusions is that some effects observed may be due to the use of the human gene in a hamster cell line.
It is not clear as to why there are an increased number of centrosomes in XRCC3-defective cells. Centrosome amplification can arise via five different routes, as previously summarized (26,27). In the first pathway, endoreduplication of centrosomes is caused by both DNA and centrosomes continuing the cell cycle without entering mitosis (27). A second possibility is that the increase in centrosome number arises from fragmentation of centrosomes, rather than duplication (28). Thirdly, aberrant centrosome numbers could be a result of failure of cytokinesis (29). This would lead to binucleated cells with increased numbers of centrosomes and polyploidy. In the fourth route, the centrosome cycle is uncoupled from DNA synthesis and mitosis. This has especially been observed in cells arrested in the G1/S phases of the cell cycle by hydroxyurea or aphidicholin. The fifth and last pathway suggests that centrosome amplification takes place in G2 during a G2 block induced by DNA damage (26). It is also a possibility that cells with aberrant numbers of centrosomes arise from mixture of the various models described above.
It has been shown by others that BRCA2 inactivation leads to an increased number of centrosomes and hampering of cytokinesis in mouse embryonic fibroblasts and HeLa cells (7). This resulted in high frequencies of binucleated cells (7). In addition, BRCA2 was found to be localized to the cytokinetic midbody. Here, we wanted to study whether cells with compromised XRCC3 function also showed the same aberrations as BRCA2 mutants concerning cytokinesis, by scoring cells with two equally large nuclei. It was found that D213N and T241M had higher frequencies of binucleated cells than control AA8 and WT cells, indicating problems with cleavage. The increased numbers of multinucleated cells in the XRCC3 variants could be explained either by the binucleated population progressing into another mitosis or by the amplification of centrosomes resulting in multipolar mitotic spindles and inhibited cleavage. A ploidy checkpoint could also be involved because the difference in frequency of the multinuclear relative to binucleated cells is greater for the parental AA8 and WT cells than the other cell types.
The mitotic defect in XRCC3-deficient cells found here is in line with an earlier report showing that human HCT116 cells, carrying a defect in XRCC3, demonstrate endoreduplication (27). Although the XRCC3-defective HCT116 cells fail to form normal RAD51 foci in response to damage, they are far from as sensitive to MMC as the irs1SF cells. One explanation for the different phenotypes could be that the HCT116 cells are known to have functional mitotic and p53-dependent checkpoints (30), in contrast to the CHO cells used here. However, the lack of profound MMC sensitivity in HCT116 XRCC3 knockout cells, when compared with irs1SF cells, is most likely to be explained by the fact that the parental HCT116 cell line is defective for homologous recombination (31), due to a mutation in the MRE11 gene (32). Thus, knockout of XRCC3 in a cell line already defective in homologous recombination does not provide a suitable model to study the role of XRCC3 in homologous recombination. Nevertheless, although the XRCC3T241M protein complements the additional recombination defect in HCT116 cells, it was reported not to complement endoreduplication, which may result in increased number of centrosomes (27). Our results that the XRCC3 T241M does not complement the XRCC3 defect that results in binucleated cells are in line with the results found in the HCT116 XRCC3 knockout cells.
An interesting observation in Schizosaccharomyces pombe is that sites of initiation of replication and homologous recombination hot spots are in close vicinity to each other (33). Furthermore, it has been proposed that XRCC3 is associated with replication protein A (RPA) and Rad52 in mammalian cells and that endoreduplication caused by XRCC3 defects could be a result of inhibited RPA function (27). Endoreduplication could play a role in the centrosome amplification in the variant cells observed in this study; however, it does not alone explain the increase in centrosome numbers observed here. In any case, the previous results on a mitotic defect of the XRCC3 T241M-deficient cells are consistent with our current observation.
It is not clear as to why the XRCC3 T241M, and not D213N, is linked with a possible increased cancer risk. One reason could be that the function of XRCC3 in homologous recombination is vital to avoid DNA damage that channel cells into apoptosis. It is well known that BRCA2−/− knockout mice die from apoptosis during embryogenesis and that this is relieved by an additional knockout of p53 (34,35). Thus, an additional defect in a gate-keeping gene (e.g. p53) to avoid apoptosis is often required for tumour development in cells with a defective caretaking gene (e.g. BRCA2) (36). In line with this, we found a high level of spontaneous apoptosis in irs1SF and D213N cells (Fig. 5). In contrast, the lower spontaneous apoptotic rates in the XRCC3 T241M mutant can be explained by a proficiency in recombination that would result in a low level of DNA damage following replication. In addition, we show that XRCC3 T241M still has a defect in cytokinesis leading to binucleated cells. We suggest that inability of the XRCC3 T241M allele to complement the centrosome amplification defect (Fig. 4) (27) and decreased apoptotic rates (Fig. 5) provides a combination that may result in aberrant cells not entering apoptosis. This may cause genetic instability and lead to an increased susceptibility to various cancers.
MATERIALS AND METHODS
Cell lines
The irs1SF cell line is an XRCC3-deficient derivative of the AA8 CHO cell line (20,21). The D213N, T241M and WT stable cell lines were derived from irs1SF by transfection with the corresponding variant forms of the human XRCC3 gene, expressed from the pcDNA3.1 vector (10) (Table 1). At least two individual clones were used in all the assays and the results pooled to avoid clonogenic variation. All cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 9% fetal calf serum, penicillin–streptomycin (90 U/ml; DMEM) at 37°C under an atmosphere containing 5% CO2.
Antibodies
Primary antibodies used were monoclonal anti-α-tubulin IgG N356 (Amersham) and rabbit anti-γ-tubulin IgG (Sigma). Secondary antibodies used were Alexa 488-conjugated donkey-anti mouse IgG (Molecular Probes) and Alexa 555-conjugated donkey anti rabbit IgG (Molecular Probes).
Immunofluorescence
Cells were seeded on cover slips and grown for 48 h in DMEM. The medium was removed and the cover slips were rinsed once in 37°C PHEM buffer (1 mm MgCl2.6H2O, 10 mm EGTA, 60 mm PIPES, 25 mm HEPES, pH 6.9) and fixed in 3.7% paraformaldehyde in PHEM for 15 min in room temperature. The cells were then rinsed and permeabilized with 0.25% Triton X-100 in phosphate-buffered saline (PBS) and incubated for 5 min before adding ice-cold methanol for 10 min at −20°C. Prior to incubation with the primary antibody, the cells were rinsed three times with PBS and treated for 30 min with 20% fetal bovine serum in 37°C. After rinsing once in PBS containing 0.05% Tween-20, the cover slips were incubated with anti-α-tubulin (Amersham), diluted 1:1000 in 3% bovine serum albumin in PBS at 4°C over night. Thereafter, the cells were briefly rinsed in 0.05% Tween-20 in PBS and incubated for 60 min with the primary anti-γ-tubulin antibody (Sigma) in 37°C. After rinsing three times in 0.05% Tween-20 in PBS, the cover slips were incubated with the secondary antibodies for 45 min at room temperature in the dark. The cells were rinsed three times in 0.05% Tween-20 in PBS and DNA was counterstained with 1 µg/ml TO PRO (Molecular Probes) for 30 min before the cover slips were mounted with Slow Fade (Molecular Probes). Images were obtained with a Zeiss LSM 510 inverted confocal microscope with a Planapochromat 63/NA 1.4 oil immersion objective and excitation wavelengths of 488, 546 and 630 nm. Through-focus maximum projection images were acquired from optical sections 0.5 µm apart and with a section thickness of 1.0 µm. Images were processed with Adobe PhotoShop (Abacus Inc.).
Scoring of centrosomes
Series for scoring of cells in a Zeiss fluorescence microscope were treated as above but with the following alterations: the culture medium was removed and the cells were fixated in paraformaldehyde in PBS and incubated on ice for 15 min before being treated with 0.25% Triton X-100 in PBS and then in methanol. These cells were incubated for 60 min with the primary anti-γ-tubulin antibody (Sigma) in 37°C and rinsed before addition of the secondary antibody. DNA was counterstained with DAPI for 10 min at room temperature before being mounted on glass slides as above. In this study, there were two separate clones of each variant cell line, D213N and T241M and wild-type. The experiments were run with the separate clones. As there was no significant difference between the clones, the results were pooled together. The frequencies of centrosomes in the different cell lines were determined by scoring 50 mitoses and 300 interphase cells per cover slip in encoded series. The number of centrosomes per cell was calculated as the number of approximately 1 µm large dots at least 2 µm apart (distance between centre to centre of the centrosomes).
Scoring apoptosis
These samples used for centrosome counting were also used for scoring of apoptosis. Five hundred cells per encoded slide were counted and cells were classified as apoptotic when apoptotic bodies were visible.
Apoptosis was also measured using the Annexin-V assay. For this assay, 5×105 cells were seeded and cultured overnight. The cells were pelleted by centrifugation and resuspended for apoptosis analysis with FITC-conjugated Annexin-V and propidium iodine (PI) (ApoTarget, Biosource International) according to manufacturer's protocol. Samples were analysed by flow cytometry (Becton-Dickenson FACSort, 488 nm laser), and percentage of apoptotic cells was determined by the fraction of live cells (PI-negative) bound with FITC-conjugated Annexin-V.
We also determined the cell cycle distribution as an additional method to determine apoptosis. For this assay, 1×106 cells were washed, recovered by centrifugation, fixed with 70% ethanol and incubated at 4°C overnight. The cells were then washed in PBS three times, resuspended in PI (50 µg/ml) and RNase (0.1 U/ml) and incubated at 4°C overnight. Samples were analysed using flow cytometry at 488 nm laser, and the level of apoptosis was determined as the fraction of cells in the sub-G1 peak.
Scoring of binucleated cells
Cells were grown as described above, rinsed with Hank's balanced salt solution (Gibco), trypsinized and fixed in methanol–acetic acid (3:1) in centrifuge tubes. The tubes were centrifuged for 5 min at 190g, the supernatant was discarded and new methanol–acetic acid was added. This was repeated twice before the tubes were refrigerated for at least 12 h. Glass slides were prepared in 99% ethanol and rinsed with water, then 10 µl of the cell suspensions were added and the slides allowed to dry over night. The cells were stained for 5 min in May–Grunwald solution and in 4% Gurr's Giemsa for 4 min, rinsed in water, dried and mounted with DPX before scoring in a Leitz microscope. Five hundred cells per encoded slide were counted and divided into four categories: cells with one nucleus, binucleated cells with nuclei of similar size, binucleated cells with one nucleus being smaller and cells with more than two nuclei. Micronuclei were not counted (counted nuclei were not less than one-fourth of the size of the normal nucleus diameter).
Western blotting
Transfectants for CHO cell lines irs1SF and AA8 were cultured, washed, trypsinized and pelleted by centrifugation. Cells were mixed with 200 µl cold RIPA buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS in PBS) and 2 mM phenyl methyl sulfonyl fluoride. The mixture was then centrifuged at 14 000g for 10 min at 4°C, protein (supernatant) was recovered, mixed with electrophoresis buffer, aliquoted and stored at −70°C until use. The protein concentration was determined using Bradford reagent. Equal amounts of protein from irs1SF transfectants, AA8 and human HEK293 cell lines were heated to 100°C and subjected to electrophoresis on a 10% SDS–PAGE gel at 160 V for 1.4 h, along with a high molecular weight protein marker (BioRad). The gel was placed in cold transfer buffer (20 mm Tris–base, 0.2 m glycine, 200 ml methanol in water) for 10 min, and protein transfer onto PVDF membrane was performed using a Biorad Trans Blot Semi-dry Transfer Cell at 10 V for 30 min. The membrane was incubated in blocking solution overnight, washed with PBS-T three times and incubated with monoclonal mouse anti-XRCC3 primary antibody (Abcam®, 1:200 dilution in 3% milk in PBS-T) overnight. The membrane was washed three times and incubated with sheep polyclonal anti-mouse antibody (Abcam) for 1 h. Actin was visualized by polyclonal rabbit anti-actin primary antibody (Sigma, 1:2500 dilution in 3% milk in PBS-T) overnight. The membrane was washed three times and incubated with polyclonal anti-rabbit HRP antibody (Cell Signaling) for 1 h. The membrane was washed in PBS-T three times and the XRCC3 protein was detected using ECL detection reagent (Amersham Pharmacia Biotech) according to manufacturer's protocol, followed by autoradiography.
Statistical analysis
All P-values were calculated using two-sided Student's t-test.
ACKNOWLEDGEMENTS
We thank the Swedish Cancer Society, the Swedish Children's Cancer Foundation, the Swedish Research Council, the Swedish Pain Relief Foundation, the Jeansson's Foundation, the British Breast Cancer Campaign and Yorkshire Cancer Research for supporting this work financially.
Conflict of Interest statement. None declared.
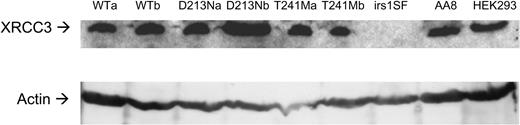
Figure 1. Western blot to detect XRCC3 protein and actin in transfected cell lines. The AA8 hamster and HEK293 human cell line were controls for the hamster and human XRCC3 proteins, respectively.
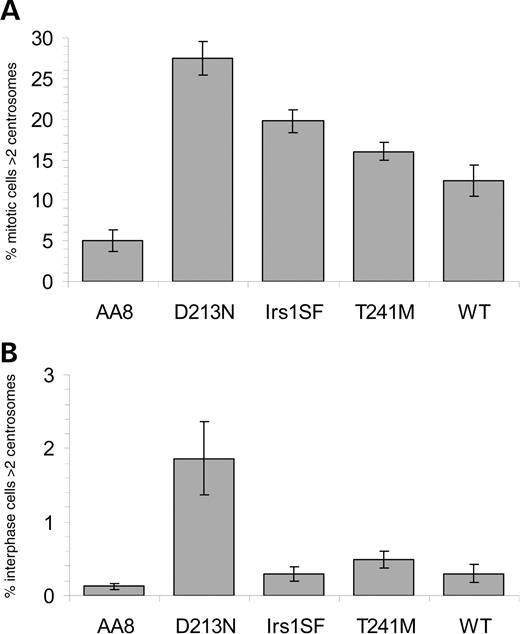
Figure 2. XRCC3 deficient and variant cells have significantly higher frequency of mitotic cells with increased centrosome numbers compared with parental cells. (A) Percentage mitotic cells with more than two centrosomes. (B) Percentage interphase cells with more than two centrosomes. The average (columns) and standard errors (bars) of eight to 10 experiments are depicted.
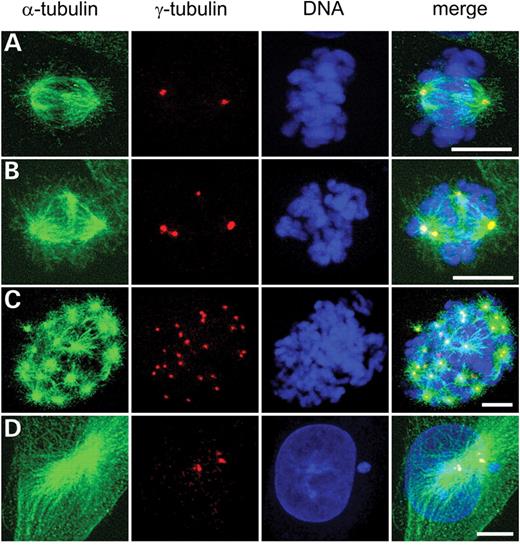
Figure 3. Aberrant mitotic spindles in XRCC3-deficient irs1SF cells. DNA (blue), centrosomes (red) and microtubules (green). (A) Normal mitotic spindle with two centrosomes. (B) Tetrapolar mitotic spindle. (C) Multipolar spindle with >20 active centrosomes. (D) Interphase cell with three centrosomes. Bar=10 µm.
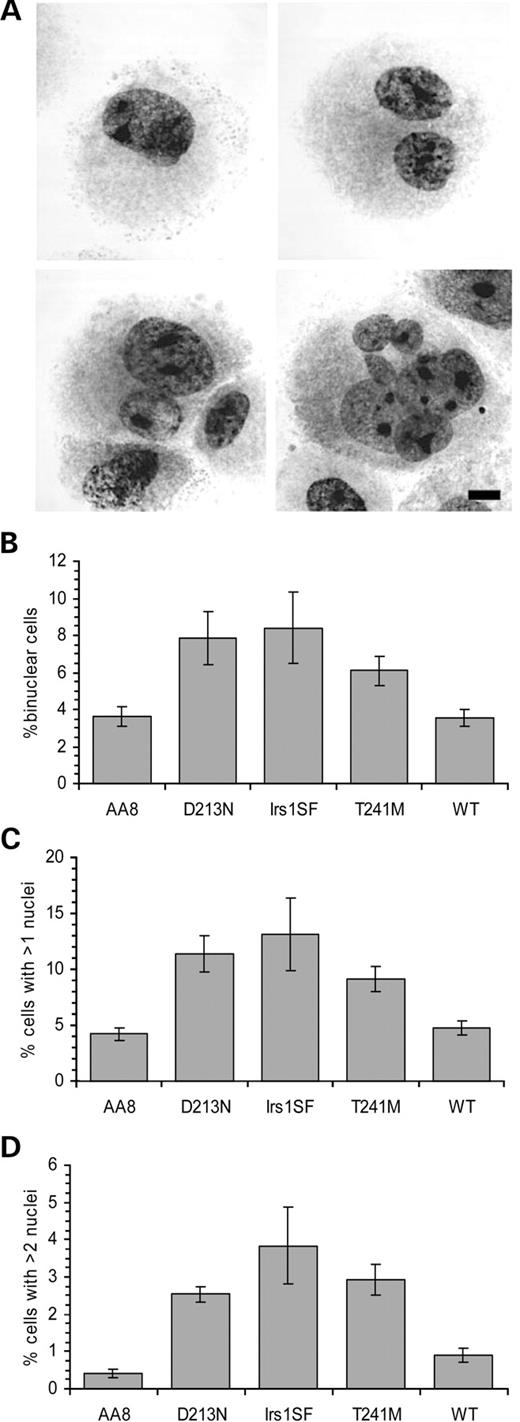
Figure 4. XRCC3 variant cells have more binucleated cells than controls. (A) Top left: normal, mononucleated cell. Top right: binucleated cell with equally sized nuclei. Bottom left: binuceated cell with unequal sized nuclei. Bottom right: multinucleated cell. (B) Percentage binucleated cells with equally sized nuclei, indicating cells with cleavage failure. (C) Percentage of cells that carry more than one nuclei regardless of size. (D) Percentage multinucleated cells, indicating cells that have gone through a mitosis with a multipolar spindle. The average (columns) and standard errors (bars) of six experiments are depicted.
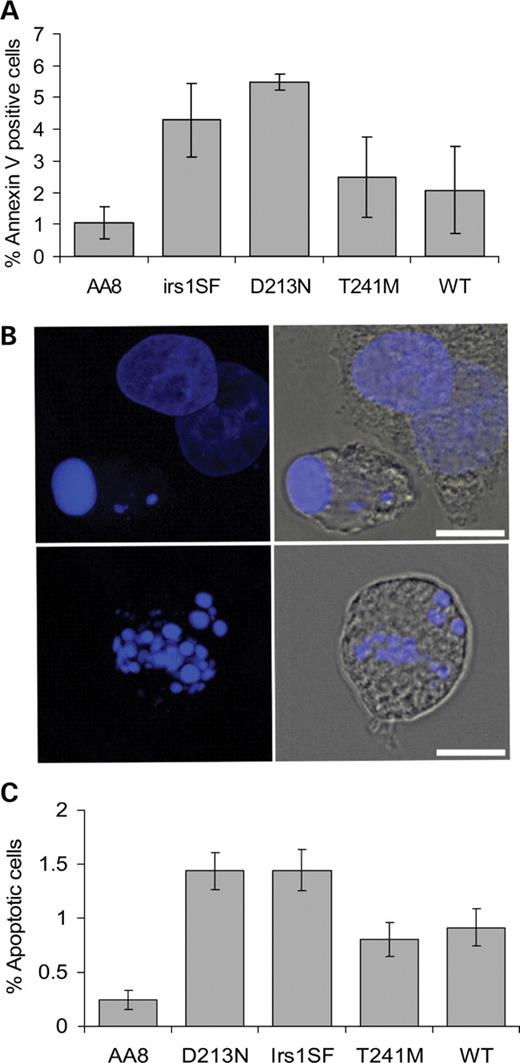
Figure 5. XRCC3 T241M has increased apoptotic level when compared with parental AA8 cells, but lower level than XRCC3 D213 N. (A) Apoptosis was assessed by Annexin-V. The means of apoptotic cells (columns) and standard deviations (error bars) of four to six experiments are shown. (B) Appearance of apoptotic cells when stained with DAPI. Top and bottom left: apoptotic bodies shown in blue. Top and bottom right: the cytoplasm (grey) surrounding the apoptotic bodies. Bars=10 µm. (C) Percentage apoptotic cells. The average (columns) and standard errors (bars) of nine to 16 experiments are depicted.
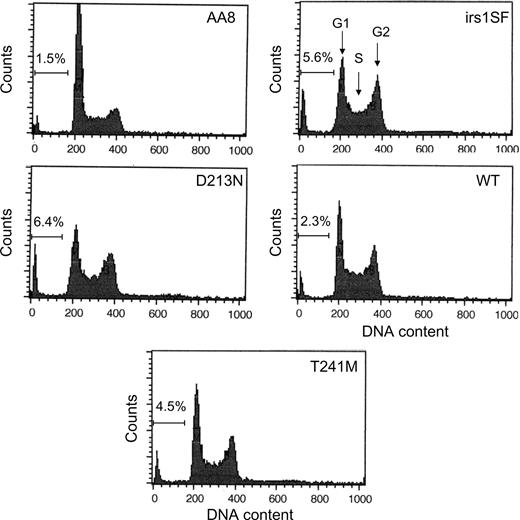
Figure 6. Cell cycle profiles in wild-type and XRCC3-deficient cells. The DNA content in fixed cells was analysed with PI. An increased population of sub-G1 cells in irs1SF and hXRCC3 D213N expressing irs1SF cells indicates an increase in apoptotic cells.
Genotype and origin of Chinese hamster cell lines used in this study
| Cell line | Genotype | Defect /modification | Origin | Reference |
|---|---|---|---|---|
| D213 N | XRCC3− | irs1SF complemented with the hXRCC3 D213 N gene | irs1SF | (10) |
| T241M | XRCC3+ | irs1SF complemented with the hXRCC3 T241M gene | irs1SF | (10) |
| irs1SF | XRCC3− | XRCC3−, deficient in homologous recombination | AA8 | (21) |
| WT | XRCC3+ | irs1SF complemented with the hXRCC3 gene | irs1SF | (10) |
| AA8 | wt | Wild-type Chinese hamster | Ovary | (21) |
| Cell line | Genotype | Defect /modification | Origin | Reference |
|---|---|---|---|---|
| D213 N | XRCC3− | irs1SF complemented with the hXRCC3 D213 N gene | irs1SF | (10) |
| T241M | XRCC3+ | irs1SF complemented with the hXRCC3 T241M gene | irs1SF | (10) |
| irs1SF | XRCC3− | XRCC3−, deficient in homologous recombination | AA8 | (21) |
| WT | XRCC3+ | irs1SF complemented with the hXRCC3 gene | irs1SF | (10) |
| AA8 | wt | Wild-type Chinese hamster | Ovary | (21) |
Genotype and origin of Chinese hamster cell lines used in this study
| Cell line | Genotype | Defect /modification | Origin | Reference |
|---|---|---|---|---|
| D213 N | XRCC3− | irs1SF complemented with the hXRCC3 D213 N gene | irs1SF | (10) |
| T241M | XRCC3+ | irs1SF complemented with the hXRCC3 T241M gene | irs1SF | (10) |
| irs1SF | XRCC3− | XRCC3−, deficient in homologous recombination | AA8 | (21) |
| WT | XRCC3+ | irs1SF complemented with the hXRCC3 gene | irs1SF | (10) |
| AA8 | wt | Wild-type Chinese hamster | Ovary | (21) |
| Cell line | Genotype | Defect /modification | Origin | Reference |
|---|---|---|---|---|
| D213 N | XRCC3− | irs1SF complemented with the hXRCC3 D213 N gene | irs1SF | (10) |
| T241M | XRCC3+ | irs1SF complemented with the hXRCC3 T241M gene | irs1SF | (10) |
| irs1SF | XRCC3− | XRCC3−, deficient in homologous recombination | AA8 | (21) |
| WT | XRCC3+ | irs1SF complemented with the hXRCC3 gene | irs1SF | (10) |
| AA8 | wt | Wild-type Chinese hamster | Ovary | (21) |
References
Venkitaraman, A.R. (
Tutt, A., Bertwistle, D., Valentine, J., Gabriel, A., Swift, S., Ross, G., Griffin, C., Thacker, J. and Ashworth, A. (
Tutt, A., Gabriel, A., Bertwistle, D., Connor, F., Paterson, H., Peacock, J., Ross, G. and Ashworth, A. (
Xu, X., Weaver, Z., Linke, S.P., Li, C., Gotay, J., Wang, X.W., Harris, C.C., Ried, T. and Deng, C.X. (
Hsu, L.C. and White, R.L. (
Hsu, L.C., Doan, T.P. and White, R.L. (
Daniels, M.J., Wang, Y., Lee, M. and Venkitaraman, A.R. (
Pierce, A.J., Johnson, R.D., Thompson, L.H. and Jasin, M. (
Griffin, C.S., Simpson, P.J., Wilson, C.R. and Thacker, J. (
Rafii, S., Lindblom, A., Reed, M., Meuth, M. and Cox, A. (
Kuschel, B., Auranen, A., McBride, S., Novik, K.L., Antoniou, A., Lipscombe, J.M., Day, N.E., Easton, D.F., Ponder, B.A., Pharoah, P.D. et al. (
Jacobsen, N.R., Raaschou-Nielsen, O., Nexo, B., Wallin, H., Overvad, K., Tjonneland, A. and Vogel, U. (
Seedhouse, C., Faulkner, R., Ashraf, N., Das-Gupta, E. and Russell, N. (
Benhamou, S., Tuimala, J., Bouchardy, C., Dayer, P., Sarasin, A. and Hirvonen, A. (
Jacobsen, N.R., Nexo, B.A., Olsen, A., Overvad, K., Wallin, H., Tjonneland, A. and Vogel, U. (
Figueiredo, J.C., Knight, J.A., Briollais, L., Andrulis, I.L. and Ozcelik, H. (
Thacker, J. (
Araujo, F.D., Pierce, A.J., Stark, J.M. and Jasin, M. (
Lengauer, C., Kinzler, K.W. and Vogelstein, B. (
Tebbs, R.S., Zhao, Y., Tucker, J.D., Scheerer, J.B., Siciliano, M.J., Hwang, M., Liu, N., Legerski, R.J. and Thompson, L.H. (
Fuller, L.F. and Painter, R.B. (
Schultz, N. and Onfelt, A. (
Hinz, J.M., Helleday, T. and Meuth, M. (
Martin, S.J., Reutelingsperger, C.P., McGahon, A.J., Rader, J.A., van Schie, R.C., LaFace, D.M. and Green, D.R. (
Yamada, N.A., Hinz, J.M., Kopf, V.L., Segalle, K.D. and Thompson, L.H. (
Dodson, H., Bourke, E., Jeffers, L.J., Vagnarelli, P., Sonoda, E., Takeda, S., Earnshaw, W.C., Merdes, A. and Morrison, C. (
Yoshihara, T., Ishida, M., Kinomura, A., Katsura, M., Tsuruga, T., Tashiro, S., Asahara, T. and Miyagawa, K. (
Hut, H.M., Lemstra, W., Blaauw, E.H., Van Cappellen, G.W., Kampinga, H.H. and Sibon, O.C. (
Vogel, C., Kienitz, A., Hofmann, I., Muller, R. and Bastians, H. (
Mohindra, A., Hays, L.E., Phillips, E.N., Preston, B.D., Helleday, T. and Meuth, M. (
Giannini, G., Ristori, E., Cerignoli, F., Rinaldi, C., Zani, M., Viel, A., Ottini, L., Crescenzi, M., Martinotti, S., Bignami, M. et al. (
Segurado, M., Gomez, M. and Antequera, F. (
Ludwig, T., Chapman, D.L., Papaioannou, V.E. and Efstratiadis, A. (
Connor, F., Bertwistle, D., Mee, P.J., Ross, G.M., Swift, S., Grigorieva, E., Tybulewicz, V.L. and Ashworth, A. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}