




Abstract
The near completeness of human chromosome sequences is facilitating accurate characterization and assessment of all classes of genomic variation. Particularly, using the DNA reference sequence as a guide, genome scanning technologies, such as microarray-based comparative genomic hybridization (array CGH) and genome-wide single nucleotide polymorphism (SNP) platforms, have now enabled the detection of a previously unrecognized degree of larger-sized (non-SNP) variability in all genomes. This heterogeneity can include copy number variations (CNVs), inversions, insertions, deletions and other complex rearrangements, most of which are not detected by standard cytogenetics or DNA sequencing. Although these genomic alterations (collectively termed structural variants or polymorphisms) have been described previously, mainly through locus-specific studies, they are now known to be more global in occurrence. Moreover, as just one example, CNVs can contain entire genes and their number can correlate with the level of gene expression. It is also plausible that structural variants may commonly influence nearby genes through chromosomal positional or domain effects. Here, we discuss what is known of the prevalence of structural variants in the human genome and how they might influence phenotype, including the continuum of etiologic events underlying monogenic to complex diseases. Particularly, we highlight the newest studies and some classic examples of how structural variants might have adverse genetic consequences. We also discuss why analysis of structural variants should become a vital step in any genetic study going forward. All these progresses have set the stage for a golden era of combined microscopic and sub-microscopic (cytogenomic)-based research of chromosomes leading to a more complete understanding of the human genome.
INTRODUCTION
In the past few years, several studies have identified a previously uncharacterized prevalence of structural variants of DNA along chromosomes in the size range of 1 kb or greater, adding to the catalog of variants in the human genome (Table 1). Namely, sub-microscopic (usually less than ∼3 Mb) copy number variations (CNVs) and inversions have been found to occur in every genome studied at high frequencies when compared with the equivalent classes of cytogenetically detectable rearrangements (1–8). Similar findings are also now being made more readily in disease gene studies.
These discoveries have come somewhat later than the description (and generation of comprehensive maps) of single nucleotide polymorphisms (SNPs) (9,10), microsatellites (11,12) and minisatellites (13), as well as catalogs of cytogenetically detectable heteromorphisms and rearrangements, because of limits of resolution in the technology at that time. However, new developments in genome-wide scanning methodologies using genomic clone and oligonucleotide-based arrays occurring in parallel with the availability of a reference human genome sequence now provide opportunity to generate advanced maps of structural variation in worldwide populations. Moreover, next generation sequencing technologies and computational comparisons of sequences from different sources will yield a vast number of variants primarily in the <1 kb size range that have not been described previously. Comprehensive reviews describing the discovery and properties of, in particular, CNVs, but also other structural variants, have been published recently (14–19). Here, we highlight the latest findings, with a particular emphasis on the new sub-microscopic variants being increasingly described in the ∼1 kb to ∼3 Mb size range and how they may influence phenotype or be involved in disease.
STRUCTURAL VARIATION INFLUENCING PHENOTYPE
Changes in DNA that affect gene function (often through affecting dosage) can have a deleterious effect on the reproductive fitness of an organism, and in some cases represent lethal mutations. In these circumstances, the variants would eventually be destined to disappear, but they can often exist in a heterozygous form for many generations. In between these extremes of selectively neutral variants and lethal mutations lie variants that can influence physiological, biochemical, morphological and pathological variation in the human population. Recent descriptions of numerous ‘gene-sized’ (the average size of a gene being ∼70 kb) sub-microscopic structural variants in all genomes have generated significant excitement in the field (20–25), because (i) it was presumed they probably should exist for the same reasons as SNPs and microscopic variants; (ii) their sheer size (often affecting hundreds to thousands of nucleotides of DNA) increases the likelihood that the alteration is, in fact, a genomic lesion explaining disease outcomes; (iii) as such, some will also be shown to predispose to disease either directly or in combination with other variants and factors and (iv) some will provide substrate for evolutionary change. The description of all variants will be important for many wide-ranging reasons, better resolving a more completely annotated reference genome sequence to understanding implications in pharmacogenomics and clinical diagnostic testing.
It has been well established in many classic (26–33) as well as in more recent studies of monogenic disease (34), oligogenetic disease (35–41), and most recently in complex disease that the study of such chromosome rearrangements can be the most rapid approach to identify candidate susceptibility loci and genes (that then need to be confirmed in other samples). For complex diseases (note that in some cases, these were the Mendelian sub-forms of complex disease demonstrating the same phenotypic endpoint), examples include: in autism, X-chromosome deletions led to the identification of the neuroligin NLGN3 and NLGN4 genes (42); in schizophrenia, a familial chromosome 1 translocation led to the discovery of the DISC1 and DISC2 genes (43); in dyslexia, distinct chromosomes 3 and 15 translocations led to the discovery of ROBO1 and DYXC1, respectively (44,45); in severe speech and language disorder, a chromosome 7q31 translocation pinpointed the FOXP2 gene (46); in Tourette syndrome, a de novo inversion led to SLITRK1 involvement (47); in severe expressive language delay, microduplication of the Williams–Beuren syndrome locus on chromosome 7q11.23 (48) and in early onset Parkinson and Alzheimer's disease, duplications of SNCA and APP on chromosomes 4 and 21, respectively, have been shown to be causative (49,50).
Indeed, the primary message of this review is to increase the awareness of the necessity for including steps for screening for structural variants in genetic experiments. This was exquisitely demonstrated in a recent study showing that copy number polymorphism in the FCGR3 gene predisposes to glomerulonephritis in humans and rats (51). Preliminary data suggest that dozens of CNVs alone will be found in a given genome when assessed using comprehensive scanning methodologies. The sub-microscopic variants will be intermediate in size and frequency in comparison to occurrence of cytogenetically detectable and smaller (<1 kb) polymorphisms. These aberrations can be hundreds of kilobases long, having important implications for the potential effect they may exert on genes and transcriptional regulation (Fig. 1). Moreover, a large number of CNVs have been shown to contain one or more entire coding transcripts (1,2,52,53). In studies where these genes have been characterized, there seems to be a direct correlation between increases in gene copy number and increased levels of mRNA (53–56). Polymorphic deletions containing entire genes have also been described, where a fraction of the population are homozygous for the deletion allele and, therefore, do not have the gene present in their genome (6,8,57). Most of the genes in this category belong to gene families or are recently duplicated in evolutionary history, and this may increase the tolerance for null alleles.
Specific categories of genes seem over-represented in CNVs including those important for interaction with the surrounding environment, such as olfaction and response to external stimuli (19,58). Examples of such polymorphic genes include glutathione S-transferase genes (59,60), cytochrome P450 genes (61–65) and the complement component C4 (66). In each case, changes to gene copy number have been shown to give rise to concomitant changes in the level of enzyme activity, with phenotypic consequences. Another example is the CCL3L1 gene, where the increased copy number has been shown to be protective against HIV infection (56).
Inversions represent another class of structural variation (Table 1), but knowledge of their prevalence in the human genome is more limited. This is partly due to a lack of technologies for robust and inexpensive discovery of such balanced rearrangements. In addition, preliminary data indicate that inversion variants are less abundant than CNVs in the human genome (4). However, there are a number of well-documented cases where inversion variants can be associated with disease predisposition, primarily in microdeletion syndromes. In these instances, the inversion variant need not be a direct cause of the disease, but instead it can act as a risk factor for microdeletion to occur in the offspring, as appears to be the case in Williams–Beuren (67), Angelman (68) and Sotos syndromes (69).
POTENTIAL LONG-RANGE (POSITION) EFFECTS OF STRUCTURAL VARIANTS ON GENES
As discussed, structural variants can affect dosage by directly interrupting genes, but it is important to appreciate that they can have an equivalent effect at a distance (in an indirect manner) (Fig. 1). Although genes only represent a small portion (<3%) of the human genome and there are hundreds of putative ‘gene deserts’, sometimes millions of base pairs in size (70–72), there is now substantial evidence that regulatory elements of genes can reside up to a million base pairs or more away (Fig. 1; Table 2) (Supplementary Material, Table S1). Thus, structural variants cannot be presumed to be selectively neutral because they encompass only non-coding segments, but instead a careful assessment of nearby genes that may be affected via a ‘position effect’ mechanism also needs to be considered.
Position effect refers to the alteration of a gene's expression pattern as a result of a change in its genomic location or chromatin environment. This phenomenon has been most extensively studied in Drosophila (73) and yeast (74), but an increasing number of examples in humans have been reported, including a variety of developmental disorders such as aniridia (75–78), holoprosencephaly (79–81), campomelic dysplasia (82–90), thalassemias (91–94), X–Y sex reversal (95,96) and others (Supplementary Material, Table S1). Position effects can be caused by a variety of mechanisms. These include translocation of a gene into a heterochromatic region resulting in the methylation of promoter regions and consequent down-regulation of expression, chromosome breakage (translocations, inversions, deletions and duplications) that separates a gene from some or all of its transcriptional control elements or otherwise alters gene expression (94), or genomic rearrangements that bring a gene into close proximity to a positive regulatory element (97). In recent reviews (80,97), several additional examples of congenital abnormalities resulting in either obvious or postulated position effects in humans have been reported. Here, we call attention to some studies from the past few years, highlighting how the structural variants can be involved in disease through different mechanisms of action (Table 2) (additional historical studies are summarized in Supplementary Material, Table S1).
In most cases, the effect of genomic rearrangement on gene expression has been inferred, rather than observed directly. This is often due to the unavailability of appropriate tissue or developmental timing of expression that would render gene expression analysis impossible. As an example, a translocation that disrupts the HDAC9 gene at 7p21.1 has its reciprocal breakpoint on chromosome 1, ∼500 kb from the TGFB2 gene. The patient carrying this translocation has Peter's anomaly, a defect of the anterior chamber of the eye, and as Tgfb2 null mice have very similar developmental eye defects, and therefore, the authors consider a position effect at TGFB2, rather than HDAC9 disruption, to be the most likely underlying pathology (98). A more complex example is a 23–25 kb deletion and 340 kb insertion at the deletion point, 67 kb 3′ to the SOX3 gene, found in a patient with X-linked recessive hypoparathyroidism (99). It is presumed that down-regulation of SOX3 results in the phenotype, as SOX3 has been observed to be expressed in the developing parathyroid of mouse embryos.
In other recent examples, Wakui et al. (100) report a patient with a large deletion located just 3′ of the ALX4 gene having atypical manifestation of Potocki–Shaffer syndrome. Beysen et al. (101) describe a number of patients with Blepharophimosis syndrome (one of the syndromes most frequently reported to be associated with position effects in humans), each having a microdeletion near the FOXL2 gene. Lee et al. (102) describe a patient with an atypical phenotype (spastic paraplegia type II with axonal neuropathy) because of a duplication near the PLP1 gene, deletions and duplications of which usually result in Pelizaeus–Merzbacher syndrome (103). Mild forms of campomelic dysplasia, a skeletal malformation syndrome, have also been reported, as a result of balanced translocations near the SOX9 gene in three different patients [two with simple reciprocal translocations and one with a complex translocation (82,83)]. Ellison et al. (104) report a patient with a ring (X) chromosome that is presumed to cause a down-regulation of the SHOX gene, resulting in short stature in that patient, although a significant amount of Xp and Xq material is also deleted, including several other genes. Finally, in an extreme case carrying a de novo t(6;7) (p21.1;q36) reciprocal translocation exhibiting both holoprosencephaly and cleidocranial dysplasia, there are two apparent position effect mutations in the same individual: the 7q36 breakpoint mapping 15 kb telomeric to the 5′ end of Sonic Hedgehog causes holoprosencephaly and the 6p21.1 breakpoint mapping 800 kb upstream of CBFA1 (RUNX2) causes cleidocranial dysplasia (80).
It is striking that a majority of genes reported to be affected by apparent position effects in humans are involved in developmental syndromes. This could be due to ascertainment bias, as phenotypes in these patients tend to be either atypical or unusually mild. Alternatively, it could be that other classes of genes, e.g. those encoding enzymes, are much more tolerant to positional silencing or down-regulation and that individuals with such rearrangements thus escape clinical notice. It has also been suggested that large ‘gene deserts’ often found around the developmental genes (71) may serve as enhanced targets for chromosomal rearrangements (105). Notwithstanding, the take home message from these studies and others is that the structural variant need not only affect what we usually define as the classical gene unit to have an effect; proximal and distal genes also need to be considered.
MEDICAL AND BIOLOGICAL SIGNIFICANCE
With the ability to recognize dozens of sub-microscopic variants in all genomes, a hierarchical paradigm of how to differentiate the manifestation of disease- (or phenotype-) associated changes needs to be considered (Fig. 2). For example, in some cases, a structural variant correlates directly with the disease, such as the case in dosage-related microdeletions and duplications that cause genomic disorders and in other cases described earlier (106). Family-based studies can demonstrate whether a change is de novo or has been inherited and, in the latter case, whether there are likely to be associated phenotypic consequences. However, there are numerous examples of lack of penetrance or variable expression of phenotype in inherited chromosomal rearrangements (107), requiring analysis be extended to a larger population of controls. Other factors such as the genomic context (e.g. types of genes and likelihood to be affected by position effect), the heritable stability of the variant (it could predispose to other mitotic or meiotic rearrangements), influence of other variants and possible parent-of-origin effects (e.g. imprinted regions) all need to be considered when evaluating the effect at the genic level.
Databases cataloging large-scale genotype and phenotype correlations (17,70,108) will be increasingly important to help discern how these changes might cause phenotypic or functional outcomes (Table 3 provides web links). For example, the genome-wide initiatives including the Database of Chromosomal Imbalance and Phenotypes in Humans using Ensembl Resources (DECIPHER) and the Developmental Genome Anatomy Project (DGAP) promise to bridge the gap from DNA sequence to medical genetic outcomes, but the databases are still sparsely populated. Other efforts, including the Mendelian Cytogenetics Network Online Database, the Chromosome Abnormality Database, the European Collection of Cell Cultures, Coriell Cell Repositories, NIGMS Human Genetic Cell Repository and the Mitelman Database of chromosome aberrations in cancer, among others, provide catalogs of samples with karyotypes and phenotypes. The Database of Genomic Variants and the Human Structural Variation Database house information on structural variants that are generally not known to cause disease. It is also worth noting that there are large data sets of SNPs and microsatellites from disease mapping studies (Supplementary Material, Tables S2 and S3) that could be analyzed for their putative CNV content in a manner similar to what has been done in autism (109) and HapMap samples (6,8).
CONCLUSIONS AND FUTURE STUDIES
The complexity of variation in the human genome continues to be unraveled, providing opportunity to explain genetic contributions to disease in a more comprehensive manner. Going forward into the next few years, studies examining the role of sub-microscopic structural variation will become a predominant theme because of significant advances in technology allowing for the scanning of genomes at relatively high resolution. In fact, on the basis of the numbers of discoveries and impact alone in the past 2 years, it could be argued that we have entered a ‘cytogenomic’ era for discovery in human genetics. In large-scale population-based whole genome association studies (Supplementary Material, Table S3), and in any disease gene study, a component of assessing structural variation content should be incorporated. However, comprehending the contribution of these variants will require the understanding of wide-ranging data from simple presence or absence (in cases and controls) to the position and context in the genome (Fig. 2). It will be important to determine the new mutation rate (25,110) of these variants across the genome, including the heterochromatic regions. The next frontier will be to fully catalog all the structural variants in the ∼1 kb to 3 Mb size range discussed here, but also all other variations in the 1 bp to 1 kb size range (Table 1), which will probably be best discerned through personalized genome (re-)sequencing (111). Coupling all of this information to large cohorts of meticulously phenotyped sample collections and corresponding databases would provide insight toward understanding the etiology of many unresolved diseases.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
The work was supported by funds from The Centre for Applied Genomics, Hospital for Sick Children, Genome Canada/Ontario Genomics Institute, the Canadian Institutes of Health Research (CIHR) and the McLaughlin Centre for Molecular Medicine. L.F. is supported by the Swedish Medical Research Council, C.R.M. by The Hospital for Sick Children Research Training Centre and S.W.S. is an Investigator of CIHR and International Scholar of the Howard Hughes Medical Institute.
Conflict of Interest statement. None declared.
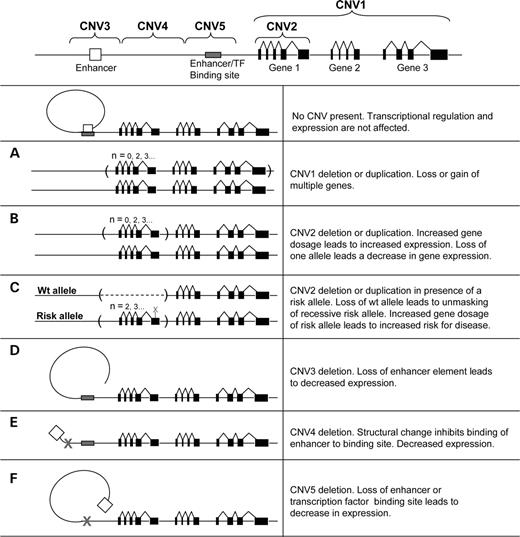
Figure 1. CNV influencing gene dosage and expression and disease. There are a number of mechanisms by which CNVs potentially could have an effect on gene expression and phenotypic traits. Well-documented examples involve regions with multiple genes deleted in microdeletion syndromes and microduplication syndromes (A), where there is a direct correlation between genotype and phenotype. Copy number polymorphisms where a gene is located entirely within a region that varies in copy number (B) have also been described to show a direct correlation between gene copy number and gene expression. Another mechanism by which CNVs may have an influence on disease phenotype is if the remaining copy harbors a risk allele that becomes apparent only in the hemizygous state (C). The opposite scenario may also occur, with an increased number of copies harboring a risk allele, causing a concurrent increase in disease susceptibility. It can also be hypothesized that CNVs may affect gene expression without directly changing the gene copy number. Gains or losses affecting the regulatory elements or promoter regions can also be important contributors to differences in gene expression. This could involve either loss of an element of transcriptional regulation (D and F) or a loss/gain changing the structural properties of DNA inhibiting enhancer interaction, chromatin structure or access of transcription factors to their binding sites (E). Interaction and additive models with any of these scenarios combined or in combination with any type of variation at other loci can be expected to be the cause of more complex genetic traits.
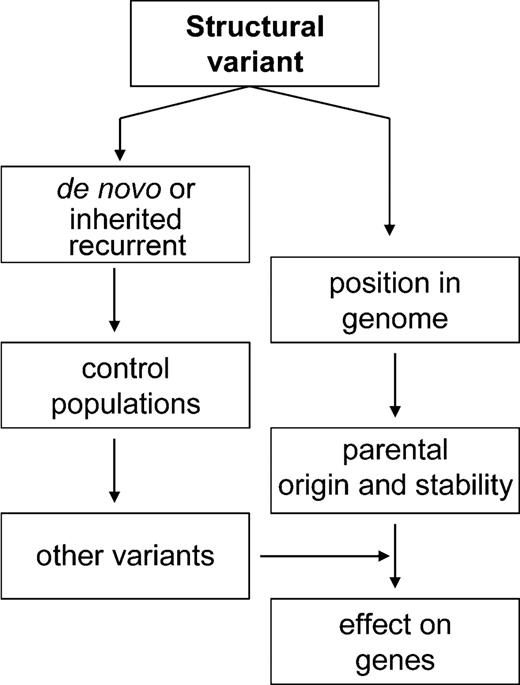
Figure 2. Genetic and genomic considerations of assessing the influence of structural variants on gene expression and phenotype/disease manifestation.
Genetic variation in the human genome
| Variation type | Definition | Frequency (if known) in the human genome | References |
|---|---|---|---|
| SNP | Single base pair variation found in >1% of chromosomes in a given population | ∼10 million SNPs in the human population | (112–114) |
| Insertion/deletion variant (InDel) | Deletion or insertion of a segment of DNA. Includes small polymorphic changes and large chromosomal aberrations. InDels >1 kb in size are often also called CNVs | ∼1 million insertion/deletions polymorphisms >1 bp in the human genome | (115,116) |
| Microsatellite (e.g. CAn repeats) | Sequences containing variable numbers of 1–6 bp repeats totaling <200 bp in length | >1 million microsatellites in the human genome, accounting for ∼3% of the sequence | (114,117,118) |
| Minisatellite and variable numbers of tandem repeats (VNTRs) | Polymorphic sequence containing 20–50 copies of 6–100 bp repeats | ∼150 000 minisatellites, of which ∼20% are polymorphic | (13,119,120) |
| Multisite variant (MSV) | Single nucleotide variant with complex characteristics due to CNV or gene conversion | The number of MSVs is currently unknown | (121) |
| Intermediate-sized structural variant (ISV) | Gain or loss of a DNA sequence >8 kb in size also includes inversion breakpoints | 297 ISVs were identified using a fosmid library from a single genome | (4) |
| CNV; copy number polymorphism (CNP); large-scale CNV | Copy number change >1 kb. If the frequency is >1%, it is called a CNP. LCVs are CNVs ∼50 kb in size or greater | The frequency of CNVs in the human genome is unknown. Estimates of larger CNVs (>50 kb) | (1,2,19) |
| Inversion | Rearrangement causing a segment of DNA to be present in reverse orientation | Estimates of microscopically detectable inversion frequencies are 0.12–0.7% (pericentric) and 0.1–0.5% (paracentric); sub-microscopic unknown | (122,123) |
| Translocation | Rearrangement in which a DNA fragment is attached to different chromosome | 1/500 is heterozygous for a reciprocal translocation and 1/1000 for Robertsonian translocations | (122–124) |
| Unbalanced rearrangements | Rearrangements which lead to a net gain or loss of DNA are referred to as unbalanced | Unbalanced rearrangements occur in ∼1/1500 live births | (125) |
| Variation type | Definition | Frequency (if known) in the human genome | References |
|---|---|---|---|
| SNP | Single base pair variation found in >1% of chromosomes in a given population | ∼10 million SNPs in the human population | (112–114) |
| Insertion/deletion variant (InDel) | Deletion or insertion of a segment of DNA. Includes small polymorphic changes and large chromosomal aberrations. InDels >1 kb in size are often also called CNVs | ∼1 million insertion/deletions polymorphisms >1 bp in the human genome | (115,116) |
| Microsatellite (e.g. CAn repeats) | Sequences containing variable numbers of 1–6 bp repeats totaling <200 bp in length | >1 million microsatellites in the human genome, accounting for ∼3% of the sequence | (114,117,118) |
| Minisatellite and variable numbers of tandem repeats (VNTRs) | Polymorphic sequence containing 20–50 copies of 6–100 bp repeats | ∼150 000 minisatellites, of which ∼20% are polymorphic | (13,119,120) |
| Multisite variant (MSV) | Single nucleotide variant with complex characteristics due to CNV or gene conversion | The number of MSVs is currently unknown | (121) |
| Intermediate-sized structural variant (ISV) | Gain or loss of a DNA sequence >8 kb in size also includes inversion breakpoints | 297 ISVs were identified using a fosmid library from a single genome | (4) |
| CNV; copy number polymorphism (CNP); large-scale CNV | Copy number change >1 kb. If the frequency is >1%, it is called a CNP. LCVs are CNVs ∼50 kb in size or greater | The frequency of CNVs in the human genome is unknown. Estimates of larger CNVs (>50 kb) | (1,2,19) |
| Inversion | Rearrangement causing a segment of DNA to be present in reverse orientation | Estimates of microscopically detectable inversion frequencies are 0.12–0.7% (pericentric) and 0.1–0.5% (paracentric); sub-microscopic unknown | (122,123) |
| Translocation | Rearrangement in which a DNA fragment is attached to different chromosome | 1/500 is heterozygous for a reciprocal translocation and 1/1000 for Robertsonian translocations | (122–124) |
| Unbalanced rearrangements | Rearrangements which lead to a net gain or loss of DNA are referred to as unbalanced | Unbalanced rearrangements occur in ∼1/1500 live births | (125) |
Genetic variation in the human genome
| Variation type | Definition | Frequency (if known) in the human genome | References |
|---|---|---|---|
| SNP | Single base pair variation found in >1% of chromosomes in a given population | ∼10 million SNPs in the human population | (112–114) |
| Insertion/deletion variant (InDel) | Deletion or insertion of a segment of DNA. Includes small polymorphic changes and large chromosomal aberrations. InDels >1 kb in size are often also called CNVs | ∼1 million insertion/deletions polymorphisms >1 bp in the human genome | (115,116) |
| Microsatellite (e.g. CAn repeats) | Sequences containing variable numbers of 1–6 bp repeats totaling <200 bp in length | >1 million microsatellites in the human genome, accounting for ∼3% of the sequence | (114,117,118) |
| Minisatellite and variable numbers of tandem repeats (VNTRs) | Polymorphic sequence containing 20–50 copies of 6–100 bp repeats | ∼150 000 minisatellites, of which ∼20% are polymorphic | (13,119,120) |
| Multisite variant (MSV) | Single nucleotide variant with complex characteristics due to CNV or gene conversion | The number of MSVs is currently unknown | (121) |
| Intermediate-sized structural variant (ISV) | Gain or loss of a DNA sequence >8 kb in size also includes inversion breakpoints | 297 ISVs were identified using a fosmid library from a single genome | (4) |
| CNV; copy number polymorphism (CNP); large-scale CNV | Copy number change >1 kb. If the frequency is >1%, it is called a CNP. LCVs are CNVs ∼50 kb in size or greater | The frequency of CNVs in the human genome is unknown. Estimates of larger CNVs (>50 kb) | (1,2,19) |
| Inversion | Rearrangement causing a segment of DNA to be present in reverse orientation | Estimates of microscopically detectable inversion frequencies are 0.12–0.7% (pericentric) and 0.1–0.5% (paracentric); sub-microscopic unknown | (122,123) |
| Translocation | Rearrangement in which a DNA fragment is attached to different chromosome | 1/500 is heterozygous for a reciprocal translocation and 1/1000 for Robertsonian translocations | (122–124) |
| Unbalanced rearrangements | Rearrangements which lead to a net gain or loss of DNA are referred to as unbalanced | Unbalanced rearrangements occur in ∼1/1500 live births | (125) |
| Variation type | Definition | Frequency (if known) in the human genome | References |
|---|---|---|---|
| SNP | Single base pair variation found in >1% of chromosomes in a given population | ∼10 million SNPs in the human population | (112–114) |
| Insertion/deletion variant (InDel) | Deletion or insertion of a segment of DNA. Includes small polymorphic changes and large chromosomal aberrations. InDels >1 kb in size are often also called CNVs | ∼1 million insertion/deletions polymorphisms >1 bp in the human genome | (115,116) |
| Microsatellite (e.g. CAn repeats) | Sequences containing variable numbers of 1–6 bp repeats totaling <200 bp in length | >1 million microsatellites in the human genome, accounting for ∼3% of the sequence | (114,117,118) |
| Minisatellite and variable numbers of tandem repeats (VNTRs) | Polymorphic sequence containing 20–50 copies of 6–100 bp repeats | ∼150 000 minisatellites, of which ∼20% are polymorphic | (13,119,120) |
| Multisite variant (MSV) | Single nucleotide variant with complex characteristics due to CNV or gene conversion | The number of MSVs is currently unknown | (121) |
| Intermediate-sized structural variant (ISV) | Gain or loss of a DNA sequence >8 kb in size also includes inversion breakpoints | 297 ISVs were identified using a fosmid library from a single genome | (4) |
| CNV; copy number polymorphism (CNP); large-scale CNV | Copy number change >1 kb. If the frequency is >1%, it is called a CNP. LCVs are CNVs ∼50 kb in size or greater | The frequency of CNVs in the human genome is unknown. Estimates of larger CNVs (>50 kb) | (1,2,19) |
| Inversion | Rearrangement causing a segment of DNA to be present in reverse orientation | Estimates of microscopically detectable inversion frequencies are 0.12–0.7% (pericentric) and 0.1–0.5% (paracentric); sub-microscopic unknown | (122,123) |
| Translocation | Rearrangement in which a DNA fragment is attached to different chromosome | 1/500 is heterozygous for a reciprocal translocation and 1/1000 for Robertsonian translocations | (122–124) |
| Unbalanced rearrangements | Rearrangements which lead to a net gain or loss of DNA are referred to as unbalanced | Unbalanced rearrangements occur in ∼1/1500 live births | (125) |
Selected recently published examples of potential position effects caused by structural variants
| Indication | OMIM | Locus | Gene(s) involved | Distance from gene | Type of rearrangement | Effect on gene(s) | Reference | Comments |
|---|---|---|---|---|---|---|---|---|
| Severe speech and language disorder | 608636 | 7q31.1 | FOXP2 | At least 680 kb 5′ | Balanced translocation | Postulated downregulation | (70) | |
| Blepharophimosis syndrome (BPES) | 110100 | 3q22.3 | FOXL2 | 101–231 kb 5′; 28.7 kb 3′ | Four different microdeletions (126 kb to 1.9 Mb in size) 5′; 188 kb microdeletion 3′ | Postulated downregulation | (101) | |
| Campomelic dysplasia | 114290 | 17q24.3 | SOX9 | 400 and 900 kb 5′; 1.3 Mb down-stream (complex case) | Two balanced translocations and one complex balanced translocation | Postulated downregulation | (82,83) | Mild acampomelic phenotype in two patients |
| Peter's anomaly | 604229 | 1q41 | TGFB2 | 500 kb 3′ | Balanced translocation | Postulated downregulation | (98) | |
| Potocki–Shaffer syndrome | 601224 | 11p11.2 | ALX4 | >15 kb 3′ | ∼1.37 Mb deletion | Postulated downregulation | (100) | Atypical phenotype (parietal foramina) |
| Short stature | 312865 | Xp22.33 | SHOX | 250–350 kb 5′ | Ring (X) with deletion of 700–900 kb of Xp and Xq pseudoautosomal regions | Postulated downregulation | (104) | Multiple other genes deleted |
| Spastic paraplegia type II with axonal neuropathy | 312920 | Xq22.2 | PLP1 | 135–185 kb 3′ | 100–150 kb duplication | Postulated down-regulation | (102,126) | Unusual phenotype; PLP1 duplications and deletions lead to Pelizaeus–Merzbacher syndrome |
| Townes–Brocks syndrome | 107480 | 16q12.1 | SALL1 | >180 kb 5′ | Balanced translocation | Postulated down-regulation | (127) | |
| X-linked recessive hypoparathyroidism | 307700 | Xq27.1 | SOX3 | 67 kb 3′ | Deletion of ∼25 kb with insertion of ∼340 kb | Postulated down-regulation | (99) |
| Indication | OMIM | Locus | Gene(s) involved | Distance from gene | Type of rearrangement | Effect on gene(s) | Reference | Comments |
|---|---|---|---|---|---|---|---|---|
| Severe speech and language disorder | 608636 | 7q31.1 | FOXP2 | At least 680 kb 5′ | Balanced translocation | Postulated downregulation | (70) | |
| Blepharophimosis syndrome (BPES) | 110100 | 3q22.3 | FOXL2 | 101–231 kb 5′; 28.7 kb 3′ | Four different microdeletions (126 kb to 1.9 Mb in size) 5′; 188 kb microdeletion 3′ | Postulated downregulation | (101) | |
| Campomelic dysplasia | 114290 | 17q24.3 | SOX9 | 400 and 900 kb 5′; 1.3 Mb down-stream (complex case) | Two balanced translocations and one complex balanced translocation | Postulated downregulation | (82,83) | Mild acampomelic phenotype in two patients |
| Peter's anomaly | 604229 | 1q41 | TGFB2 | 500 kb 3′ | Balanced translocation | Postulated downregulation | (98) | |
| Potocki–Shaffer syndrome | 601224 | 11p11.2 | ALX4 | >15 kb 3′ | ∼1.37 Mb deletion | Postulated downregulation | (100) | Atypical phenotype (parietal foramina) |
| Short stature | 312865 | Xp22.33 | SHOX | 250–350 kb 5′ | Ring (X) with deletion of 700–900 kb of Xp and Xq pseudoautosomal regions | Postulated downregulation | (104) | Multiple other genes deleted |
| Spastic paraplegia type II with axonal neuropathy | 312920 | Xq22.2 | PLP1 | 135–185 kb 3′ | 100–150 kb duplication | Postulated down-regulation | (102,126) | Unusual phenotype; PLP1 duplications and deletions lead to Pelizaeus–Merzbacher syndrome |
| Townes–Brocks syndrome | 107480 | 16q12.1 | SALL1 | >180 kb 5′ | Balanced translocation | Postulated down-regulation | (127) | |
| X-linked recessive hypoparathyroidism | 307700 | Xq27.1 | SOX3 | 67 kb 3′ | Deletion of ∼25 kb with insertion of ∼340 kb | Postulated down-regulation | (99) |
Selected recently published examples of potential position effects caused by structural variants
| Indication | OMIM | Locus | Gene(s) involved | Distance from gene | Type of rearrangement | Effect on gene(s) | Reference | Comments |
|---|---|---|---|---|---|---|---|---|
| Severe speech and language disorder | 608636 | 7q31.1 | FOXP2 | At least 680 kb 5′ | Balanced translocation | Postulated downregulation | (70) | |
| Blepharophimosis syndrome (BPES) | 110100 | 3q22.3 | FOXL2 | 101–231 kb 5′; 28.7 kb 3′ | Four different microdeletions (126 kb to 1.9 Mb in size) 5′; 188 kb microdeletion 3′ | Postulated downregulation | (101) | |
| Campomelic dysplasia | 114290 | 17q24.3 | SOX9 | 400 and 900 kb 5′; 1.3 Mb down-stream (complex case) | Two balanced translocations and one complex balanced translocation | Postulated downregulation | (82,83) | Mild acampomelic phenotype in two patients |
| Peter's anomaly | 604229 | 1q41 | TGFB2 | 500 kb 3′ | Balanced translocation | Postulated downregulation | (98) | |
| Potocki–Shaffer syndrome | 601224 | 11p11.2 | ALX4 | >15 kb 3′ | ∼1.37 Mb deletion | Postulated downregulation | (100) | Atypical phenotype (parietal foramina) |
| Short stature | 312865 | Xp22.33 | SHOX | 250–350 kb 5′ | Ring (X) with deletion of 700–900 kb of Xp and Xq pseudoautosomal regions | Postulated downregulation | (104) | Multiple other genes deleted |
| Spastic paraplegia type II with axonal neuropathy | 312920 | Xq22.2 | PLP1 | 135–185 kb 3′ | 100–150 kb duplication | Postulated down-regulation | (102,126) | Unusual phenotype; PLP1 duplications and deletions lead to Pelizaeus–Merzbacher syndrome |
| Townes–Brocks syndrome | 107480 | 16q12.1 | SALL1 | >180 kb 5′ | Balanced translocation | Postulated down-regulation | (127) | |
| X-linked recessive hypoparathyroidism | 307700 | Xq27.1 | SOX3 | 67 kb 3′ | Deletion of ∼25 kb with insertion of ∼340 kb | Postulated down-regulation | (99) |
| Indication | OMIM | Locus | Gene(s) involved | Distance from gene | Type of rearrangement | Effect on gene(s) | Reference | Comments |
|---|---|---|---|---|---|---|---|---|
| Severe speech and language disorder | 608636 | 7q31.1 | FOXP2 | At least 680 kb 5′ | Balanced translocation | Postulated downregulation | (70) | |
| Blepharophimosis syndrome (BPES) | 110100 | 3q22.3 | FOXL2 | 101–231 kb 5′; 28.7 kb 3′ | Four different microdeletions (126 kb to 1.9 Mb in size) 5′; 188 kb microdeletion 3′ | Postulated downregulation | (101) | |
| Campomelic dysplasia | 114290 | 17q24.3 | SOX9 | 400 and 900 kb 5′; 1.3 Mb down-stream (complex case) | Two balanced translocations and one complex balanced translocation | Postulated downregulation | (82,83) | Mild acampomelic phenotype in two patients |
| Peter's anomaly | 604229 | 1q41 | TGFB2 | 500 kb 3′ | Balanced translocation | Postulated downregulation | (98) | |
| Potocki–Shaffer syndrome | 601224 | 11p11.2 | ALX4 | >15 kb 3′ | ∼1.37 Mb deletion | Postulated downregulation | (100) | Atypical phenotype (parietal foramina) |
| Short stature | 312865 | Xp22.33 | SHOX | 250–350 kb 5′ | Ring (X) with deletion of 700–900 kb of Xp and Xq pseudoautosomal regions | Postulated downregulation | (104) | Multiple other genes deleted |
| Spastic paraplegia type II with axonal neuropathy | 312920 | Xq22.2 | PLP1 | 135–185 kb 3′ | 100–150 kb duplication | Postulated down-regulation | (102,126) | Unusual phenotype; PLP1 duplications and deletions lead to Pelizaeus–Merzbacher syndrome |
| Townes–Brocks syndrome | 107480 | 16q12.1 | SALL1 | >180 kb 5′ | Balanced translocation | Postulated down-regulation | (127) | |
| X-linked recessive hypoparathyroidism | 307700 | Xq27.1 | SOX3 | 67 kb 3′ | Deletion of ∼25 kb with insertion of ∼340 kb | Postulated down-regulation | (99) |
Examples of databases and resources for studies of structural variation
| Name | Host | Description | Website |
|---|---|---|---|
| Database of Genomic Variants | The Centre for Applied Genomics, The Hospital for Sick Children, Toronto | A comprehensive summary of human large-scale genomic variants with information about frequency and their relation to genes, segmental duplications and genome assembly gaps | projects.tcag.ca/variation/ |
| Human Structural Variation Database | Genome Sciences, University of Washington | A catalog of human genomic polymorphisms ascertained by experimental and computational analyses | humanparalogy.gs.washingtonedu/structuralvariation/ |
| Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER) | Wellcome Trust Sanger Institute | A database of sub-microscopic chromosomal imbalances with links to resulting phenotypes | www.sanger.ac.uk/postgenomics/decipher/ |
| Human Genome Epidemiology Network (HuGENet) | Centers for Disease Control and Prevention | Committed to assessing the impact of human genome variation on population health and how genetic information can be used to improve health and prevent disease | www.cdc.gov/genomics/hugenet/default.htm |
| Developmental Genome Anatomy Project (DGAP) | Harvard University | Database of balanced chromosomal rearrangements critical to development | www.bwhpathology.org/dgap/ |
| Mendelian Cytogenetics Network Online Database | Wilhelm Johannsen Centre for Functional Genome Research | A collection of disease-associated balanced chromosomal rearrangements | www.mcndb.org/index.jsp |
| Chromosome Abnormality Database (CAD) | NHS, UK | A collection of both constitutional and acquired abnormal karyotypes reported by UK Regional Cytogenetics Centers | www.ukcad.org.uk/cocoon/ukcad/ |
| The European Collection of Cell Cultures (ECACC) | ECACC and the Health Protection Agency | A cell-culture collection to service the research community consisting of over 40 000 cell lines representing 45 different species | www.ecacc.org.uk/ |
| Coriell Cell Respository | National Institutes of Health (NIH) | Provides essential research reagents to the scientific community by establishing, verifying, maintaining and distributing cells cultures and DNA derived from cell cultures | locus.umdnj.edu/ccr |
| NIGMS Human Genetic Cell Repository | National Institute of General Medical Sciences | Supplies scientists with the materials for accelerating disease gene discovery with highly characterized, viable and contaminant-free cell cultures and DNA samples | locus.umdnj.edu/nigms/ |
| Mitelman Database of Chromosome Aberrations in Cancer | National Cancer Institute | Relates chromosomal aberrations to tumor characteristics, based on either individual cases or associations | cgap.nci.nih.gov/chromosomes/mitelman |
| Name | Host | Description | Website |
|---|---|---|---|
| Database of Genomic Variants | The Centre for Applied Genomics, The Hospital for Sick Children, Toronto | A comprehensive summary of human large-scale genomic variants with information about frequency and their relation to genes, segmental duplications and genome assembly gaps | projects.tcag.ca/variation/ |
| Human Structural Variation Database | Genome Sciences, University of Washington | A catalog of human genomic polymorphisms ascertained by experimental and computational analyses | humanparalogy.gs.washingtonedu/structuralvariation/ |
| Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER) | Wellcome Trust Sanger Institute | A database of sub-microscopic chromosomal imbalances with links to resulting phenotypes | www.sanger.ac.uk/postgenomics/decipher/ |
| Human Genome Epidemiology Network (HuGENet) | Centers for Disease Control and Prevention | Committed to assessing the impact of human genome variation on population health and how genetic information can be used to improve health and prevent disease | www.cdc.gov/genomics/hugenet/default.htm |
| Developmental Genome Anatomy Project (DGAP) | Harvard University | Database of balanced chromosomal rearrangements critical to development | www.bwhpathology.org/dgap/ |
| Mendelian Cytogenetics Network Online Database | Wilhelm Johannsen Centre for Functional Genome Research | A collection of disease-associated balanced chromosomal rearrangements | www.mcndb.org/index.jsp |
| Chromosome Abnormality Database (CAD) | NHS, UK | A collection of both constitutional and acquired abnormal karyotypes reported by UK Regional Cytogenetics Centers | www.ukcad.org.uk/cocoon/ukcad/ |
| The European Collection of Cell Cultures (ECACC) | ECACC and the Health Protection Agency | A cell-culture collection to service the research community consisting of over 40 000 cell lines representing 45 different species | www.ecacc.org.uk/ |
| Coriell Cell Respository | National Institutes of Health (NIH) | Provides essential research reagents to the scientific community by establishing, verifying, maintaining and distributing cells cultures and DNA derived from cell cultures | locus.umdnj.edu/ccr |
| NIGMS Human Genetic Cell Repository | National Institute of General Medical Sciences | Supplies scientists with the materials for accelerating disease gene discovery with highly characterized, viable and contaminant-free cell cultures and DNA samples | locus.umdnj.edu/nigms/ |
| Mitelman Database of Chromosome Aberrations in Cancer | National Cancer Institute | Relates chromosomal aberrations to tumor characteristics, based on either individual cases or associations | cgap.nci.nih.gov/chromosomes/mitelman |
Examples of databases and resources for studies of structural variation
| Name | Host | Description | Website |
|---|---|---|---|
| Database of Genomic Variants | The Centre for Applied Genomics, The Hospital for Sick Children, Toronto | A comprehensive summary of human large-scale genomic variants with information about frequency and their relation to genes, segmental duplications and genome assembly gaps | projects.tcag.ca/variation/ |
| Human Structural Variation Database | Genome Sciences, University of Washington | A catalog of human genomic polymorphisms ascertained by experimental and computational analyses | humanparalogy.gs.washingtonedu/structuralvariation/ |
| Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER) | Wellcome Trust Sanger Institute | A database of sub-microscopic chromosomal imbalances with links to resulting phenotypes | www.sanger.ac.uk/postgenomics/decipher/ |
| Human Genome Epidemiology Network (HuGENet) | Centers for Disease Control and Prevention | Committed to assessing the impact of human genome variation on population health and how genetic information can be used to improve health and prevent disease | www.cdc.gov/genomics/hugenet/default.htm |
| Developmental Genome Anatomy Project (DGAP) | Harvard University | Database of balanced chromosomal rearrangements critical to development | www.bwhpathology.org/dgap/ |
| Mendelian Cytogenetics Network Online Database | Wilhelm Johannsen Centre for Functional Genome Research | A collection of disease-associated balanced chromosomal rearrangements | www.mcndb.org/index.jsp |
| Chromosome Abnormality Database (CAD) | NHS, UK | A collection of both constitutional and acquired abnormal karyotypes reported by UK Regional Cytogenetics Centers | www.ukcad.org.uk/cocoon/ukcad/ |
| The European Collection of Cell Cultures (ECACC) | ECACC and the Health Protection Agency | A cell-culture collection to service the research community consisting of over 40 000 cell lines representing 45 different species | www.ecacc.org.uk/ |
| Coriell Cell Respository | National Institutes of Health (NIH) | Provides essential research reagents to the scientific community by establishing, verifying, maintaining and distributing cells cultures and DNA derived from cell cultures | locus.umdnj.edu/ccr |
| NIGMS Human Genetic Cell Repository | National Institute of General Medical Sciences | Supplies scientists with the materials for accelerating disease gene discovery with highly characterized, viable and contaminant-free cell cultures and DNA samples | locus.umdnj.edu/nigms/ |
| Mitelman Database of Chromosome Aberrations in Cancer | National Cancer Institute | Relates chromosomal aberrations to tumor characteristics, based on either individual cases or associations | cgap.nci.nih.gov/chromosomes/mitelman |
| Name | Host | Description | Website |
|---|---|---|---|
| Database of Genomic Variants | The Centre for Applied Genomics, The Hospital for Sick Children, Toronto | A comprehensive summary of human large-scale genomic variants with information about frequency and their relation to genes, segmental duplications and genome assembly gaps | projects.tcag.ca/variation/ |
| Human Structural Variation Database | Genome Sciences, University of Washington | A catalog of human genomic polymorphisms ascertained by experimental and computational analyses | humanparalogy.gs.washingtonedu/structuralvariation/ |
| Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER) | Wellcome Trust Sanger Institute | A database of sub-microscopic chromosomal imbalances with links to resulting phenotypes | www.sanger.ac.uk/postgenomics/decipher/ |
| Human Genome Epidemiology Network (HuGENet) | Centers for Disease Control and Prevention | Committed to assessing the impact of human genome variation on population health and how genetic information can be used to improve health and prevent disease | www.cdc.gov/genomics/hugenet/default.htm |
| Developmental Genome Anatomy Project (DGAP) | Harvard University | Database of balanced chromosomal rearrangements critical to development | www.bwhpathology.org/dgap/ |
| Mendelian Cytogenetics Network Online Database | Wilhelm Johannsen Centre for Functional Genome Research | A collection of disease-associated balanced chromosomal rearrangements | www.mcndb.org/index.jsp |
| Chromosome Abnormality Database (CAD) | NHS, UK | A collection of both constitutional and acquired abnormal karyotypes reported by UK Regional Cytogenetics Centers | www.ukcad.org.uk/cocoon/ukcad/ |
| The European Collection of Cell Cultures (ECACC) | ECACC and the Health Protection Agency | A cell-culture collection to service the research community consisting of over 40 000 cell lines representing 45 different species | www.ecacc.org.uk/ |
| Coriell Cell Respository | National Institutes of Health (NIH) | Provides essential research reagents to the scientific community by establishing, verifying, maintaining and distributing cells cultures and DNA derived from cell cultures | locus.umdnj.edu/ccr |
| NIGMS Human Genetic Cell Repository | National Institute of General Medical Sciences | Supplies scientists with the materials for accelerating disease gene discovery with highly characterized, viable and contaminant-free cell cultures and DNA samples | locus.umdnj.edu/nigms/ |
| Mitelman Database of Chromosome Aberrations in Cancer | National Cancer Institute | Relates chromosomal aberrations to tumor characteristics, based on either individual cases or associations | cgap.nci.nih.gov/chromosomes/mitelman |
References
Iafrate, A.J., Feuk, L., Rivera, M.N., Listewnik, M.L., Donahoe, P.K., Qi, Y., Scherer, S.W. and Lee, C. (
Sebat, J., Lakshmi, B., Troge, J., Alexander, J., Young, J., Lundin, P., Maner, S., Massa, H., Walker, M., Chi, M. et al. (
Sharp, A.J., Locke, D.P., McGrath, S.D., Cheng, Z., Bailey, J.A., Vallente, R.U., Pertz, L.M., Clark, R.A., Schwartz, S., Segraves, R. et al. (
Tuzun, E., Sharp, A.J., Bailey, J.A., Kaul, R., Morrison, V.A., Pertz, L.M., Haugen, E., Hayden, H., Albertson, D., Pinkel, D. et al. (
Feuk, L., Macdonald, J.R., Tang, T., Carson, A.R., Li, M., Rao, G., Khaja, R. and Scherer, S.W. (
Conrad, D.F., Andrews, T.D., Carter, N.P., Hurles, M.E. and Pritchard, J.K. (
Hinds, D.A., Kloek, A.P., Jen, M., Chen, X. and Frazer, K.A. (
McCarroll, S.A., Hadnott, T.N., Perry, G.H., Sabeti, P.C., Zody, M.C., Barrett, J.C., Dallaire, S., Gabriel, S.B., Lee, C., Daly, M.J. et al. (
Altshuler, D., Brooks, L.D., Chakravarti, A., Collins, F.S., Daly, M.J. and Donnelly, P. (
The International HapMap Consortium (
Murray, J.C., Buetow, K.H., Weber, J.L., Ludwigsen, S., Scherpbier-Heddema, T., Manion, F., Quillen, J., Sheffield, V.C., Sunden, S., Duyk, G.M. et al. (
Kong, A., Gudbjartsson, D.F., Sainz, J., Jonsdottir, G.M., Gudjonsson, S.A., Richardsson, B., Sigurdardottir, S., Barnard, J., Hallbeck, B., Masson, G. et al. (
Nakamura, Y., Leppert, M., O'Connell, P., Wolff, R., Holm, T., Culver, M., Martin, C., Fujimoto, E., Hoff, M., Kumlin, E. et al. (
Buckley, P.G., Mantripragada, K.K., Piotrowski, A., Diaz de Stahl, T. and Dumanski, J.P. (
Freeman, J.L., Perry, G.H., Feuk, L., Redon, R., McCarroll, S.A., Althshuler, D.M., Aburatani, H., Jones, K., Tyler-Smith, C., Hurles, M.E. et al.(
Stankiewicz, P. and Lupski, J.R. (
Bugge, M., Bruun-Petersen, G., Brondum-Nielsen, K., Friedrich, U., Hansen, J., Jensen, G., Jensen, P.K., Kristoffersson, U., Lundsteen, C., Niebuhr, E. et al. (
Lupski, J.R. and Stankiewicz, P. (
Feuk, L., Carson, A.R. and Scherer, S.W. (
van Ommen, G.J. (
Cawthon, R.M., Weiss, R., Xu, G.F., Viskochil, D., Culver, M., Stevens, J., Robertson, M., Dunn, D., Gesteland, R., O'Connell, P. et al. (
Call, K.M., Glaser, T., Ito, C.Y., Buckler, A.J., Pelletier, J., Haber, D.A., Rose, E.A., Kral, A., Yeger, H., Lewis, W.H. et al. (
Royer-Pokora, B., Kunkel, L.M., Monaco, A.P., Goff, S.C., Newburger, P.E., Baehner, R.L., Cole, F.S., Curnutte, J.T. and Orkin, S.H. (
Monaco, A.P., Neve, R.L., Colletti-Feener, C., Bertelson, C.J., Kurnit, D.M. and Kunkel, L.M. (
Burghes, A.H., Logan, C., Hu, X., Belfall, B., Worton, R.G. and Ray, P.N. (
Friend, S.H., Bernards, R., Rogelj, S., Weinberg, R.A., Rapaport, J.M., Albert, D.M. and Dryja, T.P. (
Gessler, M., Poustka, A., Cavenee, W., Neve, R.L., Orkin, S.H. and Bruns, G.A. (
Wallace, M.R., Marchuk, D.A., Andersen, L.B., Letcher, R., Odeh, H.M., Saulino, A.M., Fountain, J.W., Brereton, A., Nicholson, J., Mitchell, A.L. et al. (
Vissers, L.E., Veltman, J.A., van Kessel, A.G. and Brunner, H.G. (
Rosenberg, C., Knijnenburg, J., Bakker, E., Vianna-Morgante, A.M., Sloos, W., Otto, P.A., Kriek, M., Hansson, K., Krepischi-Santos, A.C., Fiegler, H. et al. (
Schoumans, J., Ruivenkamp, C., Holmberg, E., Kyllerman, M., Anderlid, B.M. and Nordenskjold, M. (
de Vries, B.B., Pfundt, R., Leisink, M., Koolen, D.A., Vissers, L.E., Janssen, I.M., Reijmersdal, S., Nillesen, W.M., Huys, E.H., Leeuw, N. et al. (
Shaw-Smith, C., Redon, R., Rickman, L., Rio, M., Willatt, L., Fiegler, H., Firth, H., Sanlaville, D., Winter, R., Colleaux, L. et al. (
Miyake, N., Shimokawa, O., Harada, N., Sosonkina, N., Okubo, A., Kawara, H., Okamoto, N., Kurosawa, K., Kawame, H., Iwakoshi, M. et al. (
Bauters, M., Van Esch, H., Marynen, P. and Froyen, G. (
Lugtenberg, D., de Brouwer, A.P., Kleefstra, T., Oudakker, A.R., Frints, S.G., Schrander-Stumpel, C.T., Fryns, J.P., Jensen, L.R., Chelly, J., Moraine, C. et al. (
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I.C., Soderstrom, H., Giros, B., Leboyer, M., Gillberg, C. et al. (
Millar, J.K., Wilson-Annan, J.C., Anderson, S., Christie, S., Taylor, M.S., Semple, C.A., Devon, R.S., Clair, D.M., Muir, W.J., Blackwood, D.H. et al. (
Taipale, M., Kaminen, N., Nopola-Hemmi, J., Haltia, T., Myllyluoma, B., Lyytinen, H., Muller, K., Kaaranen, M., Lindsberg, P.J., Hannula-Jouppi, K. et al. (
Hannula-Jouppi, K., Kaminen-Ahola, N., Taipale, M., Eklund, R., Nopola-Hemmi, J., Kaariainen, H. and Kere, J. (
Lai, C.S., Fisher, S.E., Hurst, J.A., Vargha-Khadem, F. and Monaco, A.P. (
Abelson, J.F., Kwan, K.Y., O'Roak, B.J., Baek, D.Y., Stillman, A.A., Morgan, T.M., Mathews, C.A., Pauls, D.L., Rasin, M.R., Gunel, M. et al. (
Somerville, M.J., Mervis, C.B., Young, E.J., Seo, E.J., del Campo, M., Bamforth, S., Peregrine, E., Loo, W., Lilley, M., Perez-Jurado, L.A. et al. (
Singleton, A.B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., Hulihan, M., Peuralinna, T., Dutra, A., Nussbaum, R. et al. (
Rovelet-Lecrux, A., Hannequin, D., Raux, G., Le Meur, N., Laquerriere, A., Vital, A., Dumanchin, C., Feuillette, S., Brice, A., Vercelletto, M. et al. (
Aitman, T.J., Dong, R., Vyse, T.J., Norsworthy, P.J., Johnson, M.D., Smith, J., Mangion, J., Roberton-Lowe, C., Marshall, A.J., Petretto, E. et al. (
Groot, P.C., Mager, W.H. and Frants, R.R. (
Hollox, E.J., Armour, J.A. and Barber, J.C. (
Aldred, P.M., Hollox, E.J. and Armour, J.A. (
Linzmeier, R.M. and Ganz, T. (
Gonzalez, E., Kulkarni, H., Bolivar, H., Mangano, A., Sanchez, R., Catano, G., Nibbs, R.J., Freedman, B.I., Quinones, M.P., Bamshad, M.J. et al. (
Sprenger, R., Schlagenhaufer, R., Kerb, R., Bruhn, C., Brockmoller, J., Roots, I. and Brinkmann, U. (
Nguyen, D.Q., Webber, C. and Ponting, C.P. (
Hayes, J.D. and Strange, R.C. (
McLellan, R.A., Oscarson, M., Alexandrie, A.K., Seidegard, J., Evans, D.A., Rannug, A. and Ingelman-Sundberg, M. (
Rao, Y., Hoffmann, E., Zia, M., Bodin, L., Zeman, M., Sellers, E.M. and Tyndale, R.F. (
Koppens, P.F., Hoogenboezem, T. and Degenhart, H.J. (
Dalen, P., Dahl, M.L., Ruiz, M.L., Nordin, J. and Bertilsson, L. (
Mitsunaga, Y., Kubota, T., Ishiguro, A., Yamada, Y., Sasaki, H., Chiba, K. and Iga, T. (
Ingelman-Sundberg, M. (
Chung, E.K., Yang, Y., Rennebohm, R.M., Lokki, M.L., Higgins, G.C., Jones, K.N., Zhou, B., Blanchong, C.A. and Yu, C.Y. (
Osborne, L.R., Li, M., Pober, B., Chitayat, D., Bodurtha, J., Mandel, A., Costa, T., Grebe, T., Cox, S., Tsui, L.C. et al. (
Gimelli, G., Pujana, M.A., Patricelli, M.G., Russo, S., Giardino, D., Larizza, L., Cheung, J., Armengol, L., Schinzel, A., Estivill, X. et al. (
Visser, R., Shimokawa, O., Harada, N., Kinoshita, A., Ohta, T., Niikawa, N. and Matsumoto, N. (
Scherer, S.W., Cheung, J., MacDonald, J.R., Osborne, L.R., Nakabayashi, K., Herbrick, J.A., Carson, A.R., Parker-Katiraee, L., Skaug, J., Khaja, R. et al. (
Ovcharenko, I., Loots, G.G., Nobrega, M.A., Hardison, R.C., Miller, W. and Stubbs, L. (
Hillier, L.W., Graves, T.A., Fulton, R.S., Fulton, L.A., Pepin, K.H., Minx, P., Wagner-McPherson, C., Layman, D., Wylie, K., Sekhon, M. et al. (
Schotta, G., Ebert, A., Dorn, R. and Reuter, G. (
Tham, W.H. and Zakian, V.A. (
Crolla, J.A. and van Heyningen, V. (
Lauderdale, J.D., Wilensky, J.S., Oliver, E.R., Walton, D.S. and Glaser, T. (
Fantes, J., Redeker, B., Breen, M., Boyle, S., Brown, J., Fletcher, J., Jones, S., Bickmore, W., Fukushima, Y., Mannens, M. et al. (
Crolla, J.A., Cross, I., Atkey, N., Wright, M. and Oley, C.A. (
Belloni, E., Muenke, M., Roessler, E., Traverso, G., Siegel-Bartelt, J., Frumkin, A., Mitchell, H.F., Donis-Keller, H., Helms, C., Hing, A.V. et al. (
Fernandez, B.A., Siegel-Bartelt, J., Herbrick, J.A., Teshima, I. and Scherer, S.W. (
Wallis, D.E., Roessler, E., Hehr, U., Nanni, L., Wiltshire, T., Richieri-Costa, A., Gillessen-Kaesbach, G., Zackai, E.H., Rommens, J. and Muenke, M. (
Velagaleti, G.V., Bien-Willner, G.A., Northup, J.K., Lockhart, L.H., Hawkins, J.C., Jalal, S.M., Withers, M., Lupski, J.R. and Stankiewicz, P. (
Erdel, M., Lane, A.H., Fresser, F., Probst, P., Utermann, G. and Scherer, G. (
Wagner, T., Wirth, J., Meyer, J., Zabel, B., Held, M., Zimmer, J., Pasantes, J., Bricarelli, F.D., Keutel, J., Hustert, E. et al. (
Ninomiya, S., Isomura, M., Narahara, K., Seino, Y. and Nakamura, Y. (
Foster, J.W., Dominguez-Steglich, M.A., Guioli, S., Kowk, G., Weller, P.A., Stevanovic, M., Weissenbach, J., Mansour, S., Young, I.D., Goodfellow, P.N. et al. (
Pop, R., Conz, C., Lindenberg, K.S., Blesson, S., Schmalenberger, B., Briault, S., Pfeifer, D. and Scherer, G. (
Wirth, J., Wagner, T., Meyer, J., Pfeiffer, R.A., Tietze, H.U., Schempp, W. and Scherer, G. (
Wunderle, V.M., Critcher, R., Hastie, N., Goodfellow, P.N. and Schedl, A. (
Pfeifer, D., Kist, R., Dewar, K., Devon, K., Lander, E.S., Birren, B., Korniszewski, L., Back, E. and Scherer, G. (
Romao, L., Osorio-Almeida, L., Higgs, D.R., Lavinha, J. and Liebhaber, S.A. (
Higgs, D.R., Wood, W.G., Jarman, A.P., Sharpe, J., Lida, J., Pretorius, I.M. and Ayyub, H. (
Driscoll, M.C., Dobkin, C.S. and Alter, B.P. (
Barbour, V.M., Tufarelli, C., Sharpe, J.A., Smith, Z.E., Ayyub, H., Heinlein, C.A., Sloane-Stanley, J., Indrak, K., Wood, W.G. and Higgs, D.R. (
McElreavey, K., Vilain, E., Barbaux, S., Fuqua, J.S., Fechner, P.Y., Souleyreau, N., Doco-Fenzy, M., Gabriel, R., Quereux, C., Fellous, M. et al. (
McElreavy, K., Vilain, E., Abbas, N., Costa, J.M., Souleyreau, N., Kucheria, K., Boucekkine, C., Thibaud, E., Brauner, R., Flamant, F. et al. (
Kleinjan, D.J. and van Heyningen, V. (
David, D., Cardoso, J., Marques, B., Marques, R., Silva, E.D., Santos, H. and Boavida, M.G. (
Bowl, M.R., Nesbit, M.A., Harding, B., Levy, E., Jefferson, A., Volpi, E., Rizzoti, K., Lovell-Badge, R., Schlessinger, D., Whyte, M.P. et al. (
Wakui, K., Gregato, G., Ballif, B.C., Glotzbach, C.D., Bailey, K.A., Kuo, P.L., Sue, W.C., Sheffield, L.J., Irons, M., Gomez, E.G. et al. (
Beysen, D., Raes, J., Leroy, B.P., Lucassen, A., Yates, J.R., Clayton-Smith, J., Ilyina, H., Brooks, S.S., Christin-Maitre, S., Fellous, M. et al. (
Lee, J.A., Madrid, R.E., Sperle, K., Ritterson, C.M., Hobson, G.M., Garbern, J., Lupski, J.R. and Inoue, K. (
Muncke, N., Wogatzky, B.S., Breuning, M., Sistermans, E.A., Endris, V., Ross, M., Vetrie, D., Catsman-Berrevoets, C.E. and Rappold, G. (
Ellison, J.W., Tekin, M., Sikes, K.S., Yankowitz, J., Shapiro, L., Rappold, G.A. and Neely, K.E. (
Lettice, L.A. and Hill, R.E. (
Inoue, K. and Lupski, J.R. (
Ravnan, J.B., Tepperberg, J.H., Papenhausen, P., Lamb, A.N., Hedrick, J., Eash, D., Ledbetter, D.H. and Martin, C.L. (
Ioannidis, J.P., Gwinn, M., Little, J., Higgins, J.P., Bernstein, J.L., Boffetta, P., Bondy, M., Bray, M.S., Brenchley, P.E., Buffler, P.A. et al. (
Yu, C.E., Dawson, G., Munson, J., D'Souza, I., Osterling, J., Estes, A., Leutenegger, A.L., Flodman, P., Smith, M., Raskind, W.H. et al. (
Repping, S., van Daalen, S.K., Brown, L.G., Korver, C.M., Lange, J., Marszalek, J.D., Pyntikova, T., van der Veen, F., Skaletsky, H., Page, D.C. et al. (
Shendure, J., Mitra, R.D., Varma, C. and Church, G.M. (
International Human Genome Sequencing Consortium (
Kruglyak, L. and Nickerson, D.A. (
Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W. et al. (
Weber, J.L., David, D., Heil, J., Fan, Y., Zhao, C. and Marth, G. (
Dawson, E., Chen, Y., Hunt, S., Smink, L.J., Hunt, A., Rice, K., Livingston, S., Bumpstead, S., Bruskiewich, R., Sham, P. et al. (
Litt, M. and Luty, J.A. (
Ellegren, H. (
Jeffreys, A.J., Wilson, V. and Thein, S.L. (
Naslund, K., Saetre, P., von Salome, J., Bergstrom, T.F., Jareborg, N. and Jazin, E. (
Fredman, D., White, S.J., Potter, S., Eichler, E.E., Dunnen, J.T. and Brookes, A.J. (
Van Dyke, D.L., Weiss, L., Roberson, J.R. and Babu, V.R. (
Gardner, R.J.M. and Sunderland, G.R. (
Warburton, D. (
Nussbaum, R.L., McInnes, R.R. and Willard, H.F. (
Lee, J.A., Cheung, S.W., Ward, P.A., Inoue, K. and Lupski, J.R. (
Marlin, S., Blanchard, S., Slim, R., Lacombe, D., Denoyelle, F., Alessandri, J.L., Calzolari, E., Drouin-Garraud, V., Ferraz, F.G., Fourmaintraux, A. et al. (

{kind=link}
{kind=link}