




Abstract
Mutations in genes encoding both DJ-1 and pten-induced kinase 1 (PINK1) are independently linked to autosomal recessive early-onset familial forms of Parkinson's disease (PD). We here report identification of a family with PD patients harboring novel heterozygous missense mutations in both PINK1 and DJ-1 genes encoding DJ-1A39S and PINK1P399L, respectively. In transfected cells, DJ-1 interacts with PINK1. PINK1P399L is less stable than the wild-type protein and is degraded via the ubiquitin-mediated proteasomal pathway. Expression of wild-type DJ-1 increased steady-state levels of PINK1, whereas expression of DJ-1A39S reduced steady-state levels of PINK1. Furthermore, co-expression of wild-type DJ-1 and PINK1 suppresses neurotoxin 1-methyl-4-phenylpyridinium (MPP+)-induced death of dopaminergic SH-SY5Y cells. In contrast, co-expression of PD-associated DJ-1A39S and PINK1P399L significantly potentiated susceptibility of SH-SY5Y cells to MPP+-induced cell death. This study reports the first case of autosomal recessive PD with digenic inheritance and demonstrates that DJ-1 and PINK1 physically associate and collaborate to protect cells against stress via complex formation.
INTRODUCTION
Parkinson's disease (PD) is the most frequent neurodegenerative movement disorder characterized by age-dependent resting tremor, muscular rigidity and akinesia (1,2). The pathological hallmarks of PD patients include progressive loss of dopaminergic neurons in the substantia nigra pars compacta as well as the presence of ubiquitin-positive Lewy neurites and Lewy bodies in the remaining neurons. Majority of the PD cases appear to be sporadic. However, specific genetic defects are linked to familial form of PD that resemble idiopathic PD. Mutations in at least six genes are individually linked to familial form of PD, including autosomal dominant mutations in α-synuclein, uchL1 and LRRK2 and autosomal recessive mutations in parkin, PINK1 and DJ-1 (3–6). Characterization of these genes has provided important insights into the pathogenesis of PD. For example, α-synuclein is a major structural component of Lewy bodies in PD (7). PD-associated α-synuclein mutant proteins show an increased propensity to self-aggregate to form oligomeric species and Lewy body-like fibrils comparing to wild-type α-synuclein, directly linking the disease-associated α-synuclein mutant proteins to PD pathology (8,9).
Mutations in the parkin, DJ-1 and PINK1 genes are associated with autosomal recessive early-onset familial form of PD (10–12). Fifty percent of autosomal recessive early-onset familial PD cases harbor mutations in parkin gene. PINK1 mutations appear to be the second common genetic cause in autosomal recessive PD after parkin, found in 8–15% early-onset PD cases (10,13,14). Nevertheless, DJ-1 mutation is rare, presenting perhaps <1% in early-onset PD cases (15). Interestingly, both homozygous and heterozygous mutations of the parkin, DJ-1 and PINK1 genes are found to associate with PD cases (16–18). The mechanism for PD caused by these heterozygous mutations is not known.
Functional studies indicate that the parkin gene encodes an E3 ligase in ubiquitin–proteasome pathway (19–21). Most of PD-associated mutations in parkin tend to either impair the interaction of parkin with ubiquitin conjugating enzymes (E2s) or substrate or inhibit its E3 ligase activity. In contrast to parkin, our knowledge of DJ-1 and PINK1 is very limited. DJ-1 protein likely functions in antagonizing oxidative stress (22,23). PINK1 is a putative serine/threonine kinase with undefined function and substrate (10,24). One important question is whether parkin, DJ-1 and PINK1 function collaboratively under normal and pathological conditions.
In this study, we provide the genetic and biochemical evidences indicating that DJ-1 and PINK1 are genetically linked and physically associated. Our results further show that DJ-1 and PINK1 collaborate to protect cells against oxidative stress.
RESULTS
Digenic mutations of DJ-1 and PINK1 in early-onset familial PD
We identified a Chinese family that included two individuals suffering recessive early-onset forms of PD (Fig. 1A). The proband (II-2, age 44) and her sister (II-1, age 46) showed resting tremor of the left leg initially at age 26 and 27, respectively. Disease progressively developed in all limbs, resulting in difficulty in walking, bradykinesia and postural instability. The symptoms were improved after sleeping and relieved by oral administration of L-dopa. Clinical examination revealed increased tension of neck muscle and upper limb muscles of the patients comparing to normal people. Other genetically related family members of the proband (I-2, II-3 and II-4) showed no clinically detectable abnormality at age 68, 42 and 36, respectively. The father of the proband (I-1), whose family members recalled having a weak tremor in both hands, died at age 58 with cardiopulmonary disease without clinical diagnosis of PD.
Sequence analysis detected no mutation in any exon of the parkin gene (data not shown). A heterozygous missense mutation, G115T, in exon 3 of DJ-1 changing the 39th alanine to serine (A39S) was found in individuals II-1, II-2, II-3 and II-4 (Fig. 1B). The mother (I-2) of proband had normal DJ-1. The DJ-1 G115T variation in this family is likely inherited from their father (I-1), although we could not definitively trace its origin because I-1 had no sibling. This notion is further supported by linkage analysis, showing that individuals II-1, II-2 and II-3 have the same haplotypes at DJ-1 loci (Fig. 2A). Individual II-4 harbors a cross within a region of DJ-1 locus on the chromosome originated from I-1. The cross, however, unlikely affects DJ-1 gene of individual II-4 because he carries the same DJ-1 mutation as his siblings (Fig. 2A). Further sequence analysis of PINK1 in this family also revealed a heterozygous missense mutation, C1196T, in exon 6 among individuals I-2, II-1, II-2 and II-3, changing proline to lysine at the 399th amino acid of PINK1 protein (P399L). No sequence change was found in PINK1 of II-4 (Fig. 1B). Individuals carrying PINK1C1196T inherit the same haplotype at PINK1 locus from I-2, indicating that PINK1C1196T originates from individual I-2 in the family (Fig. 2C). The distribution of changes in DJ-1 and PINK1 genes in the family was further verified by restriction fragment length analysis (RFLP) analysis (Fig. 1C and D). Individuals II-1 and II-2, who are clinically diagnosed with PD, are both heterozygous for DJ-1G115T and PINK1C1196T. In contrast, individuals with either DJ-1G115T alone (II-4) or PINK1C1196T alone (I-2) are clinically normal. Thus, digenic mutations of DJ-1G115T and PINK1C1196T are likely associated with PD pathogenesis in this family. Both DJ-1G115T and PINK1C1196T are novel mutations that are not reported in the literatures. DJ-1G115T and PINK1C1196T were not detected in 240 chromosomes from unrelated 120 ethnically matched patients with sporadic PD and 568 chromosomes from 284 ethnically matched clinically normal individuals, further supporting a pathological role of these two mutations in PD (data not shown). Nevertheless, individual II-3 also is heterozygous for DJ-1G115T and PINK1C1196T but shows no clinical PD features. RT–PCR detected the presence of single transcript of DJ-1, parkin and PINK1 in lymphocytes of each individual of this family (Fig. 2D). Moreover, sequence analysis of the PCR products revealed no additional mutation within coding sequences of DJ-1, parkin and PINK1 (data not shown). The results suggest that a normal parkin transcript is made by each individual of the family. Therefore, the disease caused by DJ-1 and PINK1 mutants is likely the lack of penetrance in individual II-3. Similar observations have been reported in families with pathogenic α-synuclein mutations (25). Nevertheless, we cannot exclude possible roles of other genetic factors in preventing II-3 from developing into disease. It is of interest to note that the affected individuals (II-1 and II-2) carry identical haplotypes that are different from their unaffected siblings at the loci of parkin and PINK1 (Fig. 2B and C).
An alanine at residue 39 of DJ-1 (DJ-1A39) is conserved among humans, monkeys, rats, mice, hamsters and zebrafish, but not in Drosophila and Caenorhabditiselegans (Fig. 3). Variation of pathogenic mutations in different species has been reported in several PD-linked genes, including DJ-1, PINK1 and α-synuclein (26–28). DJ-1A39 is located in the third β-sheet (β3) of DJ-1, in which β3 and β4 form a β-hairpin structure and participate in dimerization of DJ-1 (29,30). The A39S mutation may affect dimerization of DJ-1, as suggested for an L166P mutation (12,30). Proline 399 of PINK1 (PINK1P399) is located in the predicted kinase domain and conserved among humans, dogs, rats, mice, chicks, mosquitoes and Drosophila (Fig. 3). It is possible that PINK1P399L has altered PINK1 kinase activity. Thus, DJ-1A39 and PINK1P399 may play important functional roles in the respective proteins.
Physical association of DJ-1 and PINK1
Digenic inheritance of PD by DJ-1 and PINK1 mutations indicates a functional interaction between these two genes. Therefore, we examined potential association of DJ-1 and PINK1 proteins. SH-SY5Y neuroblastoma cells and HEK293 cells (data not shown) expressing myc-tagged DJ-1 and flag-tagged PINK1 in various combinations were lysed and immunoprecipitated with a monoclonal anti-myc antibody followed by immunoblotting with a polyclonal anti-flag antibody. Both wild-type PINK1 (PINK1wt) and PINK1P399L were co-immunoprecipitated with both wild-type DJ-1 (DJ-1wt) or DJ-1A39S proteins (Fig. 4A, upper panel). Reciprocal immunoprecipitation showed both DJ-1wt and DJ-1A39S co-precipitated PINK1wt and PINK1P399L (Fig. 4A, lower panel). Interestingly, a protein at the size of endogenous DJ-1 that was recognized by a monoclonal anti-DJ-1 antibody was co-precipitated with PINK1 (Fig. 4A, lower panel), suggesting that exogenous PINK1 interacts with endogenous DJ-1. To further define the DJ-1 binding site on PINK1, we co-expressed DJ-1 with four deletion variants of PINK1 in SH-SY5Y cells (Fig. 4B). Cell lysates were used to perform co-immunoprecipitation assay. A fragment containing N-terminal 253 amino acid residuals of the PINK1 failed to co-precipitate DJ-1 (Fig. 4C and D). In contrast, a fragment containing PINK1 N-terminal 334 amino acid residuals was robustly co-immunoprecipitated with DJ-1 (Fig. 4C and D). Moreover, DJ-1 also co-precipitated two other fragments with overlapping PINK1 amino acid residual 253–334. Together, both wild-type and mutant DJ-1 proteins can complex with either wild-type or mutant PINK1 through PINK1 amino acid residuals 253–334, a motif located at the N-terminal part of the kinase domain.
DJ-1 regulates stability of PINK1 in cells
To investigate functional regulation of DJ-1 and PINK1, we co-expressed DJ-1wt or DJ-1A39S with PINK1wt or PINK1P399L and observed their steady-state levels. Our preliminary studies indicate that PINK1 protein is unstable and hard to be detected by direct immunoblotting analysis without treatment of proteasome inhibitors (data not shown). We therefore analyzed steady-state level of PINK1 using immunoprecipitation followed by immunoblotting. PINK1 protein variants were immunoprecipitated with a monoclonal anti-flag antibody from equal amount cell lysates followed by immunoblotting with a polyclonal anti-flag antibody. PINK1P399L was seen at consistently lower levels than PINK1wt (Fig. 5A). The results are consistent with a recent report that PD-associated PINK1 mutant proteins are less stable than their wild-type counterpart (24). Expression of DJ-1wt increased the steady-state level of both PINK1wt and PINK1P399L by ∼50 and 40%, respectively (Fig. 5A and B). In contrast, expression of DJ-1A39S reduced the steady-state levels of both PINK1wt and PINK1P399L (Fig. 5A and B). The steady-state level of PINK1P399L was reduced ∼30% compared with that of PINK1wt in the presence of co-expressed DJ-1A39S (31.5±5%, n=3, *P<0.05, Student's t-test). Likewise, steady-state level of PINK1wt is also significantly reduced with co-expressing DJ-1A39S (20.6±3%, n=3, *P<0.05, Student's t-test). However, the steady-state level of DJ-1wt was only slightly higher than that of DJ-1A39S (Fig. 5A). These results indicate that DJ-1wt enhances PINK1 stability, whereas DJ-1A39S potentiates PINK1 degradation.
We next asked whether PINK1 was degraded by the ubiquitin–proteasome pathway. SH-SY5Y cells stably expressing PINK1wt or DJ-1wt were treated with the proteasome inhibitor MG132 or with the protease inhibitors leupeptin or NH4Cl. PINK1 accumulation was observed in cells treated with MG132 in the presence or absence of the protein synthesis inhibitor cycloheximide. In contrast, little PINK1 accumulation was seen in cells treated with either leupeptin or NH4Cl (Fig. 6A). These results suggest that PINK1 is degraded via the proteasomal pathway. Interestingly, the treatments described above had little effect on DJ-1 levels in cells. Consistent with these findings, PINK1wt is heavily ubiquitinated. However, PINK1P399L was less extensively ubiquitinated than the wild-type protein in cells lacking MG132 treatment, likely because PINK1P399L was rapidly degraded. In contrast, PINK1P399L was more ubiquitinated than PINK1wt in cells treated with MG132 (Fig. 6B and C). The observed PINK1 ubiquitination is unlikely a detection of other ubiquitinated proteins co-precipitated with PINK1. In this assay, cells were lysed in 2% sodium dodecyl sulfate (SDS) followed by boiling for 10 min. Under such stringent conditions, minimal non-covalent protein–protein interactions are preserved. These results indicate that PINK1P399L is degraded more efficiently than PINK1wt via the ubiquitin–proteasome pathway. Little ubiquitination of DJ-1 was detected in cells lacking MG132 treatment. Interestingly, a small portion of DJ-1wt was ubiquitinated in cells treated with MG132, indicating that a small portion of DJ-1wt was ubiquitinated and degraded via proteasome pathway.
DJ-1wt and PINK1wt, but not the PD-associated DJ-1 and PINK1 mutants, collaboratively function against 1-methyl-4-phenylpyridinium-induced oxidative stress
Expression of wild-type DJ-1 but not PD-linked DJ-1mutants protects cells from death induced by oxidative stress (12,22,31–33). To explore the functional consequences of expression of DJ-1A39S and PINK1P399L, we established SH-SY5Y cell lines stably expressing myc-tagged DJ-1 and VSVG-tagged PINK1 variants in various combinations. Expression of exogenous variants was verified by immunoblotting (Fig. 7A). Survival of stably transfected cell lines was determined following challenge with 1-methyl-4-phenylpyridinium (MPP+) for 12 h. As shown in Figure 7B, exposure of control cells transfected with empty plasmids to MPP+ resulted in reduced cell viability in a dose-dependent manner. Consistent with previous reports (12,22), expression of DJ-1wt rescued cell death induced by MPP+ treatment. Likewise, expression of PINK1wt resulted in increased cell viability. In contrast, cells expressing PD-associated DJ-1A39S or PINK1P399L showed reduced viability compared with control cells. The results indicate that both PINK1 and DJ-1 protects cells against stress, whereas their respective PD-associated mutants do not. Interestingly, co-expression of PINK1wt and DJ-1wt led to a synergistic increase in cell viability against MPP+-induced stress (P<0.001, Student's t-test; to control transfection). On the contrary, co-expression of PD-associated DJ-1A39S and PINK1P399L mutants significantly increased susceptibility of cells to death (P<0.001, Student's t-test; to control transfection), suggesting that DJ-1 and PINK1 function collaboratively to protect SH-SY5Y cells against MPP+-induced stress.
DISCUSSION
In this study, we present, to our knowledge, the first evidence of digenic inheritance of autosomal recessive early-onset familial forms of PD, demonstrate a physical association of DJ-1 and PINK1 proteins in the cell and reveal synergistic activity of DJ-1 and PINK1 in antagonizing MPP+-induced stress. Moreover, the results show that PD-associated DJ-1A39S and PINK1L399P mutants lose their ability to protect cells against MPP+-induced oxidative stress.
It is well documented that in several autosomal recessive early-onset PD cases heterozygous mutations in only one of the three autosomal recessive PD-linked genes, DJ-1, parkin and PINK, are detected (16–18). One explanation is that heterozygosity in any one of these genes increases susceptibility to PD. Our findings further indicate that some of these cases likely harbor heterozygous mutations in more than one autosomal recessive PD-linked gene. Alternatively, epigenetic inactivation of one gene allele in combination with a heterozygous mutation of another PD-associated gene may also cause disease. More importantly, this finding provides the first genetic evidence in human indicating functional interaction of PINK1 and DJ-1. The physical association of PINK1 and DJ-1 demonstrated in this study further supports this notion and suggests that these two PD-linked genes protect cell against PD-related insults likely through a common mechanism. PD pathogenic mutations of these genes likely disrupt the protective pathway resulting in eventual neuronal degeneration upon stress. This hypothesis is consistent with a recent study finding that parkin and DJ-1 become physically associated in vitro when cells undergo oxidative stress (34). It is also possible that other PD-associated genes are also functionally co-ordinated in order to maintain normal neuronal function and survival. Parkin, another gene linked to autosomal recessive early-onset familial form of PD, encodes an E3 ligase of synphilin-1, a minor O-linked glycosylated form of α-synuclein and LRRK2 (35–37). Mutations in synphilin-1, α-synuclein and LRRK2 are associated with familial form PD (3,37).
The molecular pathways regulated by DJ-1 and PINK1 and also directly linked to PD pathogenesis are not known. Both DJ-1 and PINK1 are individually indicated to protect cells from oxidative stress. DJ-1 is shown to protect cells from oxidative stress both in transfected cells and in Drosophila (12,22,38,39). Inactivation of DJ-1 in mouse and Drosophila results in mitochondrial dysfunction and sensitizing to oxidative stress (31,38–40). In transfected cells, loss of PINK1 function also potentiates the susceptibility to oxidative stress (10,41). Consistent with these findings, we found that overexpression of wild-type DJ-1 and PINK1 synergistically protect MPP+-induced cell death. In contrast, PD-associated DJ-1 and PINK1 mutants lose the anti-stress ability and even potentiate the susceptibility to MPP+-induced cell death. The increased sensitization of disease-associated PINK1 and DJ-1 mutants to oxidative stress may underlie a common mechanism for PD pathogenesis.
It remains unclear how DJ-1 and PINK1 protect neurons from undergoing oxidative stress. But, a potential clue may be their mitochondrial localization (10,42–44). Mitochondria are the main source of reactive oxygen and nitrogen species and are the cellular compartments critical for integration of intrinsic apoptosis pathways. PINK1 may play a critical role in maintain mitochondrial homeostasis. Reduced PINK1 protein stability or loss of function of PINK1 will therefore result in dysfunction of mitochondria, oxidative stress and eventual neuronal death. To support this notion, we found that PINK1 stability is decreased by PD-associated PINK1 and DJ-1 mutations.
It has been reported that DJ-1 protein forms homodimers (30,45). PD-linked mutations perturb DJ-1 dimerization, which contributes to the reduced ability of DJ-1 to combat anti-oxidative stress (34,45,46). We propose that PINK1 forms a complex with DJ-1, which renders PINK1 more stable and thereby potentiates PINK1's anti-stress activity. In contrast, PD-associated DJ-1 and PINK1 mutants could be either less stable than wild-type proteins or lose the ability to dimerize, resulting in reduced protection against stress and contributing to neurodegeneration seen in PD.
MATERIALS AND METHODS
Patients and genotyping
A two-generation family with autosomal recessive inherited PD was identified in Yunan, China. PD was clinically diagnosed in the Department of Neurology, Xiangya Hospital of Central South University. After informed written consent was obtained from all participants, blood samples were collected from five genetically related individuals.
Genotyping was essentially performed as previously described (47). Briefly, genomic DNA (gDNA) was isolated from peripheral blood cells. All exons of the parkin, PINK1 and DJ-1 genes were PCR-amplified using specific primers corresponding to the three genes. The PCR amplification products were treated with shrimp alkaline phosphatase and exonuclease I followed by direct sequencing using an ABI3100 sequencer. The gDNA corresponding to DJ-1 exon 3 (where G115T is located) and PINK1 exon 6 (where C1196T is located) were also PCR-amplified for RFLP analysis. The PCR products of DJ-1 exon 3 (249 bp) amplified from the wild-type allele were cleaved by BstNI into a 127 and a 122 bp fragments, whereas the DJ-1G115T mutant allele lost the BstNI cleavage site. The PCR product of PINK1 exon 6 (309 bp) amplified from the wild-type allele is a 309 bp fragment that cannot be cleaved by SapI. The same fragment amplified from the PINK1C1196T mutant allele was cleaved into 177 and 132 bp fragments by SapI. DNA fragments were separated on 6% non-denaturing polyacrylamide gels. The most likely haplotype was constructed by Cyrillic program (Electrotechnical Laboratory, Japan).
To detect transcripts of DJ-1, parkin and PINK1, total RNA was isolated from immortalized lymphocytes using TRIzol reagent (Invitrogen). RT–PCR was done as previously described (48). Primers used were 5′-cggaattcgccaccatggcttccaaaagagctctg-3′ (DJ-1, forward), 5′-cgggatccccgtctttaagaacaagtggagc-3′ (DJ-1, reverse); 5′-gggaattcatagtgtttgtcaggatcaac-3′ (parkin, forward), 5′-cgggatcccacgtcgaaccagtggtcccc-3′ (parkin, reverse); 5′-cggaattcgccaccatggcggtgcgacaggcg-3′ (PINK1, forward), 5′-cccaagcttcagggctgccctccatga-3′ (PINK1, reverse); 5′-aagtactccgtgtggatcgg-3′ (β-actin, forward) and 5′-aaagccatgccaatctcatc-3′ (β-actin, reverse). Entire coding sequences of DJ-1, parkin, and PINK1 were amplified followed by complete sequencing.
Sequence alignment
Amino acid sequences of DJ-1 and PINK1 from different species were retrieved from NCBI, respectively. Alignment was done with MacVector™ 7.2.2 (Accelrys).
Cell lines and plasmids
SH-SY5Y neuroblastoma cells and HEK293 cells were purchased from ATCC and maintained according to instruction. To generate stable transfectants, cDNAs encoding VSVG-tagged PINK1 variants (wild-type PINK1 and PINK1P399L) and myc-tagged DJ-1 variants (wild-type DJ-1 and DJ-1A39S) were PCR-amplified and cloned into pIRESpuro3 plasmid and pMSCVhyg retroviral plasmid (Clontech), respectively. SH-SY5Y cell pools stably expressing VSVG-tagged PINK1 variants were generated via puromycin selection (1.5 µg/ml). Double expressors were generated by further infection of PINK1 expressors with retroviruses encoding myc-tagged DJ-1 variants and selected in hygromycin (175 µg/ml). For transient transfection, cDNAs encoding myc-tagged, flag-tagged or VSVG-tagged PINK1 and myc-tagged, flag-tagged or VSVG-tagged DJ-1 were PCR generated and subcloned into pcDNA3.1(−) plasmid (Invitrogen). A plasmid encoding HA-tagged ubiquitin was kindly provided by Ze'ev Ronai. All epitope tags were added to the C-terminus of the proteins. All plasmids were sequence confirmed.
Antibodies and immunoassays
Monoclonal antibody against DJ-1 (E2.19) was purchased from Signet Laboratories. Antibodies against different tags and agarose conjugated with these antibodies were from Sigma. Immunoblotting and immunoprecipitation were as previously described (49). Briefly, cells were lysed in 1% Triton X-100 buffer (10 mm HEPES, pH 7.5, 142.5 mm KCl, 5 mm MgCl2, 1 mm EDTA) followed by centrifugation at 14 000g, 4°C for 30 min. The resulted supernatants were determined for protein concentration. Equal amount of proteins from each sample were immunoprecipitated with antibody-conjugated beads, including anti-myc agarose, anti-flag agarose and anti-VSVG agarose (Sigma) at 4°C overnight with nutation. The beads were washed with 1% Triton X-100 buffer for four times followed by boiling for 5 min in SDS loading buffer. The immunoprecipitated proteins were separated on 4–20% Tris-glycine precasted gels (Invitrogen). The separated proteins were electro-transferred onto Immuno-P membranes (Millipore). The membranes were blocked with 5% non-fat milk in phosphate-buffered saline (PBS) for 1 h, reacted with the primary antibodies diluted in 5% non-fat milk in PBS at 4°C overnight with nutation, followed by corresponding HRP-labeled secondary antibodies diluted in 5% non-fat milk and 0.1% Triton X-100 in PBS for 1 h. Immunoreacted proteins were detected by ECL (Amersham).
Treatment with MG132 (5 µM), leupeptin (50 µm), and NH4Cl (20 µm) was performed at 37°C overnight. Quantitation was done with an Alpha Imager 2200 V5.04 and analyzed with Instat 3 (GraphPad).
Protein ubiquitination in transfected cells
Ubiquitination was analyzed essentially as previously described (50). Briefly, cells transfected with various plasmid combinations were lysed in 2% SDS buffer (2% SDS, 150 mm NaCl, 10 mm Tris–HCl, pH 8.0, 2 mm sodium orthovanadate, 50 mm sodium fluoride, 1× protease inhibitors) and boiled for 10 min followed by sonication. Lysates were diluted 1:10 in dilution buffer (10 mm Tris–HCl, pH 8.0, 150 mm NaCl, 2 mm EDTA, 1% Triton), incubated at 4°C for 1 h with rotation and centrifuged at 14 000g for 30 min. The protein concentration of supernatants was determined by Dc protein assay (BioRad). Equal amounts (1–1.5 mg) of protein were used for immunoprecipitation. Immunoprecipitated proteins were washed with washing buffer (10 mm Tris–HCl, pH 8.0, 1 M NaCl, 1 mm EDTA, 1% NP-40), boiled in SDS sample buffer and separated on SDS–PAGE. Ubiquitin and precipitated proteins were immunodetected with respective antibodies.
Cell viability assay
SH-SY5Y cells stably expressing DJ-1 and PINK1 variants and controls transfected with empty plasmids were seeded in 96-well dishes (104 cells/well) overnight followed by treatment with MMP+ (Sigma) at various concentrations for 12 h. Cell viability was assayed with a CellTiter 96 MTT kit (Promega). Triplicates or quadruplicates were analyzed at every concentration in each assay. The results from three independent experiments were analyzed.
ACKNOWLEDGEMENTS
We thank Ze'ev Ronai for insightful discussion. This work is supported by grants from the National Institute of Health (Z.Z.), the Alzheimer's Disease Association (Z.Z.), the Chinese Natural Science Foundation and the Chinese Ministry of Science and Technology (T.B., K.X. and J.-H. X.).
Conflict of Interest statement. None declared.
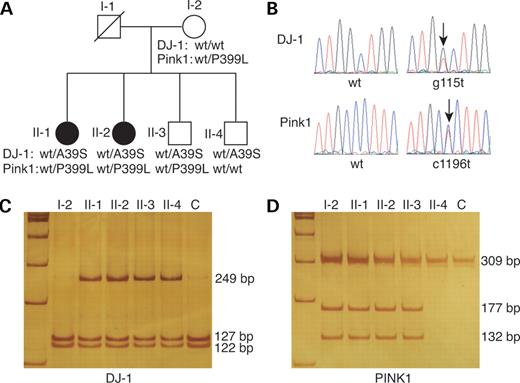
Figure 1. Association of digenic mutations of DJ-1 and PINK1 with early-onset familial PD. (A) Pedigree of a Chinese family with early-onset PD. Blackened symbols denote family members affected with parkinsonism. Genotypes of DJ-1 and PINK1 are indicated for each individual. wt, wild-type allele; A39S, DJ-1 mutant allele; P399L, PINK1 mutant allele. (B) Representative chromatogram of DJ-1 and PINK1. Arrows indicate the g115t and c1196t transitions for DJ-1 and PINK1, respectively, identified from the family. (C, D) RFLP analysis of DJ-1 and PINK1 fragments. Exon 3 of DJ-1 (C) and exon 6 of PINK1 (D) were PCR amplified from the individuals indicated on the top of the gel, followed by BstNI and SapI digestion, respectively. Wild-type allele gives two cleaved fragments (122 and 127 bp) for DJ-1 and an uncleaved 209 bp band for PINK1. An uncleaved 249 bp fragment and two cleaved fragments (177 and 132 bp) are produced for mutant alleles of DJ-1 and PINK1, respectively. (C) shows DNA from normal controls.
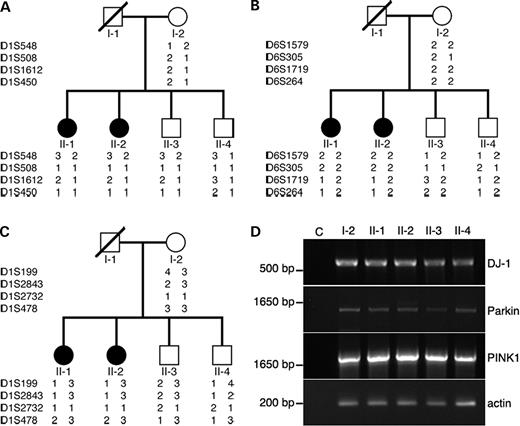
Figure 2. Haplotype and expression analysis of DJ-1, parkin and PINK1. Haplotype analysis of DJ-1 (A), parkin (B) and PINK1 (C) in the family was performed with four selected STR markers for each gene. STR markers used in these experiments are (from top to bottom) D1S548-D1S508-D1S1612-D1S450 (DJ-1), D6S1579-D6S305-D6S1719-D6S264 (parkin) and D1S199-D1S2843-D1S2732-D1S478 (PINK1). (D) Expression of DJ-1, parkin and PINK1 by individuals in the family. Coding sequences of DJ-1, parkin and PINK1 were RT–PCR amplified from lymphocyte RNAs of each individual. An actin cDNA fragment (template quantity control) and a negative control without cDNA templates (C) were included in experiments. Note: a single transcript for each of DJ-1, parkin and PINK1 genes was detected.

Figure 3. Conservation analysis of DJ-1A39 and PINK1P399. DJ-1 homologs from human, monkey, hamster, rat, mouse, zebrafish, Drosophila (dDJ-1.1 and dDJ-1.2) and C. elegans were aligned. DJ-1A39 is labeled as red (upper panel). PINK1 homologs from human, dog, rat, mouse, chick, mosquito and Drosophila (dPINK1) were aligned. PINK1P399 mutations are indicated as blue (lower panel). An asterisk indicates an identical amino acid, whereas a dot indicates a homologous amino acid across species.
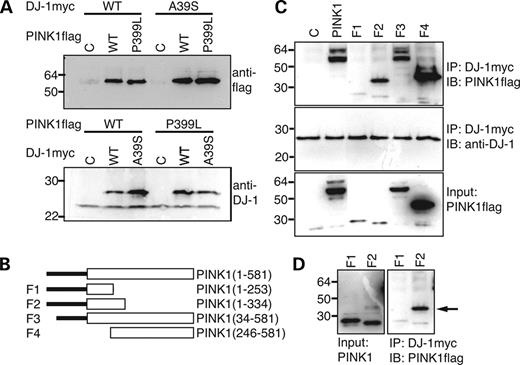
Figure 4. Physical association of DJ-1 and PINK1 in cells. (A) Co-immunoprecipitation of DJ-1 and PINK1. Upper panel: Myc-tagged wild-type DJ-1 or DJ-1A39S was co-transfected with either empty, flag-tagged wild-type PINK1 or flag-tagged PINK1P399L into SH-SY5Y cells. Cell lysates were immunoprecipitated with an anti-myc tag antibody followed by immunoblotting with an anti-flag tag antibody. Lower panel: flag-tagged wild-type PINK1 or PINK1P399L was co-transfected with either empty, myc-tagged wild-type DJ-1 or myc-tagged DJ-1A39S into SH-SY5Y cells. Cell lysates were immunoprecipitated with an anti-flag tag antibody followed by immunoblotting with an anti-DJ-1 antibody. (B) Illustration of PINK1 deletion constructs. The numbers on the right of the figure indicate amino acid composition of wild-type PINK1 (1–581) and 4 PINK1 deletion mutants (F1–F4). The numbers shown at the right side of figure indicate the amino acid residuals included in corresponding fragment. Empty box indicates the kinase domain. All constructs were flag-tagged at the C-termini. (C) Co-immunoprecipitation of DJ-1 and PINK1 deletion mutants. SH-SY5Y cells co-expressing myc-tagged DJ-1 with either control plasmid (C), flag-tagged wild-type PINK1 (PINK1) or each of the five flag-tagged PINK1 deletion mutants (F1–F4) were lysed and immunoprecipitated with an anti-myc tag antibody followed by immunoblotting with an anti-flag antibody (upper panel) or an anti-DJ-1 antibody (middle panel). Direct immunoblotting of cell lysates with an anti-flag antibody are shown as the input control (lower panel). Note: very low levels of PINK1 F1 and F2 fragments were expressed. (D) Interaction of DJ-1 and the PINK1 F3 fragment. Long exposure of Figure 4C (left panel) shows low expression of full-length PINK1 F3 fragment. However, this fragment is very efficiently co-immunoprecipitated with DJ-1 (arrow, right panel).
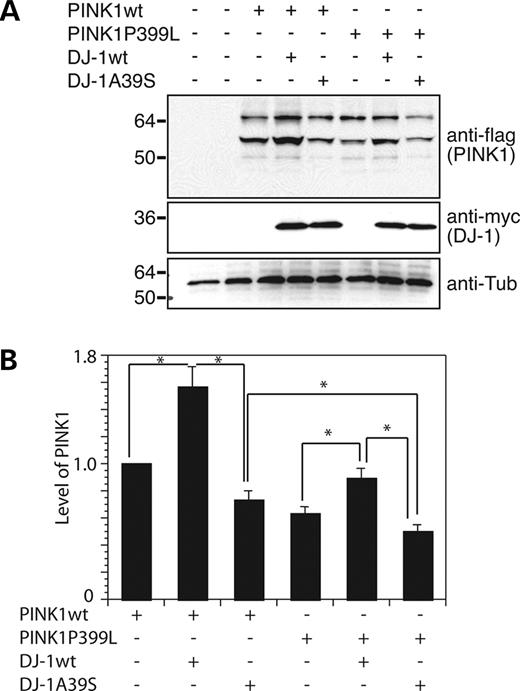
Figure 5. Expression of DJ-1 increases PINK1 levels in cells. (A) Equal amounts of lysates of cells transfected with PINK1, DJ-1 variants and their combinations were immunoprecipitated with an anti-flag antibody (to detect PINK1), followed by detection of PINK1. The same lysates were immunoblotted with anti-myc antibody, showing expression of DJ-1. Tubulin was detected to indicate equal loading. (B) Quantitation of PINK1 levels in transfected cells. The results were obtained from three independent transfections. *P<0.05 compared with the control transfectants.
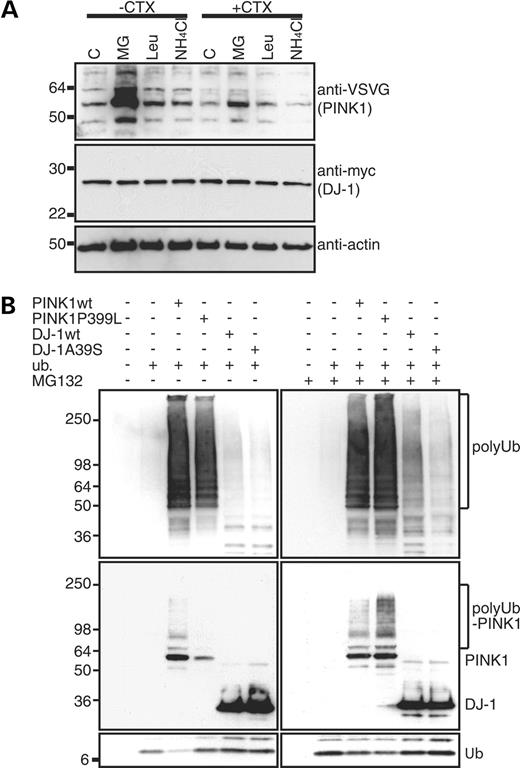
Figure 6. PINK1 degradation via the ubiquitin–proteasome pathway. (A) SH-SY5Y cells stably expressing VSVG-tagged PINK1 and myc-tagged DJ-1 treated with either solvent (C), MG132 (MG), leupeptin (Leu) or NH4Cl in the absence (−CHX) or presence (+CHX) of the protein synthesis inhibitor cycloheximide were lysed and immunoblotted to detect PINK1 (anti-VSVG) and DJ-1 (anti-myc). Actin was also detected to indicate equal loading. (B) Ubiquitination of PINK1 and DJ-1. SH-SY5Y cells were co-transfected with HA-tagged tetra-ubiquitin and flag-tagged PINK1 variants (wt and P399L) or flag-tagged DJ-1 variants (wt and A39S) followed by treatment with or without MG132. Cells were lysed, immunoprecipitated with anti-flag (M2) beads and immunoblotted with an anti-HA monoclonal antibody (to show poly-ubiquitination, upper panel) and an anti-flag polyclonal antibody (to show ubiquitination and steady-state levels of PINK1 and DJ-1, middle panel). Equal amounts of lysate were also immunoblotted with an anti-HA monoclonal antibody to show expression of tetra-ubiquitin (lower panel).
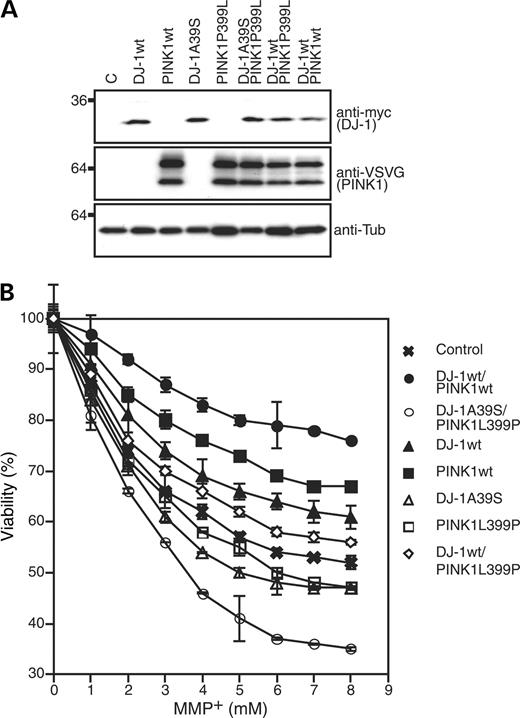
Figure 7. DJ-1 and PINK1 protect cells from MPP+-induced stress. (A) Expression of exogenous DJ-1 variants and PINK1 variants in SH-SY5Y stable expressors. Cell lysates made from SH-SY5Y cells stably expressing wild-type DJ-1 (DJ-1wt), wild-type PINK1 (PINK1wt), DJ-1A39S, PINK1P399L, DJ-1A39S and PINK1P399L, DJ-1wt and PINK1P399L and DJ-1wt and PINK1wt, as well as vector transfected controls (C) were assayed with an anti-myc tag antibody (DJ-1) or an anti-VSVG tag antibody (PINK1) to detect expression of DJ-1 and PINK1 variants. As a loading control, the membrane was also probed with an anti-tubulin antibody (anti-Tub). To detect expression of different plasmids, cells were treated with 5 µm MG132 overnight. (B) Cell survival analysis of SH-SY5Y cells expressing DJ-1 and PINK1 variants. SH-SY5Y cells stably expressing DJ-1 or/and PINK1 variants (indicated on the right of the figure) were treated with MMP+ at various concentrations indicated on the X-axis for 12 h. Cell survival was shown as a percentage of untreated controls. The results shown in the figure were obtained from three independent experiments.
References
Lang, A.E. and Lozano, A.M. (
Lang, A.E. and Lozano, A.M. (
Dawson, T.M. and Dawson, V.L. (
Paisan-Ruiz, C., Jain, S., Evans, E.W., Gilks, W.P., Simon, J., van der Brug, M., Lopez de Munain, A., Aparicio, S., Gil, A.M., Khan, N. et al. (
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., Kachergus, J., Hulihan, M., Uitti, R.J., Calne, D.B. et al. (
Spillantini, M.G., Schmidt, M.L., Lee, V.M., Trojanowski, J.Q., Jakes, R. and Goedert, M. (
Conway, K.A., Lee, S.J., Rochet, J.C., Ding, T.T., Williamson, R.E. and Lansbury, P.T., Jr (
Conway, K.A., Harper, J.D. and Lansbury, P.T. (
Valente, E.M., Abou-Sleiman, P.M., Caputo, V., Muqit, M.M., Harvey, K., Gispert, S., Ali, Z., Del Turco, D., Bentivoglio, A.R., Healy, D.G. et al. (
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y. and Shimizu, N. (
Bonifati, V., Rizzu, P., van Baren, M.J., Schaap, O., Breedveld, G.J., Krieger, E., Dekker, M.C., Squitieri, F., Ibanez, P., Joosse, M. et al. (
Li, Y., Tomiyama, H., Sato, K., Hatano, Y., Yoshino, H., Atsumi, M., Kitaguchi, M., Sasaki, S., Kawaguchi, S., Miyajima, H. et al. (
Hatano, Y., Li, Y., Sato, K., Asakawa, S., Yamamura, Y., Tomiyama, H., Yoshino, H., Asahina, M., Kobayashi, S., Hassin-Baer, S. et al. (
Lockhart, P.J., Lincoln, S., Hulihan, M., Kachergus, J., Wilkes, K., Bisceglio, G., Mash, D.C. and Farrer, M.J. (
Abou-Sleiman, P.M., Healy, D.G. and Wood, N.W. (
West, A.B. and Maidment, N.T. (
Bonifati, V., Rohe, C.F., Breedveld, G.J., Fabrizio, E., De Mari, M., Tassorelli, C., Tavella, A., Marconi, R., Nicholl, D.J., Chien, H.F. et al. (
Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., Shimizu, N., Iwai, K., Chiba, T., Tanaka, K. et al. (
Zhang, Y., Gao, J., Chung, K.K., Huang, H., Dawson, V.L. and Dawson, T.M. (
Imai, Y., Soda, M. and Takahashi, R. (
Taira, T., Saito, Y., Niki, T., Iguchi-Ariga, S.M., Takahashi, K. and Ariga, H. (
Takahashi-Niki, K., Niki, T., Taira, T., Iguchi-Ariga, S.M. and Ariga, H. (
Beilina, A., Van Der Brug, M., Ahmad, R., Kesavapany, S., Miller, D.W., Petsko, G.A. and Cookson, M.R. (
Papadimitriou, A., Veletza, V., Hadjigeorgiou, G.M., Patrikiou, A., Hirano, M. and Anastasopoulos, I. (
Abou-Sleiman, P.M., Healy, D.G., Quinn, N., Lees, A.J. and Wood, N.W. (
Valente, E.M., Salvi, S., Ialongo, T., Marongiu, R., Elia, A.E., Caputo, V., Romito, L., Albanese, A., Dallapiccola, B. and Bentivoglio, A.R. (
Polymeropoulos, M.H., Lavedan, C., Leroy, E., Ide, S.E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R. et al. (
Wilson, M.A., Collins, J.L., Hod, Y., Ringe, D. and Petsko, G.A. (
Tao, X. and Tong, L. (
Kim, R.H., Smith, P.D., Aleyasin, H., Hayley, S., Mount, M.P., Pownall, S., Wakeham, A., You-Ten, A.J., Kalia, S.K., Horne, P. et al. (
Martinat, C., Shendelman, S., Jonason, A., Leete, T., Beal, M.F., Yang, L., Floss, T. and Abeliovich, A. (
Yokota, T., Sugawara, K., Ito, K., Takahashi, R., Ariga, H. and Mizusawa, H. (
Moore, D.J., Zhang, L., Troncoso, J., Lee, M.K., Hattori, N., Mizuno, Y., Dawson, T.M. and Dawson, V.L. (
Chung, K.K., Zhang, Y., Lim, K.L., Tanaka, Y., Huang, H., Gao, J., Ross, C.A., Dawson, V.L. and Dawson, T.M. (
Shimura, H., Schlossmacher, M.G., Hattori, N., Frosch, M.P., Trockenbacher, A., Schneider, R., Mizuno, Y., Kosik, K.S. and Selkoe, D.J. (
Smith, W.W., Pei, Z., Jiang, H., Moore, D.J., Liang, Y., West, A.B., Dawson, V.L., Dawson, T.M. and Ross, C.A. (
Menzies, F.M., Yenisetti, S.C. and Min, K.T. (
Meulener, M., Whitworth, A.J., Armstrong-Gold, C.E., Rizzu, P., Heutink, P., Wes, P.D., Pallanck, L.J. and Bonini, N.M. (
Chen, L., Cagniard, B., Mathews, T., Jones, S., Koh, H.C., Ding, Y., Carvey, P.M., Ling, Z., Kang, U.J. and Zhuang, X. (
Petit, A., Kawarai, T., Paitel, E., Sanjo, N., Maj, M., Scheid, M., Chen, F., Gu, Y., Hasegawa, H., Salehi-Rad, S. et al. (
Silvestri, L., Caputo, V., Bellacchio, E., Atorino, L., Dallapiccola, B., Valente, E.M. and Casari, G. (
Canet-Aviles, R.M., Wilson, M.A., Miller, D.W., Ahmad, R., McLendon, C., Bandyopadhyay, S., Baptista, M.J., Ringe, D., Petsko, G.A. and Cookson, M.R. (
Zhang, L., Shimoji, M., Thomas, B., Moore, D.J., Yu, S.W., Marupudi, N.I., Torp, R., Torgner, I.A., Ottersen, O.P., Dawson, T.M. et al. (
Olzmann, J.A., Brown, K., Wilkinson, K.D., Rees, H.D., Huai, Q., Ke, H., Levey, A.I., Li, L. and Chin, L.S. (
Moore, D.J., Zhang, L., Dawson, T.M. and Dawson, V.L. (
Xia, K., Wu, L., Liu, X., Xi, X., Liang, D., Zheng, D., Cai, F., Pan, Q., Long, Z., Dai, H. et al. (
Zhang, Z., Vuori, K., Reed, J.C. and Ruoslahti, E. (
Zhang, Z., Hartmann, H., Do, V.M., Abramowski, D., Sturchler-Pierrat, C., Staufenbiel, M., Sommer, B., van de Wetering, M., Clevers, H., Saftig, P. et al. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}