



Abstract
Neurofibromas are common tumors found in neurofibromatosis type 1 (NF1) patients. These complex tumors are composed of Schwann cells, mast cells, fibroblasts and perineurial cells embedded in collagen that provide a lattice for tumor invasion. Genetic studies demonstrate that in neurofibromas, nullizygous loss of Nf1 in Schwann cells and haploinsufficiency of Nf1 in non-neuronal cells are required for tumorigenesis. Fibroblasts are a major cellular constituent in neurofibromas and are a source of collagen that constitutes ∼50% of the dry weight of the tumor. Here, we show that two of the prevalent heterozygous cells found in neurofibromas, mast cells and fibroblasts interact directly to contribute to tumor phenotype. Nf1+/− mast cells secrete elevated concentrations of the profibrotic transforming growth factor-beta (TGF-β). In response to TGF-β, both murine Nf1+/− fibroblasts and fibroblasts from human neurofibromas proliferate and synthesize excessive collagen, a hallmark of neurofibromas. We also establish that the TGF-β response occurs via hyperactivation of a novel Ras-c-abl signaling pathway. Genetic or pharmacological inhibition of c-abl reverses fibroblast proliferation and collagen synthesis to wild-type levels.These studies identify a novel molecular target to inhibit neurofibroma formation.
INTRODUCTION
The classic concept of tumor suppressor genes is that functional inactivation of both alleles in the tumorigenic cell is sufficient for tumor progression (1). More recently, studies in a range of tumor suppressor genes including p53 and p27 indicate that loss of a single gene copy in the tumorigenic cell is also sufficient for tumor formation in mice but at a reduced latency compared with mice that have homozygous loss (2,3). Although homozygous or heterozygous inactivation of multiple tumor suppressor genes in tumorigenic cells have been described in great detail, the functional consequences of haploinsufficiency of tumor suppressors within lineages in the tumor microenvironment have received limited attention.
Mutations in the NF1 tumor suppressor gene cause neurofibromatosis type 1 (NF1), one of the most common human genetic disorders (4,5). NF1 encodes a GTPase activating protein (GAP) for p21ras (Ras) called neurofibromin. Individuals with NF1 have a wide range of malignant and non-malignant manifestations, including the pathognomonic cutaneous neurofibromas and plexiform neurofibromas (6). Neurofibromas are complex tumors composed of Schwann cells, mast cells, endothelial cells, pericytes and fibroblasts (7). Recent studies utilizing genetically engineered mice demonstrated that nullizygosity of Nf1 in Schwann cells, the tumorigenic cell of origin, was necessary but not sufficient for tumorigenesis and that haploinsufficiency of Nf1 (Nf1+/−) in non-neuronal lineages of the tumor microenvironment was required for tumor progression (8). Although these genetic data indicate an essential role for the tumor microenvironment in neurofibroma development, a detailed molecular understanding of how this heterotypic network of Nf1+/− cells functions within the neurofibroma microenvironment is lacking. Dissection of these cellular interactions and identification of alterations in discrete Ras effector pathways in the different non-neuronal Nf1+/− cells within the neurofibromas are required for developing targeted experimental therapeutics.
Fibroblasts are one of the major cell types in neurofibromas and secrete interstitial collagen, which accounts for 50% of the weight of neurofibromas (9). The secretion of collagen and subsequent extracellular matrix remodeling in the emerging tumor are a fundamental event that provides a cellular and protein infrastructure to allow tumor invasion and the recruitment of blood vessels (10). Recent studies in murine models have shown that inflammatory mast cells secrete growth factors to stimulate fibroblast proliferation, collagen synthesis and migration (11,12). This is consistent with the observation that activated mast cells secrete proinflammatory growth factors that promote fibrosis including basic fibroblast growth factor (bFGF), platelet derived growth factor (PDGF) and transforming growth factor-beta (TGF-β).
TGF-β activation of its receptor has recently been shown to be critical for the initiation, maintenance and termination of fibrosis in the alteration of the extracellular matrix and tumor progression in stromal cells of epithelial cancers (10). Additionally, although Smad-dependent signaling is responsible for the expression of numerous TGF-β target genes, c-abl and Pak2 have recently been linked to chemical and mechanical-induced pathological fibrosis in a Smad-independent fashion (13,14). However, the network of upstream effectors that mediate the TGF-β signals to c-abl and Pak2 and in the context of cancers is incompletely understood.
Given the role of non-neuronal Nf1+/− cell lineages in neurofibroma formation and interactions between inflammatory cells and fibroblasts in altering the extracellular matrix, we sought to determine the molecular interaction between Nf1+/− mast cells and fibroblasts in order to identify molecular targets which disrupt these processes. Here, we show that Nf1+/− fibroblasts have increased proliferation, migration and collagen synthesis at physiologically relevant concentrations of TGF-β, which is secreted by Nf1+/− mast cells. Further, we identify Ras-dependent hyperactivation of c-abl as the underlying determinant of the Nf1+/− fibroblast phenotype in response to TGF-β. Collectively, these studies identify a novel interaction between Ras and c-abl as well as a molecular target and therapeutic agent that could be useful in disrupting the tumor-initiating interactions between two cellular constituents within the neurofibroma microenvironment.
RESULTS
Haploinsufficiency of Nf1 in mast cells promotes Nf1+/− fibroblast collagen synthesis, proliferation and migration
Mast cells promote fibrosis and remodeling of the extracellular matrix in defined murine models of human tumors to facilitate tumor progression (11,12,15,16). Given the high density of mast cells and collagen content in human and mouse neurofibromas, we tested whether mast cell conditioned media (MCCM) from Nf1+/− or wild-type (WT) mast cells would alter the proliferation, collagen synthesis or migration of Nf1+/− fibroblasts. WT MCCM induced a modest increase in collagen synthesis by both WT and Nf1+/− fibroblasts (Fig. 1A). In contrast, Nf1+/− MCCM promoted a 50% increase in collagen synthesis by WT fibroblasts, and a 3-fold increase in collagen synthesis by Nf1+/− fibroblasts (Fig. 1A).
![Measurement of bioactivity of MCCM on fibroblast cellular functions. (A) Bioactivity of WT and Nf1+/− MCCM on collagen production was examined by [3H]proline incorporation. Results represent the mean±SEM from three replicate dishes in one of five independent experiments with similar results. *P<0.001 for comparison of Nf1+/− versus WT fibroblast collagen production mediated by Nf1+/− MCCM using the Student's t-test. **P<0.05 for comparison of WT fibroblast collagen synthesis in response to Nf1+/− MCCM versus WT MCCM using the Student's t-test. In five independent experiments, Nf1+/− MCCM promotes a 3.1±1-fold increase in [3H]proline incorporation in Nf1+/− fibroblasts versus WT fibroblasts, whereas WT MCCM does not increase [3H]proline incorporation above background (1.1±0.3). (B) Quiescent, mitotic inactivated WT and Nf1+/− fibroblasts were scraped with a pipette tip, and then stimulated with WT or Nf1+/− MCCM. The number of migrating cells were scored by counting cells in 10 high-power fields/dish. (C) Quantitative evaluation of migrating Nf1+/− and WT fibroblasts in response to MCCM. Data represent the mean±SEM of three replicate dishes in one of five independent experiments with similar results. *P<0.01 for comparison of the number of WT versus Nf1+/− fibroblast migration mediated by Nf1+/− MCCM using the Student's t-test. **P<0.05 for comparison of the number of WT fibroblasts migrating to Nf1+/− MCCM versus that of WT fibroblasts in response to WT MCCM. In five independent experiments, Nf1+/− MCCM promotes a 3.1±1-fold increase in migration of Nf1+/− fibroblasts. WT MCCM does not increase the migration of WT or Nf1+/− fibroblasts above background levels (1.2±0.2). (D) 105 WT or Nf1+/− fibroblasts and 105 WT or Nf1+/− mast cells were admixed in a three-dimensional collagen lattice and lattice contraction was measured. Data represent the mean±SEM of three replicate dishes from one of five independent experiments with similar results. *P<0.01 comparing collagen contraction of Nf1+/− fibroblasts in lattices with Nf1+/− mast cells versus WT mast cells. **P<0.01 comparing collagen contraction of Nf1+/− fibroblasts with WT fibroblasts in response to the same stimulus. (E) Representative lattices containing the indicated populations and a ring indicating the area of the contracted lattice is shown. The baseline area of each lattice is 961 mm2.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/15/16/10.1093/hmg/ddl165/2/m_ddl16501.jpeg?Expires=1716415637&Signature=g4QDleQFD~HbrX3AC-SJYrO-wGwBH1QsI5dNt--h1D~ZntppdV3MtP3WZuWSMUBSYOBjHbh1DOpFECs731XwDCdaA4VFcUYs16NemPJ3z2GWoKYGm5bMyvQNtTctyQzBssfP6MGi~iLkvfs2f2aVwNRYjyo7Wkq~ZL1lAaz8FVqG~desOh9zMVDG0PRQ8WDiSjfGsG1jPJqbO5Hx1hT5PrK66hm2v~O95W-eWlZmDR-Yum3abh6165yZ8bh16ybQqS~Je6idrSqtb652J3BgG0ds6UzLXMcMSPZdoVm381aBqIUdfQmDklWADFl4nid~GxHaWIOOFPZ6AlD8mrKlUw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Measurement of bioactivity of MCCM on fibroblast cellular functions. (A) Bioactivity of WT and Nf1+/− MCCM on collagen production was examined by [3H]proline incorporation. Results represent the mean±SEM from three replicate dishes in one of five independent experiments with similar results. *P<0.001 for comparison of Nf1+/− versus WT fibroblast collagen production mediated by Nf1+/− MCCM using the Student's t-test. **P<0.05 for comparison of WT fibroblast collagen synthesis in response to Nf1+/− MCCM versus WT MCCM using the Student's t-test. In five independent experiments, Nf1+/− MCCM promotes a 3.1±1-fold increase in [3H]proline incorporation in Nf1+/− fibroblasts versus WT fibroblasts, whereas WT MCCM does not increase [3H]proline incorporation above background (1.1±0.3). (B) Quiescent, mitotic inactivated WT and Nf1+/− fibroblasts were scraped with a pipette tip, and then stimulated with WT or Nf1+/− MCCM. The number of migrating cells were scored by counting cells in 10 high-power fields/dish. (C) Quantitative evaluation of migrating Nf1+/− and WT fibroblasts in response to MCCM. Data represent the mean±SEM of three replicate dishes in one of five independent experiments with similar results. *P<0.01 for comparison of the number of WT versus Nf1+/− fibroblast migration mediated by Nf1+/− MCCM using the Student's t-test. **P<0.05 for comparison of the number of WT fibroblasts migrating to Nf1+/− MCCM versus that of WT fibroblasts in response to WT MCCM. In five independent experiments, Nf1+/− MCCM promotes a 3.1±1-fold increase in migration of Nf1+/− fibroblasts. WT MCCM does not increase the migration of WT or Nf1+/− fibroblasts above background levels (1.2±0.2). (D) 105 WT or Nf1+/− fibroblasts and 105 WT or Nf1+/− mast cells were admixed in a three-dimensional collagen lattice and lattice contraction was measured. Data represent the mean±SEM of three replicate dishes from one of five independent experiments with similar results. *P<0.01 comparing collagen contraction of Nf1+/− fibroblasts in lattices with Nf1+/− mast cells versus WT mast cells. **P<0.01 comparing collagen contraction of Nf1+/− fibroblasts with WT fibroblasts in response to the same stimulus. (E) Representative lattices containing the indicated populations and a ring indicating the area of the contracted lattice is shown. The baseline area of each lattice is 961 mm2.
We next tested whether Nf1+/− or WT mast cells produce growth factors which augment fibroblast migration, utilizing a wound healing assay, as fibroblasts are often recruited to an emerging tumor (10). A representative photomicrograph of mitomycin C treated Nf1+/− or WT fibroblasts, which had migrated into the wound in response to either WT MCCM or Nf1+/− MCCM, is shown in Figure 1B, and quantitative data from one of five independent experiments, each of which was conducted in triplicate cultures, are shown in Figure 1C. Nf1+/− MCCM significantly enhanced Nf1+/− fibroblast migration when compared with WT MCCM.
In emerging tumors, fibroblasts proliferate, secrete collagen and other extracellular matrix proteins. The consequence is one of altered cell–cell interactions and increased availability of growth factors, which generally enhance the tumor-promoting function of other cell types within the microenvironment (10).
Genetic studies have demonstrated that kit-ligand is secreted by fibroblasts and functions as a paracrine growth factor to mediate mast cell and fibroblast interactions (17). An established in vitro system that recapitulates fibroblast activity within the tumor microenvironment is fibroblast-mediated reorganization of three-dimensional collagen lattices (17–19). We established three-dimensional lattice assays to test the effect of Nf1 haploinsufficiency on mast cell and fibroblast interactions in extracellular matrix remodeling. In these experiments, a 2×2 checkerboard grid design was established where WT or Nf1+/− fibroblasts were incubated alone or with WT or Nf1+/− mast cells for 24 h. The quantitative results from one of five independent experiments containing three replicates per condition are summarized in Figure 1D, and the results of representative lattices are shown in Figure 1E. Cultures containing Nf1+/− fibroblasts show increased contraction when compared with WT fibroblasts irregardless of the mast cell genotype. Similarly, Nf1+/− mast cells induce an increase in contraction in cultures containing WT fibroblasts when compared with lattices containing WT mast cells. However, lattices containing Nf1+/− mast cells and Nf1+/− fibroblasts have the highest kinetics of collagen contraction. Collectively, these data define a synergistic interaction between Nf1+/− mast cells and Nf1+/− fibroblasts which promote fibrosis and remodeling of the extracellular matrix.
TGF-β mediates the hypersensitivity of Nf1+/− fibroblasts to generate collagen synthesis in response to Nf1+/− MCCM
To identify mast cell specific factors that would represent candidates for altering Nf1+/− fibroblast function, we conducted semiquantitative proteomic arrays (data not shown) and quantitative ELISAs (Fig. 2A). Three profibrotic growth factors (TGF-β, PDGF-AB and bFGF) were increased in Nf1+/− MCCM compared with WT controls. PDGF has been previously found in elevated concentrations in neurofibromas and Nf1+/− fibroblasts have increased sensitivity to pharmacological concentrations of PDGF (20,21). However, a role of TGF-β in regulating the function of Nf1+/− cell lineages has not been studied. In solid tumors, TGF-β is a critical regulator of fibroblast synthesis, deposition and remodeling of the extracellular matrix (10). On the basis of these prior observations, we tested the effects of TGF-β on the above described fibroblast assays. Nf1+/− fibroblasts had increased TGF-β-mediated proliferation (Fig. 2B), collagen synthesis (Fig. 2C) and migration (Fig. 2D) when compared with WT syngeneic fibroblasts over a range of concentrations TGF-β. Nf1+/− fibroblasts also had increased proliferation and migration in response to maximum pharmacological concentrations of PDGF or bFGF when compared with WT controls (data not shown). However, in contrast to TGF-β, PDGF and bFGF did not alter Nf1+/− fibroblast proliferation or migration at growth factor concentrations detected in Nf1+/− MCCM, either alone or in combination (data not shown).
![Quantification of the concentrations of profibrotic growth factors in MCCM and the effect of TGF-β on fibroblast proliferation, collagen synthesis and migration. (A) The concentration of murine bFGF, PDGF-AB, TGF-β and IL-6 in WT and Nf1+/− MCCM was determined by ELISA. Results represent the mean±SEM of five independent cultures. *P<0.001 for Nf1+/− versus WT MCCM by Student's t test. (B) About 104 WT or Nf1+/− fibroblasts were starved, stimulated with indicated concentrations TGF-β and [3H]thymidine incorporation was measured. Results represent the mean±SEM of three replicates from one of five independent experiments with similar results. *P<0.01 for Nf1+/− versus WT fibroblast proliferation. (C) Collagen synthesis of WT and Nf1+/− fibroblasts was examined by [3H]proline incorporation in response to 1 ng/ml TGF-β. Results represent the mean±SEM of three replicates from one of five independent experiments with similar results. *P<0.001 for comparison of Nf1+/− versus WT fibroblasts. In five independent experiments, Nf1+/− fibroblasts had a 2.7±0.4-fold increase in [3H]proline incorporation at the 0.1 ng/ml concentration of TGF-β and a 2.2±0.1-fold increase in [3H]proline incorporation in response to 1.0 ng/ml of TGF-β when compared with WT controls. (D) TGF-β-mediated fibroblast migration was measured by a wound healing assay. Data represent mean±SEM of three replicates from one of five independent experiments with similar results. *P<0.01 comparing migration of Nf1+/− fibroblasts with WT fibroblasts in response to TGF-β. In five independent experiments, Nf1+/− fibroblasts had a 2.8±0.4 and a 2.3±0.3-fold increases in migration at the 0.1 and 1 ng/ml concentrations of TGF-β when compared with WT fibroblasts. (E) About 5×104 WT and Nf1+/− fibroblasts were pre-incubated with a neutralizing antibody to TGF-β or vehicle, stimulated with 1 ng/ml of TGF-β or MCCM and [3H]proline incorporation was measured. Results represent the mean±SEM of three replicates from one of five independent experiments with similar results. *P<0.01 comparing TGF-β-stimulated proline incorporation of Nf1+/− fibroblast with WT fibroblasts in the absence of TGF-β neutralizing antibody. **P<0.01 comparing proline incorporation of Nf1+/− fibroblasts or WT fibroblasts in the presence of TGF-β neutralizing antibodies with genotype equivalent controls treated with vehicle only following TGF-β stimulation. ***P<0.01 comparing proline incorporation of Nf1+/− fibroblasts with proline incorporation of WT fibroblasts in response to Nf1+/− MCCM. ****P<0.01 comparing Nf1+/− MCCM-mediated proline incorporation of Nf1+/− fibroblasts in cultures containing TGF-β neutralizing antibodies versus vehicle control. In five independent experiments, there was a 2.4–2.5±0.3 fold increase in [3H]proline incorporation in Nf1+/− fibroblasts when compared with WT fibroblasts following TGF-β stimulation or Nf1+/− MCCM stimulation. The increase in Nf1+/− MCCM-mediated [3H]proline incorporation was reduced to background levels in the presence of the anti-TGF-β antibody (1.1±0.1).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/15/16/10.1093/hmg/ddl165/2/m_ddl16502.jpeg?Expires=1716415637&Signature=nlawKq2zCe71OGVKpFRV97P9sm9FhKt8gKobQfs03fDAAKJWUvYemg2t4My0KFVt-cag~sRboZxLbbZo3Wxd7wTMhiqCebFYqO9CyOC7vcC5~4R-Hc5RJhOiuzv~HpJFuq9oL81pdbMHmemaUx5upROO3wsAIFUqKu5lNR2ePBpfMoj1dxOPX13j0sD1lT7mjMNclT9h9ZWfOAOFPW0Gm~m0ZM2Muym4Fefw2NjQBZCkygwmKJjCCbWkXGdAteNosjbLLzcrIOfOwQ61pE7FONgQhXXsDX8R-qkK2ezB8OuWvRoCxhIBFy2d-wnBwhZp7tfiEAB75A27-gFUKX6GPw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Quantification of the concentrations of profibrotic growth factors in MCCM and the effect of TGF-β on fibroblast proliferation, collagen synthesis and migration. (A) The concentration of murine bFGF, PDGF-AB, TGF-β and IL-6 in WT and Nf1+/− MCCM was determined by ELISA. Results represent the mean±SEM of five independent cultures. *P<0.001 for Nf1+/− versus WT MCCM by Student's t test. (B) About 104 WT or Nf1+/− fibroblasts were starved, stimulated with indicated concentrations TGF-β and [3H]thymidine incorporation was measured. Results represent the mean±SEM of three replicates from one of five independent experiments with similar results. *P<0.01 for Nf1+/− versus WT fibroblast proliferation. (C) Collagen synthesis of WT and Nf1+/− fibroblasts was examined by [3H]proline incorporation in response to 1 ng/ml TGF-β. Results represent the mean±SEM of three replicates from one of five independent experiments with similar results. *P<0.001 for comparison of Nf1+/− versus WT fibroblasts. In five independent experiments, Nf1+/− fibroblasts had a 2.7±0.4-fold increase in [3H]proline incorporation at the 0.1 ng/ml concentration of TGF-β and a 2.2±0.1-fold increase in [3H]proline incorporation in response to 1.0 ng/ml of TGF-β when compared with WT controls. (D) TGF-β-mediated fibroblast migration was measured by a wound healing assay. Data represent mean±SEM of three replicates from one of five independent experiments with similar results. *P<0.01 comparing migration of Nf1+/− fibroblasts with WT fibroblasts in response to TGF-β. In five independent experiments, Nf1+/− fibroblasts had a 2.8±0.4 and a 2.3±0.3-fold increases in migration at the 0.1 and 1 ng/ml concentrations of TGF-β when compared with WT fibroblasts. (E) About 5×104 WT and Nf1+/− fibroblasts were pre-incubated with a neutralizing antibody to TGF-β or vehicle, stimulated with 1 ng/ml of TGF-β or MCCM and [3H]proline incorporation was measured. Results represent the mean±SEM of three replicates from one of five independent experiments with similar results. *P<0.01 comparing TGF-β-stimulated proline incorporation of Nf1+/− fibroblast with WT fibroblasts in the absence of TGF-β neutralizing antibody. **P<0.01 comparing proline incorporation of Nf1+/− fibroblasts or WT fibroblasts in the presence of TGF-β neutralizing antibodies with genotype equivalent controls treated with vehicle only following TGF-β stimulation. ***P<0.01 comparing proline incorporation of Nf1+/− fibroblasts with proline incorporation of WT fibroblasts in response to Nf1+/− MCCM. ****P<0.01 comparing Nf1+/− MCCM-mediated proline incorporation of Nf1+/− fibroblasts in cultures containing TGF-β neutralizing antibodies versus vehicle control. In five independent experiments, there was a 2.4–2.5±0.3 fold increase in [3H]proline incorporation in Nf1+/− fibroblasts when compared with WT fibroblasts following TGF-β stimulation or Nf1+/− MCCM stimulation. The increase in Nf1+/− MCCM-mediated [3H]proline incorporation was reduced to background levels in the presence of the anti-TGF-β antibody (1.1±0.1).
To determine whether TGF-β presence in Nf1+/− MCCM is required to promote collagen synthesis by Nf1+/− fibroblasts, neutralizing antibodies to TGF-β were added to Nf1+/− fibroblast cultures stimulated with Nf1+/− MCCM prior to scoring collagen synthesis (22). In five independent experiments, the Nf1+/− fibroblast collagen synthesis was reduced to a basal level by the addition of TGF-β neutralizing antibodies. A representative experiment is shown in Figure 2E.
Murine (Nf1+/−) and NF1 human-derived fibroblasts hyperactivate p21ras in response to TGF-β
TGF-β activates a number of downstream signaling pathways, including SMAD proteins, MAPK, PI3-K and Rho-GAPs (2,13,14,23,24). To examine whether Ras is hyperactivated in Nf1+/− fibroblasts exposed to TGF-β, and whether hyperactivation of Ras is sufficient to modulate the elevated TGF-β-mediated collagen synthesis observed in Nf1+/− fibroblasts, Nf1+/− and WT fibroblasts were transduced with a retrovirus encoding the GAP domains of NF1 (MSCV-NF1 GRD-pac). Previous studies have established that the NF1 GRD sequences are necessary and sufficient to correct Ras-mediated hyperproliferation of primary Nf1 deficient cells (21,25,26). Control constructs included empty vector (MSCV-pac) and an inactivating GAP activity mutation (MSCV-GRD1276 Arg-pac) (27). Following puromycin selection, the transduced fibroblasts were stimulated with TGF-β, and Ras activity was examined using a Ras pulldown assay. Figure 3A shows a representative experiment (left panel) and the summary of three independent experiments (right panel). Basal levels of Ras-GTP were similar in all experimental groups. Following stimulation, Nf1+/− fibroblasts transduced with the reporter transgene or the construct containing the 1276 Arg mutation have approximately a 3-fold increase in Ras activity when compared with WT cells. However, expression of the GRD domains in Nf1+/− fibroblasts corrects the elevated Ras activity of Nf1+/− fibroblasts to WT levels. These data provide genetic evidence that neurofibromin functions as a GAP for Ras downstream of TGF-β stimulation in primary fibroblasts.
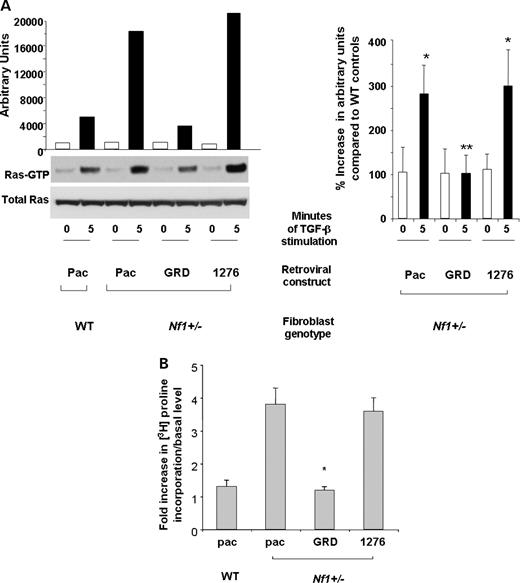
TGF-β-mediated Ras activity and collagen synthesis in Nf1+/− fibroblasts expressing the GRDs of NF1. (A) Nf1+/− fibroblasts were transduced with retroviral constructs expressing a selectable marker and the GRDs of NF1, the GRDs containing a point mutation that inactivates GAP activity (1276) or a construct encoding only the reporter transgene (Pac). WT cells were transduced with the Pac construct. Ras activity in the indicated populations at basal levels and 5 min following stimulation with TGF-β (left panel) and mean activation from three independent experiments (right panel) are shown. (B) Nf1+/− and WT fibroblasts were transduced with the indicated constructs. Following antibiotic selection, transduced cells were stimulated with TGF-β and proline incorporation was measured. Results represent the mean±SEM of three replicates from four independent experiments. *P<0.01 for comparison of Nf1+/− fibroblasts transduced with the Pac construct in response to TGF-β versus WT fibroblasts. **P<0.01 comparing TGF-β-mediated proline incorporation of Nf1+/− fibroblasts transduced with the GRD construct with Nf1+/− fibroblasts expressing the Pac or 1276 constructs using the Student's t-test.
To determine whether hyperactivation of Ras effector pathways are biochemically linked to the pathological collagen synthesis in Nf1+/− fibroblasts, fibroblasts transduced with the GRD, pac or 1276 Arg constructs were stimulated with TGF-β, and collagen synthesis was assessed. Nf1+/− fibroblasts transduced with the reporter transgene or the transgene containing the 1276 Arg mutation had a 2–4-fold increase in collagen synthesis when compared with WT cells. However, the proline incorporation of Nf1+/− fibroblasts transduced with the GRD construct was comparable to WT control fibroblasts (Fig. 3B). A similar correction in proliferation and migration was observed following transduction of the GRD but not the 1276 Arg construct (data not shown). Collectively, these biochemical and cellular data support the hypothesis that the gain in function in TGF-β-mediated signals in Nf1+/− fibroblasts is mediated by hyperactivation of p21ras.
To verify that the cellular functions and signaling motifs observed in murine fibroblasts were conserved in fibroblasts from neurofibromas collected from NF1 patients, primary fibroblasts were isolated from cutaneous neurofibromas, and the cellular and biochemical functions were examined. Verification of the purity of the fibroblasts was established using fluorescence cytometry using a ColIA antibody that specifically recognizes human fibroblasts (Fig. 4A). Similar to murine Nf1+/− fibroblasts, there is a significant increase in TGF-β-stimulated Ras activity in fibroblasts from neurofibromas when compared with Ras activity in fibroblasts from unaffected control individuals (Fig. 4B). A representative experiment (left panel) and the mean increase in Ras-GTP activity from four independent experiments (right panel) is shown. Similarly, fibroblasts from neurofibromas that were transduced with the reporter construct or the construct containing the 1276 Arg point mutation within the GRD had an increase in both collagen synthesis and migration compared with control fibroblasts (Fig. 4C and D). Importantly, transduction of the GRD transgene into fibroblasts generated from human neurofibromas restored collagen synthesis to control fibroblast levels in response to TGF-β.
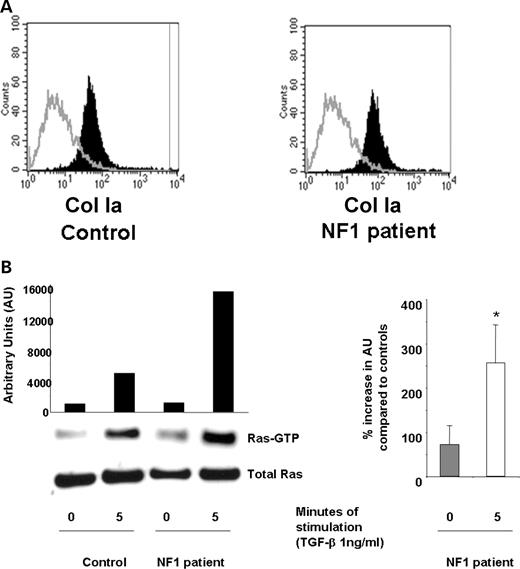
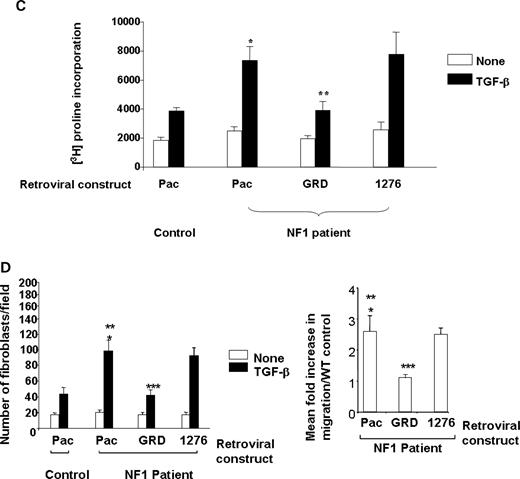
Evaluation of TGF-β-mediated Ras activity and collagen synthesis in fibroblasts from neurofibromas. (A) Fibroblasts from cutaneous neurofibromas and fibroblasts from normal volunteers were examined using an antibody to the surface antigen of Col Ia by using fluorescence cytometry. The population indicated by solid bars represents Col Ia-positive cells and the population indicated by open bars indicates the fluorescence in cells following staining with an isotype control antibody. (B) Ras activity in NF1 and control fibroblasts was examined at basal levels and 5 min following TGF-β stimulation. A representative experiment (left panel) and the mean data from four independent experiments (right panel) are indicated. *P<0.001 comparing Ras-GTP activity in NF1 patients with unaffected controls using the Student's t-test. (C) Fibroblasts from NF1 patients or healthy controls were transduced and collagen synthesis was measured following a 24 h stimulation with TGF-β. Results represent the mean±SEM of three replicates from one of four independent experiments with similar results. *P<0.01 comparing proline incorporation of NF1 Pac transduced cells with control Pac transduced fibroblasts. **P<0.01 comparing proline incorporation of NF1 GRD transduced fibroblasts with NF1 pac and NF1 1276 transduced fibroblasts using the Student's t-test. (D) NF1 fibroblasts were transduced with the indicated constructs and wound healing assays were performed following stimulation with TGF-β. Left panel: results represent the mean±SEM of three replicate dishes from one of four independent experiments with similar results. *P<0.05 comparing the number of unaffected control or NF1 fibroblasts that migrate in response to TGF-β versus vehicle. **P<0.01 comparing migration of NF1 pac transduced fibroblasts with Pac control fibroblasts using the Student's t-test. ***P<0.01 comparing the migration of NF1 GRD transduced fibroblasts with NF1 pac or NF1 1276 transduced fibroblasts using the Student's t-test. Right panel: summary of the relative TGF-β-mediated migration of human NF1 fibroblasts expressing NF1 GAP sequences from four independent experiments.
Hyperactivated c-abl activity in NF1 fibroblasts is Ras-dependent and controls fibrosis
Recent studies reported that TGF-β mediates bleomycin-induced fibrosis in pulmonary fibroblasts by activation of the non-receptor tyrosine kinase, c-abl (14). Given the data that TGF-β mediates elevated Ras activity in Nf1+/− fibroblasts, we examined whether p21ras and c-abl are biochemically linked. WT and Nf1+/− fibroblasts were stimulated with TGF-β, and c-abl kinase activity was measured. Although basal levels of c-abl activity were comparable in WT and Nf1+/− fibroblasts, TGF-β-stimulated activity was significantly higher in the Nf1+/− fibroblasts at all measured time points (Fig. 5A, left panel). The mean increase in activation of c-abl of Nf1+/− fibroblasts when compared with WT fibroblasts from four independent experiments are also shown (Fig. 5A, right panel). To verify that increased c-abl activation is secondary to hyperactivation of p21ras, Nf1+/− fibroblasts transduced with either the GRD construct or the reporter construct were stimulated with TGF-β, and c-abl activity was examined. Consistent with initial studies, TGF-β-mediated c-abl activity was higher in Nf1+/− fibroblasts transduced with the reporter gene when compared with WT control cells (Fig. 5B). However, this increased activity was corrected to WT levels in cells that contained the GRD sequences, indicating that hyperactivation of c-abl was mediated through p21ras signalling.
![Effect of c-abl activation on Nf1+/− fibroblast function. (A) WT and Nf1+/− fibroblasts were stimulated with TGF-β and c-abl activity was examined using a kinase assay. Left panel: data are representative of one of four independent experiments using different primary cell lines. Right panel: mean activation of c-abl in WT and Nf1+/− fibroblasts (n=4). (B) Nf1+/− and WT fibroblasts were transduced with the indicated retroviral constructs and TGF-β-mediated c-abl activity was examined. Results are representative of one of four independent experiments. (C) Nf1+/− and WT fibroblasts were transfected with siRNAs to c-abl or with scramble siRNA sequences, and 48 h following transfection, western blotting was performed to measure endogenous c-abl. (D) Nf1+/− and WT fibroblasts were transfected with siRNA sequences to c-abl, scramble siRNA sequences or the vehicle, then stimulated with TGF-β, and [3H]proline incorporation was measured. Results show the mean±SEM from five independent experiments. *P<0.01 comparing proline incorporation of Nf1+/− fibroblasts with vehicle or scramble siRNA to Nf1+/− fibroblasts containing c-abl siRNA. (E) Nf1+/− and WT fibroblasts were cultured in the presence of TGF-β and imatinib mesylate or TGF-β and the vehicle for 24 h prior to measuring proline incorporation. Results represent mean±SEM from one of five independent experiments. *P<0.001 comparing Nf1+/− fibroblasts with WT fibroblasts following TGF-β stimulation. **P<0.001 comparing Nf1+/− fibroblasts treated with TGF-β versus Nf1+/− fibroblasts treated with TGF-β and imatinib mesylate. In five independent experiments, TGF-β mediated a 2.1±0.3-fold increase in [3H]proline incorporation of Nf1+/− fibroblasts compared with WT fibroblasts and increased incorporation was inhibited by imatinib mesylate. (F) Human fibroblasts from neurofibromas or normal donors were stimulated with TGF-β±imatinib mesylate. *P<0.001 comparing Nf1+/− fibroblasts with WT fibroblasts following TGF-β stimulation. **P<0.001 comparing fibroblasts from an NF1 patient treated with TGF-β versus NF1 patient derived fibroblasts treated with TGF-β and imatinib mesylate. (G) Nf1+/− and WT fibroblasts were stimulated with TGF-β±imatinib mesylate and c-abl activity was measured. Data are representative of one of three independent experiments with similar results. (H) Nf1+/− mast cells and fibroblasts were admixed in three-dimensional collagen lattices together with imatinib mesylate or the vehicle. Matrix contraction was measured 24 h after initiation of cultures. Data represent mean±SEM of five independent experiments with similar results. *P<0.01 comparing collagen contraction of Nf1+/− fibroblasts with WT fibroblasts incubated with WT mast cells. **P<0.01 comparing collagen contraction of Nf1+/− fibroblasts with WT fibroblasts incubated with TGF-β. ***P<0.001 comparing collagen contraction of cultures containing imatinib mesylate added to the cultures with genotypic equivalent cultures incubated in the vehicle. (I) Representative collagen lattices of the indicated genotypes and experimental treatments.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/15/16/10.1093/hmg/ddl165/2/m_ddl16505a.jpeg?Expires=1716415637&Signature=PQ-SpKlcJd7qOw4jpptgKJamvpVfZqrHlW7N6dkCL1PacTsq3Ui7KEf4dMzHJyjIUdgdft7Fsp4xWMzNRVMaVmKPM~Hq76kqPlCIreWIeCkgtvPadrFNG1kx-MX7gaVoZA1EIFX2wXbHN50zV3doxg9RUnhEjc6WQCLItaUIMyI7TGL1~nUx-p9FwaNg62JBuC24UpCowqCS6b9mIsHxwGH~aqu7NQYoFn1xSl3M0WwEOQWDhi8nqbZ8gPkU4mEmZ1jJB3HJ8liohmOcnJkryinV4cuU4B7pwsN7Hyg88tH5eNF3m6xSf1o29hh6GnknCzK5PnYH8L0jSIFDUE6CIg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Effect of c-abl activation on Nf1+/− fibroblast function. (A) WT and Nf1+/− fibroblasts were stimulated with TGF-β and c-abl activity was examined using a kinase assay. Left panel: data are representative of one of four independent experiments using different primary cell lines. Right panel: mean activation of c-abl in WT and Nf1+/− fibroblasts (n=4). (B) Nf1+/− and WT fibroblasts were transduced with the indicated retroviral constructs and TGF-β-mediated c-abl activity was examined. Results are representative of one of four independent experiments. (C) Nf1+/− and WT fibroblasts were transfected with siRNAs to c-abl or with scramble siRNA sequences, and 48 h following transfection, western blotting was performed to measure endogenous c-abl. (D) Nf1+/− and WT fibroblasts were transfected with siRNA sequences to c-abl, scramble siRNA sequences or the vehicle, then stimulated with TGF-β, and [3H]proline incorporation was measured. Results show the mean±SEM from five independent experiments. *P<0.01 comparing proline incorporation of Nf1+/− fibroblasts with vehicle or scramble siRNA to Nf1+/− fibroblasts containing c-abl siRNA. (E) Nf1+/− and WT fibroblasts were cultured in the presence of TGF-β and imatinib mesylate or TGF-β and the vehicle for 24 h prior to measuring proline incorporation. Results represent mean±SEM from one of five independent experiments. *P<0.001 comparing Nf1+/− fibroblasts with WT fibroblasts following TGF-β stimulation. **P<0.001 comparing Nf1+/− fibroblasts treated with TGF-β versus Nf1+/− fibroblasts treated with TGF-β and imatinib mesylate. In five independent experiments, TGF-β mediated a 2.1±0.3-fold increase in [3H]proline incorporation of Nf1+/− fibroblasts compared with WT fibroblasts and increased incorporation was inhibited by imatinib mesylate. (F) Human fibroblasts from neurofibromas or normal donors were stimulated with TGF-β±imatinib mesylate. *P<0.001 comparing Nf1+/− fibroblasts with WT fibroblasts following TGF-β stimulation. **P<0.001 comparing fibroblasts from an NF1 patient treated with TGF-β versus NF1 patient derived fibroblasts treated with TGF-β and imatinib mesylate. (G) Nf1+/− and WT fibroblasts were stimulated with TGF-β±imatinib mesylate and c-abl activity was measured. Data are representative of one of three independent experiments with similar results. (H) Nf1+/− mast cells and fibroblasts were admixed in three-dimensional collagen lattices together with imatinib mesylate or the vehicle. Matrix contraction was measured 24 h after initiation of cultures. Data represent mean±SEM of five independent experiments with similar results. *P<0.01 comparing collagen contraction of Nf1+/− fibroblasts with WT fibroblasts incubated with WT mast cells. **P<0.01 comparing collagen contraction of Nf1+/− fibroblasts with WT fibroblasts incubated with TGF-β. ***P<0.001 comparing collagen contraction of cultures containing imatinib mesylate added to the cultures with genotypic equivalent cultures incubated in the vehicle. (I) Representative collagen lattices of the indicated genotypes and experimental treatments.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/15/16/10.1093/hmg/ddl165/2/m_ddl16505b.jpeg?Expires=1716415637&Signature=RA2qRC07q15xKoZg7MMXIdS-Hdqb2-DxVjQ0Fbp6Hy5lDhYkVOea4I22e0vu7lWEChlndU1YNRhtFhHhleIjCPGWF-syf2iVseHL52A~2wazsc66pmMbTrzAmsXbn3o1tF185FpkgYiDJijIPiAo4LDqVZMcjxOQPoDImfMHtl-kivHWpw3WffHP-Oe6jxeen~w3voOzLIe1Izg6~09ZnbWRVWZ40I7hAn2q1vQy~DUsHHDpn~J15MfbPeEMZhtd21hV7THbZvQRadtwIr4BEfFdrBnLgLOnS2gApc7xH234jnuHh4PiCQEYQ2v6GwLWr6-cI6tVEIQtAk6EYwReRw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Effect of c-abl activation on Nf1+/− fibroblast function. (A) WT and Nf1+/− fibroblasts were stimulated with TGF-β and c-abl activity was examined using a kinase assay. Left panel: data are representative of one of four independent experiments using different primary cell lines. Right panel: mean activation of c-abl in WT and Nf1+/− fibroblasts (n=4). (B) Nf1+/− and WT fibroblasts were transduced with the indicated retroviral constructs and TGF-β-mediated c-abl activity was examined. Results are representative of one of four independent experiments. (C) Nf1+/− and WT fibroblasts were transfected with siRNAs to c-abl or with scramble siRNA sequences, and 48 h following transfection, western blotting was performed to measure endogenous c-abl. (D) Nf1+/− and WT fibroblasts were transfected with siRNA sequences to c-abl, scramble siRNA sequences or the vehicle, then stimulated with TGF-β, and [3H]proline incorporation was measured. Results show the mean±SEM from five independent experiments. *P<0.01 comparing proline incorporation of Nf1+/− fibroblasts with vehicle or scramble siRNA to Nf1+/− fibroblasts containing c-abl siRNA. (E) Nf1+/− and WT fibroblasts were cultured in the presence of TGF-β and imatinib mesylate or TGF-β and the vehicle for 24 h prior to measuring proline incorporation. Results represent mean±SEM from one of five independent experiments. *P<0.001 comparing Nf1+/− fibroblasts with WT fibroblasts following TGF-β stimulation. **P<0.001 comparing Nf1+/− fibroblasts treated with TGF-β versus Nf1+/− fibroblasts treated with TGF-β and imatinib mesylate. In five independent experiments, TGF-β mediated a 2.1±0.3-fold increase in [3H]proline incorporation of Nf1+/− fibroblasts compared with WT fibroblasts and increased incorporation was inhibited by imatinib mesylate. (F) Human fibroblasts from neurofibromas or normal donors were stimulated with TGF-β±imatinib mesylate. *P<0.001 comparing Nf1+/− fibroblasts with WT fibroblasts following TGF-β stimulation. **P<0.001 comparing fibroblasts from an NF1 patient treated with TGF-β versus NF1 patient derived fibroblasts treated with TGF-β and imatinib mesylate. (G) Nf1+/− and WT fibroblasts were stimulated with TGF-β±imatinib mesylate and c-abl activity was measured. Data are representative of one of three independent experiments with similar results. (H) Nf1+/− mast cells and fibroblasts were admixed in three-dimensional collagen lattices together with imatinib mesylate or the vehicle. Matrix contraction was measured 24 h after initiation of cultures. Data represent mean±SEM of five independent experiments with similar results. *P<0.01 comparing collagen contraction of Nf1+/− fibroblasts with WT fibroblasts incubated with WT mast cells. **P<0.01 comparing collagen contraction of Nf1+/− fibroblasts with WT fibroblasts incubated with TGF-β. ***P<0.001 comparing collagen contraction of cultures containing imatinib mesylate added to the cultures with genotypic equivalent cultures incubated in the vehicle. (I) Representative collagen lattices of the indicated genotypes and experimental treatments.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/15/16/10.1093/hmg/ddl165/2/m_ddl16505c.jpeg?Expires=1716415637&Signature=eHlpnB8nd2n3M2bUFDVLnL64xQzD7PFqC4ZWLJf-afnBX43EgzqMobL9Lm-XPEAUiHzGHS5qrAI6tt67ltUiRadvpQeKcQ4XULv7b5IVSbNyH8Tyr4VMfhk7Jx02Sxs7PxWNsi6If9zbf8wB6vKv-wXei0TD4Cf~qsFkcNZr3kkyNCowbrGQcDhwvkdOLDAi4wXC3vBuR22HfDdVVWrfu~6Oz0yrlM361rfTEAKLJmfO4l2q7EY96Z3Iw1lz755y4ZCb7-g0fmK5Th7uaCVYrcNEen0JrjGzusm0BfJN-Xk4zHte7gJTBm-QK6NR1LH497l1D6~USkfY7yVq1qnK-g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of c-abl activation on Nf1+/− fibroblast function. (A) WT and Nf1+/− fibroblasts were stimulated with TGF-β and c-abl activity was examined using a kinase assay. Left panel: data are representative of one of four independent experiments using different primary cell lines. Right panel: mean activation of c-abl in WT and Nf1+/− fibroblasts (n=4). (B) Nf1+/− and WT fibroblasts were transduced with the indicated retroviral constructs and TGF-β-mediated c-abl activity was examined. Results are representative of one of four independent experiments. (C) Nf1+/− and WT fibroblasts were transfected with siRNAs to c-abl or with scramble siRNA sequences, and 48 h following transfection, western blotting was performed to measure endogenous c-abl. (D) Nf1+/− and WT fibroblasts were transfected with siRNA sequences to c-abl, scramble siRNA sequences or the vehicle, then stimulated with TGF-β, and [3H]proline incorporation was measured. Results show the mean±SEM from five independent experiments. *P<0.01 comparing proline incorporation of Nf1+/− fibroblasts with vehicle or scramble siRNA to Nf1+/− fibroblasts containing c-abl siRNA. (E) Nf1+/− and WT fibroblasts were cultured in the presence of TGF-β and imatinib mesylate or TGF-β and the vehicle for 24 h prior to measuring proline incorporation. Results represent mean±SEM from one of five independent experiments. *P<0.001 comparing Nf1+/− fibroblasts with WT fibroblasts following TGF-β stimulation. **P<0.001 comparing Nf1+/− fibroblasts treated with TGF-β versus Nf1+/− fibroblasts treated with TGF-β and imatinib mesylate. In five independent experiments, TGF-β mediated a 2.1±0.3-fold increase in [3H]proline incorporation of Nf1+/− fibroblasts compared with WT fibroblasts and increased incorporation was inhibited by imatinib mesylate. (F) Human fibroblasts from neurofibromas or normal donors were stimulated with TGF-β±imatinib mesylate. *P<0.001 comparing Nf1+/− fibroblasts with WT fibroblasts following TGF-β stimulation. **P<0.001 comparing fibroblasts from an NF1 patient treated with TGF-β versus NF1 patient derived fibroblasts treated with TGF-β and imatinib mesylate. (G) Nf1+/− and WT fibroblasts were stimulated with TGF-β±imatinib mesylate and c-abl activity was measured. Data are representative of one of three independent experiments with similar results. (H) Nf1+/− mast cells and fibroblasts were admixed in three-dimensional collagen lattices together with imatinib mesylate or the vehicle. Matrix contraction was measured 24 h after initiation of cultures. Data represent mean±SEM of five independent experiments with similar results. *P<0.01 comparing collagen contraction of Nf1+/− fibroblasts with WT fibroblasts incubated with WT mast cells. **P<0.01 comparing collagen contraction of Nf1+/− fibroblasts with WT fibroblasts incubated with TGF-β. ***P<0.001 comparing collagen contraction of cultures containing imatinib mesylate added to the cultures with genotypic equivalent cultures incubated in the vehicle. (I) Representative collagen lattices of the indicated genotypes and experimental treatments.
To directly address the role of c-abl activation in TGF-β-mediated collagen synthesis, we knocked down endogenous c-abl expression by siRNA in Nf1+/− fibroblasts. c-abl siRNA diminished endogenous c-abl transcripts by at least 90% (Fig. 5C). Nf1+/− fibroblasts transfected with siRNA sequences to c-abl inhibited collagen synthesis (Fig. 5D), whereas transfection of scrambled control c-abl sequences or vehicle did not inhibit collagen synthesis.
Imatinib mesylate, a drug that was initially utilized because of its activity on c-abl in chronic myeloid leukemia (28), was utilized to reduce c-abl activity and collagen synthesis in our system. Addition of imatinib mesylate to cultures of murine Nf1+/− fibroblasts (Fig. 5E) or human fibroblasts from neurofibromas (Fig. 5F) was sufficient to inhibit proline incorporation to below WT levels and was associated with a decrease in c-abl activity (Fig. 5G). Finally, using the three-dimensional culture assay, matrices containing the experimental mast cell and fibroblast genotypes were established in the presence or absence of imatinib mesylate (Fig. 5H and I). Consistent with previous studies, lattices containing Nf1+/− mast cells and fibroblasts had the greatest contraction. However, addition of imatinib mesylate significantly inhibited collagen contraction, regardless of the experimental genotype. Collectively, these data demonstrate that imatinib mesylate is a potent inhibitor of collagen synthesis and the pathological interactions between Nf1+/− mast cells and Nf1+/− fibroblasts.
Imatinib mesylate inhibits TGF-β-mediated fibroblast recruitment in vivo
As a proof of concept to determine whether Nf1+/− fibroblasts have increased migration and/or proliferation in vivo in response to TGF-β or mast cell conditioned medium and to examine whether oral administration of imatinib mesylate can inhibit these processes, we utilized a matrigel plug assay to examine these functions. Matrigel plugs containing a low concentration of TGF-β were placed in the groin of Nf1+/− and WT mice. Mice were fed either 6 µm imatinib mesylate daily or the vehicle control for 1 week and plugs were then examined for the number of fibroblasts that was recruited into the plug. Representative plugs are shown in Figure 6A and B. Mean quantitative data from five mice per experimental group are shown (Fig. 6C). Consistent with in vitro studies, TGF-β and Nf1+/− MCCM mediates an increase in the recruitment of Nf1+/− fibroblasts into the matrigel plug. Further, treatment of Nf1+/− mice with imatinib mesylate reduces this process. Collectively, the in vitro and in vivo data indicate that Nf1+/− fibroblasts are hypersensitive to TGF-β-mediated growth signals through hyperactivation of Ras-c-abl signals.
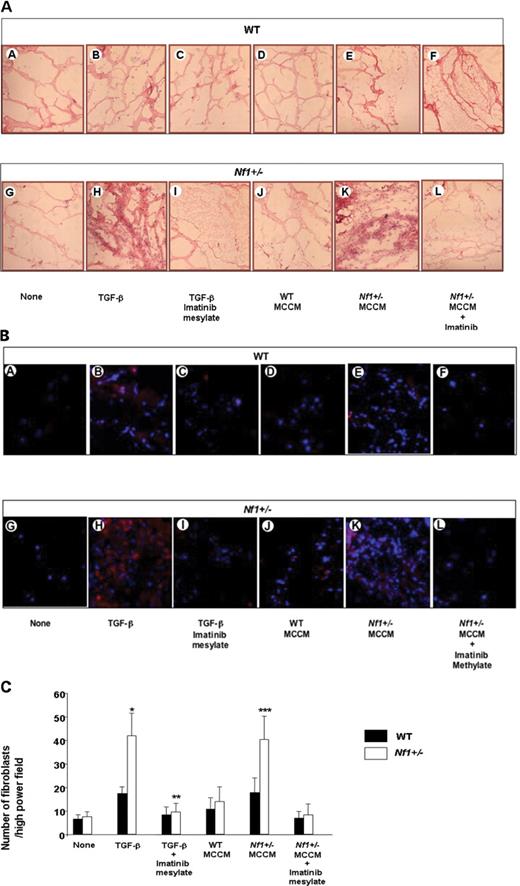
TGF-β-mediated invasion of fibroblasts in matrigel plugs. (A) Representative hematoxylin and eosin stained sections of matrigel plugs from WT and Nf1+/− mice that contained vehicle, TGF-β or MCCM as a stimulus are indicated. Sections from mice that were administered plugs containing TGF-β or MCCM as a stimulus and then fed imatinib mesylate are also indicated. (B) Merged image of PE conjugated anti-CollA positive cells and DAPI counterstain to identify the nuclei. Treatment groups are indicated. (C) Quantitative scoring of fibroblasts within matrigel plugs of the indicated genotypes and experimental treatments. Data represent the mean±SEM of 10 individual sections from five mice per experimental group. *P<0.01 comparing the number of fibroblasts per high-power field in plugs containing TGF-β harvested from WT mice with the number of fibroblasts in plug from WT mice containing vehicle. **P<0.001 comparing the number of fibroblasts per high-power field in the plug harvested from Nf1+/− mice with the number of fibroblasts in plugs from WT mice containing TGF-β. ***P<0.001 comparing fibroblasts/high-power field from Nf1+/− mice that were administered matrigel plugs containing TGF-β and then fed 200 mg/kg/day of imatinib mesylate daily with mice that were administered TGF-β containing matrigel plugs and fed the vehicle control.
DISCUSSION
Von Recklinghausen's neurofibromatosis (NF1) affects one in 3500 newborn children worldwide. In addition, because a majority of the affected individuals reach reproductive age, the introduction of mutant alleles into the population is continuously rising. The predominant pathology of NF1 is the appearance of incurable peripheral nerve tumors of a unique multicellular type called neurofibromas. These tumors contain diverse cell types including Schwann cells, fibroblasts, pericytes, mast cells, endothelial cells and perineurial cells. Studies from human tumor samples have suggested that the Schwann cell is at the origin of these tumors (29–31). Genetic studies in the mouse have confirmed these suggestions and further indicated a critical non-cell autonomous component to tumor evolution (8). Thus, our preceding genetic studies on neurofibroma models implicate additional cell types present as possible contributors to tumorigenesis and provide a valuable genetic model for studying the role of the microenvironment in cancer. The present study identifies unique and novel cellular interactions between NF1 haploinsufficient mast cells and fibroblasts and two cell types that comprise significant components of the neurofibroma tumor despite the demonstration that their heterozygous genotype is retained in tumors. Our data indicate that Nf1+/− mast cells secrete TGF-β and that, in proximity to NF1 heterozygous fibroblasts, this secretion results in induced proliferation, migration and collagen deposits. We describe the underlying mechanisms and suggest an explanation of how this unique multicellular and incurable tumor arises. This genetic paradigm provides an important example in a human disease model of how tumorigenesis may be significantly impacted by the non-tumorigenic microenvironment.
Mast cells infiltrate murine and human neurofibromas and are a potential source of growth factors that promote fibrosis, alterations of the extracellular matrix and neoangiogenesis, three key pathological processes identified in neurofibromas. Further, others have shown that mast cells initiate tumor progression in an experimental model of squamous cell carcinoma (16). Identifying the mast cell derived growth factors that alter the function of other cell lineages in the tumor and dissecting the biochemical mechanisms for increased secretion of growth factors may provide novel molecular targets.
Fibroblasts are a major constituent of neurofibromas and produce excessive amounts of collagen (9). Given that mast cells secrete multiple profibrotic growth factors following activation of the c-kit receptor, studies here focussed on determining the molecular interactions between Nf1+/− mast cells and fibroblasts. Using a combination of proteomics arrays, individual ELISAs and neutralizing antibodies to TGF-β, we determined that Nf1+/− mast cells secrete increased concentrations of TGF-β to enhance fibroblast proliferation, migration and collagen synthesis. TGF-β secretion in Nf1−/− Schwann cell conditioned media was also examined but was not detected (data not shown). Elevated levels of TGF-β in Nf1+/− MCCM is particularly interesting given that recent studies implicate TGF-β as a critical regulator of fibroblast proliferation, cytoskeletal reorganization, collagen synthesis and transformation in other human diseases, including colorectal cancer (10).
Though understanding of the TGF-β-mediated signaling network in fibroblasts is incomplete, it is clear that TGF-β activates MAPK and Rho-GTPase family members, which are downstream Ras effectors (2,14,23,32). Despite observations that NF1 is a large complex gene with 60 exons, the only ascribed function of the NF1 gene in mammalian cells is to function as a GAP for p21ras, and this function is specifically mediated through the GRD of the NF1 gene (33). Similar to prior studies in myeloid cells (25), we found that stable transduction and retroviral-mediated expression of the GRD sequences in Nf1+/− fibroblasts reduces their migration, proliferation and collagen synthesis to WT levels in response to TGF-β. Similar results were also observed in fibroblasts from neurofibromas of NF1 patients confirming that the abnormal signaling pathways that we identified in murine cells are conserved in human fibroblasts. Importantly, transduction of GRD sequences harboring a known patient mutation that inactivates GTPase function does not alter the migration, proliferation or collagen synthesis of Nf1+/− fibroblasts. These data provide direct genetic evidence that the excessive TGF-β-mediated Nf1+/− phenotypes are secondary to increased Ras activity.
Two previous studies have evaluated the role of Ras in Nf1 deficient fibroblasts with varying results. One study did not observe quantifiable differences in Ras activity between Nf1−/− murine embryonic fibroblasts and WT fibroblasts and suggested that neurofibromin was not an essential activator of Ras activity in fibroblasts (34). In contrast, Johannessen et al. (21) found that murine Nf1+/− fibroblasts had elevated PDGF-β-mediated Ras activity that was corrected by the GRD domains of Nf1 consistent studies reported here using TGF-β.
A novel observation of these studies is that hyperactivation of the p21ras oncogene in Nf1+/− cells by TGF-β stimulation is biochemically linked to increased activation of c-abl, a well known non-receptor tyrosine kinase (35). In support of this observation, we found that the cellular phenotype of Nf1+/− fibroblasts mediated by TGF-β stimulation is similar to other murine models of fibrosis associated with hyperactivation of c-abl in fibroblasts (13,14). We found that Nf1+/− fibroblasts had increased TGF-β-induced c-abl activity and that transduction of the NF1 GRD into Nf1+/− fibroblasts restored c-abl activity to WT levels. Further, introduction of the NF1 GRD into Nf1+/− fibroblasts, siRNA inhibition of c-abl or pharmacological treatment with imatinib methylate inhibited the excessive migration and collagen synthesis in response to TGF-β.
Therapeutic targeting of Ras activation in different human cancers has been problematic (36–38). Therefore, an important priority for the development of experimental therapeutics is identifying discrete Ras effector pathways that regulate the growth of the tumor initiating cell and the supporting functions of the non-tumor cells in the tumor microenvironment. On the basis of this conceptual framework, our data, together with previously published data by us and others, suggest that imatinib mesylate could be a single agent which targets multiple lineages within plexiform neurofibromas. Imatinib mesylate inhibits biochemical signals generated via c-abl, PDGF-β and c-kit receptor tyrosine kinases (28). In data presented here, administration of imatinib mesylate to NF1 patients with neurofibromas could potentially inhibit the tumor-promoting functions of mast cells and fibroblasts within the tumor microenvironment via two mechanisms. Others have demonstrated that the c-kit receptor tyrosine kinase is a central regulator of the development and function of mast cells (39). In addition, we have previously shown that Nf1+/− mast cells have an increase in kit-ligand-mediated proliferation and migration and that the recruitment of Nf1+/− mast cells to Nf1−/− Schwann cells is mediated by an elevated secretion of kit-ligand by Nf1−/− Schwann cells (26). Fibroblasts are a second source of kit-ligand and are known to be required for initiating mast cell–fibroblast interactions (17). In the present study, we show that Nf1+/− mast cells secrete increased concentrations of TGF-β and that imatinib mesylate inhibits mast cell dependent collagen synthesis and extracellular matrix remodeling. We also found that c-kit phosphorylation and kit-ligand-induced TGF-β secretion by Nf1+/− mast cells were inhibited by the addition of imatinib mesylate to the cultures (unpublished data). PDGF is also central to the proliferation and migration of vascular smooth muscle cells (VSMCs)/pericytes, and in other studies, Nf1+/− VSMCs were found to have hypersensitive growth to PDGF-BB and imatinib mesylate inhibited this activity (D. Ingram and A. Munchhof, manuscript in press, Human Molecular Genetics). Collectively, the data indicate that this single agent can inhibit multiple lineages that contribute to tumor progression within the tumor microenvironment of neurofibromas. Our experimental data together with recent trials demonstrating that imatinib mesylate has relatively low toxicity, even in young children, suggest that this FDA approved drug or drugs that have a similar spectrum of inhibition could be useful in the treatment of plexiform neurofibromas.
MATERIAL AND METHODS
Animals
Nf1+/− mice were obtained from Dr Tyler Jacks at the Massachusetts Institute of Technology (Cambridge, MA, USA) in a C57BL/6J.129 background and backcrossed for 13 generations into a C57BL/6J strain (40). These studies were conducted with the approval of the Indiana University Laboratory Animal Research Center.
Generation of primary cells and preparation of MCCM
Murine fibroblasts were prepared from 13.5 dpc (days postcoitus) embryos as described previously (25). All the fibroblasts used in the experiments were passages 2–3. NF1 patient fibroblasts and fibroblasts from unaffected adult controls were generated from cutaneous neurofibromas or healthy donor skin. Briefly, dermal samples were isolated, digested with trypsin to obtain a single cell suspension and placed in Dulbecco's minimum essential medium (DMEM) containing 10% fetal calf serum (FCS), 1% glutamine, 2% penicillin/streptomycin and cultured in a 37°C, 5% CO2 humidified incubator.
Bone marrow derived mast cells (BMMCs) were generated from 6-week-old mice as described previously (41). The homogeneity of BMMC was determined by Alcian blue–Safranin O staining (42). Furthermore, fluorescence-activated cytometric analysis (FACS) (Becton Dickinson, San Jose, CA, USA) revealed similar forward and side light scatter characteristics and the same percentage of c-kit+ expression in BMMCs for all murine experimental genotypes (data not shown). MCCM was collected from each mast cell line following 24 h starvation with RPMI1640 containing 0.5% bovine serum albumin (BSA) (Sigma-Aldrich, St Louis, MO, USA) at 37°C, 5% CO2 with modification (26). Briefly, the cells were centrifuged for 5 min at 120g and supernatants were collected and stored in aliquots at −80°C. Supernatants were then used for analysis of bioactivity in fibroblasts and for evaluation of cytokine concentrations by proteomics array and ELISA.
Measurement of cytokine secretion from mast cells
Cytokine secretion from BMMCs was measured by semiquantitative protein array (RayBiotech Inc., Norcross, GA, USA) as described previously (26). The results were confirmed quantitatively by ELISA following manufacturer's instruction (R&D Systems, MN, USA).
Flow cytometric analysis for collagen type I expression
Surface collagen type I antigen expression was evaluated by fluoresence cytometry. Fibroblasts were stained for 30 min at 4°C with 1 µg/ml of FITC-conjugated anti-ColIA monoclonal antibody (Pharmingen, San Diego, CA, USA) or an isotype control and were analyzed by FACS for CollA expression.
Fibroblast proliferation
[3H]Thymidine incorporation assay was performed to examine the fibroblast proliferation with modification (43). Briefly, fibroblasts were plated at a concentration of 4×103 cells in 96-well dishes in 200 µl of DMEM containing 1% glutamine, 10% FCS, in a 37°C, 5% CO2 humidified incubator. Culture media were then switched to serum-free DMEM or MCCM. The cells were then cultured for 24–72 h, and tritiated thymidine (PerkinElmer Life and Analytical Sciences, Boston, MA, USA) was added to cultures 6 h prior to harvest on glass fiber filters (Packard Instrument Co.) and β-emission was measured.
Wound healing assay
Confluent fibroblast monolayers were wounded by manually scraping the cells with a pipette tip as described previously (32). The wound size was measured to ensure that all wounds were of the same width. To ensure that the width change was not caused by fibroblast proliferation, 10 µg/ml of mitomycin C was added into the fibroblast culture for 2 h before wounding to block the cell mitosis. The cell culture media were then replaced with fresh media, and wound closure was monitored by time lapse microscopy.
Three-dimensional collagen lattice formation
Three-dimensional lattice cultures were performed in collagen matrices as described (43). Briefly, type I collagen from rat tail tendon (2 mg/ml in 18 mm acetic acid) was added to 35 mm dishes containing DMEM medium with 1% FCS and 0.1 m NaOH. Fibroblasts (105) were added just after collagen solution, alone or with 105 mast cells, before fibrillation and lattice formation. Lattice diameter was measured daily.
Collagen synthesis assay
Collagen production by fibroblasts incubated with mast cells (1×105/well) or TGF-β (1 ng/ml) in 24-well tissue culture plates was assessed by [3H]proline incorporation into collagenous proteins as previously described (43). Data are expressed as c.p.m. of [3H]proline incorporation in 104 fibroblasts.
Use of neutralizing antibodies to TGF-β
TGF-β neutralizing antibodies were obtained from R&D Systems, Inc. (Cat. no. AF-101-NA) and utilized as suggested by the manufacturer. The ND50 for this lot of anti-TGF-β1 antibody was previously established by the manufacturer and confirmed in pilot experiments.
Generation of recombinant retroviral plasmids
The recombinant retroviral constructs have been previously described (25). Briefly, the GAP-related domain (GRD) sequences which modulate Ras activity are under the transcriptional control of the myeloproliferative sarcoma retrovirus promoter. Constructs also contain a puromycin resistance gene, Pac. Three viruses were used in these experiments: a virus expressing the full-length NF1 GTPase-activating related domain (NF1 GRD) and pac (MSCV-NF1 GRD-pac), a virus expressing a GAP-inactive mutant of the NF1 GRD that harbors a known human mutation in the arginine finger loop (R1276P) (25) and pac (MSCV-1276P NF1 GRD-pac) and a virus expressing the selectable marker gene alone (MSCV-pac).
Retroviral transduction of fibroblasts
The transduction protocol has been previously described and was used here with minor modifications (25). Briefly, fibroblasts were transduced with supernatants of GP+E86 or GP+AM12 packaging cells in the presence of polybrene (5 µg/ml) for 48 h. Transduced cells were then cultured in 1 µg/ml of puromycin to select the tranduced cells.
c-abl kinase assays
Fibroblasts were serum starved for 48 h, then stimulated with TGF-β for the indicated time and subsequently lysed for 30 min at 4°C in 750 µl of kinase lysis buffer [50 mm Tris (pH 7.4), 150 mm NaCl, 1% Triton X, 0.1% SDS, 1% sodium deoxycholate, 0.1 TIU/ml aprotinin, 50 µg/ml PMSF, 1 mm sodium vanadate, 1 mg/ml leupeptin]. Extracts were clarified, and equivalent protein (500 µg) was incubated overnight at 4°C with anti-Abl (K12; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) or anti-Flag (Sigma-Aldrich). Immune complexes were collected with protein A–Sepharose (Sigma-Aldrich) and twice in kinase buffer [25 mm Tris (pH 7.4), 10 mm MgCl2, 1 mm DTT] prior to incubation in 40 µl kinase buffer containing 5 µm ATP, 2 µg GST-Crk and 0.5 µCi/Rxn [32P]ATP. The kinase reaction was allowed to proceed for 5 min at 37°C, stopped with 40 µl (2× concentration) Laemmli buffer and visualized by autoradiography after SDS–PAGE. Total c-abl protein was detected using an antibody from BD Biosciences (Cat. no. 554148, San Jose, CA, USA).
Transfection with siRNA
c-abl siRNA oligonucleotides (Cat. no. sc-29844) were purchased from Santa Cruz Biotechnology, Inc. Scrambled oligonucleotides (Cat. no. sc-36869) or vehicle only was used as controls. Fibroblasts were cultured in a six-well tissue culture dish to 40–50% confluency. The siRNA oligonucleotides were diluted with siRNA transfection reagents and siRNA transfection medium, and transfections were conducted as per manufacturer's recommendations (Santa Cruz). After the transfection, the cell mixture was replaced with culture medium. Cells were incubated for 48 h at 37°C before harvesting for experiments.
Detection of p21ras-GTP levels
Fibroblasts were deprived of serum for 24 h and stimulated with 1 ng/ml of TGF-β for 5 min. p21ras activation was subsequently determined using a p21ras activation pulldown assay kit (Upstate Biotechnology, Lake Placid, NY, USA) according to manufacturer's protocol and as described previously (25,44).
Matrigel plug assay
To measure fibroblast migration and growth in vivo, a matrigel plug assay was performed with modification as described previously (45). Briefly, matrigel was thawed at 4°C for 24 h and mixed with 10 ng/ml TGF-β (Gibco) or vehicle. The matrigel mixture was then injected subcutaneously into the groin of WT or Nf1+/− mice. Recipients received a daily dose of 200 mg/kg/day of imatinib mesylate or the vehicle control for 1 week. The plug was then removed on day 7 and was frozen immediately. All tissues were sectioned (5 µm thickness), mounted onto slides and stained with PE-conjugated anti-ColIA antibody and counterstained with DAPI to identify the nuclei using standard techniques. Cells were imaged using a QImaging camera and QCapture-Pro software (Fryer Company Inc., Cincinnati, OH, USA). The number of fibroblasts was determined by scoring cells in three high power fields from 10 random sections within the plug. Five independent plugs per experimental group were examined.
ACKNOWLEDGEMENTS
We thank Dr Mary Dinauer and Dr Mervin Yoder for helpful comments during the preparation of the manuscript. Dr. F. Levi-Schaffer at Department of Pharmacology, School of Pharmacy, Faculty of Medicine, The Hebrew University of Jerusalem, Jerusalem, Israel, kindly provided us detailed protocols for scoring proline incorporation. We also thank Marilyn Wales for administrative support. This work was supported by NIH R01 CA74177-06 (D.W.C.), Department of Defense (DOD) NF043032 (F.-C.Y.) and NF043019 (D.A.I.) and NINDS and DOD funds to L.F.P.
Conflict of Interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}