




Abstract
Many adult tissue stem cells, such as the cells of the hematopoietic system, gastrointestinal epithelium, brain, epidermis, mammary gland and lung have now been identified, all of them fulfilling a crucial role in supplying organisms with mature cells during normal homeostasis as well as in times of tissue generation or repair. Two unique features characterize adult stem cells: the ability to generate new pluripotent stem cells (to self-renew) and the ability to give rise to differentiated progeny that has lost its self-renewal capacity. Our understanding of the mechanisms that determine whether, where and when a stem cell will self-renew or differentiate is still limited, but recent advances have indicated that the stem cell microenvironment, or niche, provides essential cues that direct these cell fate decisions. Moreover, loss of control over these cell fate decisions might lead to cellular transformation and cancer. This review addresses the current understandings of the molecular mechanisms that regulate hematopoietic stem cell self-renewal in the niche and how leukemic transformation might change the dependency of leukemic stem cells on their microenvironment for self-renewal and survival.
INTRODUCTION: HEMATOPOIETIC AND LEUKEMIC STEM CELLS
Hematopoietic stem cells (HSCs) were first identified by Till and McCulloch in 1961 (1). Since then, HSCs are the most extensively studied adult stem cells that have been isolated (2). Nevertheless, HSCs are a rare population representing less than 0.01% of the cells in the bone marrow. This has complicated HSC research, and it is indeed surprising to realize how limited our current knowledge still is regarding mechanisms that determine whether, where and when HSCs undergo self-renewal divisions. The most reliable assays to detect and enumerate HSCs are in vivo repopulation experiments and advances in technologies have greatly helped to purify mouse HSCs (3–5). These studies have indicated that the mouse HSC is contained within the lineage negative (Lin−), Sca1+, Kit+, CD34− and side-population (SP, cells that have the ability to efflux the DNA-binding dye Hoechst 33342) compartment. Recently, it was reported that combinatorial expression of cell surface receptors of the SLAM family members precisely distinguishes mouse stem from progenitor cells (6). Thus, most of the gained knowledge from mouse HSCs in the past years relies on the possibility to isolate and quantitatively measure stem cell activity in in vivo assays. The isolation of true human HSCs still remains elusive and additional studies need to be performed in order to identify specific hematopoietic markers that will allow its purification to homogeneity. A great progress in the study of human HSCs has been made since the development of NOD/SCID and NOD/SCID-β2-microglobulin−/− xenotransplantation assays (7–9). These assays are based on the lympho-myeloid repopulating capacity of the transplanted human HSC, termed as SCID-repopulating cell (SRC), and these SRCs are contained within the Lin−CD34+CD38− population in the bone marrow (7,8). The NOD/SCID and NOD/SCID-β2-microglobulin−/− model systems have also greatly facilitated the identification of the leukemic stem cell (LSC) or SCID leukemia-initiating cell (SL-IC) (10,11). As in the normal hematopoietic system, it has been recognized that in acute myeloid leukemia (AML) the developing malignant clone comprises a heterogeneous group of cells that differ in their differentiation status and only the rare SL-IC population was capable of initiating and sustaining leukemic growth in vivo in SCID or NOD-SCID mice (11,10). These SL-ICs have self-renewal capacity as demonstrated in serial transplantation experiments, but heterogeneity exists in the self-renewal potential of different classes LSCs, which further supports the hypothesis that they are derived from normal HSCs (12).
The interaction of the stem cell with specific microenvironmental elements is thought to be a key regulatory mechanism in maintenance of its self-renewal and differentiation capacities. The concept of a ‘stem cell niche’ was first proposed in the late 1970s (13) but the HSC niche was identified in mice almost three decades later (14,15). Detailed information on the structure, composition and exact localization of the niche is only beginning to be revealed, and particularly in the human system knowledge is only emerging. Nevertheless, a number of different types of signaling and adhesion molecules that have a role in the regulation of stem cell quiescence, self-renewal and cell fate decisions have been reported (discussed in more detail subsequently). The stem cell niche has been attributed functions such as maintenance of stem cell quiescence by providing proliferation inhibiting cues. However, the niche should also provide proliferation or differentiation-inducing signals in times that high numbers of progenitors are needed that quickly can give rise to all committed cell lineages. Ultimately, the niche needs to ensure a life-long reserve of stem cells, which requires tightly controlled stem cell self-renewal divisions. It is therefore no surprise that deregulation of self-renewal of stem cells is the main cause for neoplastic or cancer diseases. Stem and cancer cells share certain signaling pathways that regulate their self-renewal which suggests that normal stem cells can in fact give rise to cancer cells. This notion has fed the concept that tumors contain rare ‘cancer stem cells’ that maintain tumor growth because of their indefinite proliferative and self-renewal potential (reviewed in 16,17). Understanding the genetic and molecular regulations of the self-renewal program and an appreciation of how perturbations in these regulations initiate proliferative diseases such as leukemia is a major challenge of the medical research.
In this review, we will discuss how normal and leukemic stem cells interact with their microenvironment, which mechanisms have been reported to be involved and how these correlate with processes such as self-renewal. Do the hematopoietic and LSCs share the same niche or do leukemic cells interact differently with their own microenvironment? Do these cells home only in the bone marrow or also in other niches? Indeed, do LSCs need a niche at all?
HSCs AND THEIR MICROENVIRONMENT
HSC quiescence, self-renewal and differentiation are regulated by both intrinsic and extrinsic mechanisms. Intrinsic mechanisms include those that affect the epigenetic state of HSCs, as it is controlled by chromatin remodellers such as polycomb group (PcG) proteins. Extrinsic mechanisms include those changes in stem cell fate that are dictated by the environment, i.e. the niche. A schematic presentation of these regulatory mechanisms and their complementary interaction is shown in Figure 1. In such a model, direct physical interaction between HSCs and their niche could, for example, be mediated by integrins and cadherins. Once localized within the niche, HSCs are in close proximity of locally secreted or membrane-bound cytokines and growth factors that dictate HSC fate by initiating specific signal transduction within the HSC.
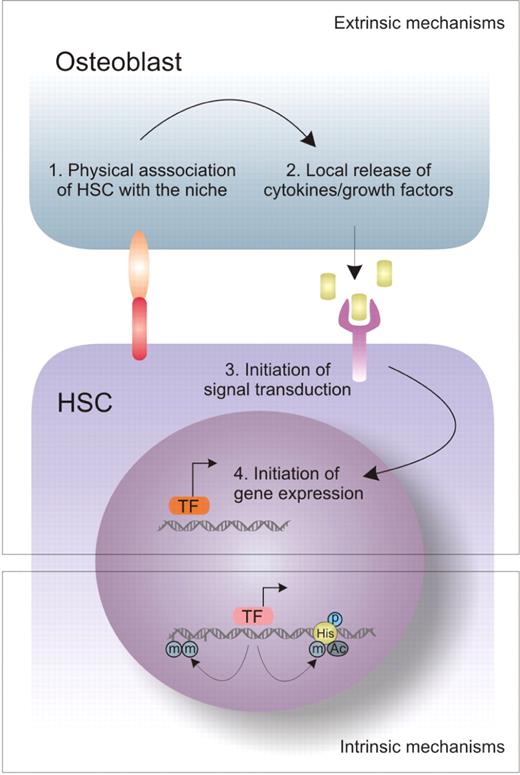
Extrinsic and intrinsic mechanisms that determine stem cell fate. HSC quiescence, self-renewal and differentiation are regulated by both intrinsic and extrinsic mechanisms. Extrinsic mechanisms include changes in stem cell fate that are dictated by the environment, i.e. niche. Once physical association between the osteoblast and the HSC has occurred, release of different growth factors will trigger diverse signal transduction pathways that will initiate expression of downstream target genes. Intrinsic mechanisms are niche-independent and, for example, they can affect the epigenetic state of HSC, as it is controlled by chromatin remodelers. (TF-transcription factors).
Physical association of the stem cell with its niche
The stem cell niche is an anatomical unit located in the endosteum within the bone marrow cavity that is composed of osteoblasts, osteoclasts and stromal fibroblasts. Various studies have shown that osteoblasts are important players in providing HSCs with extrinsic cues. Figure 2 provides an overview of molecules that have been implicated in HSC–osteoblast interactions, and these molecules will be discussed in detail in this section.
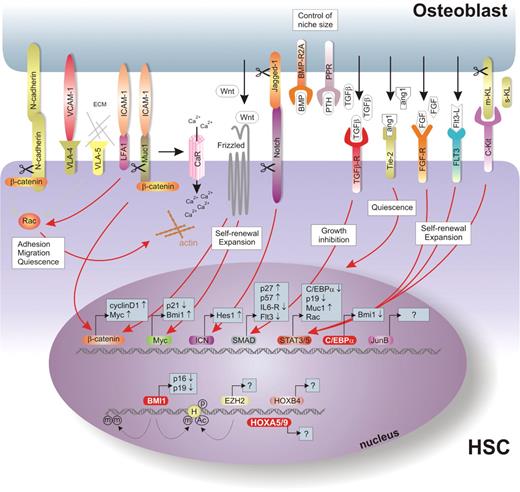
Signal transduction pathways in the HSC niche. A graphical representation of signaling pathways involved in the HSC fate in the niche. In this model, direct physical interaction between the HSC and osteoblast could be mediated by cadherins and integrins such as VLA-4 and VLA-5 (upper left section) which are involved in processes like adhesion and migration of the stem cell. Once appropriately localized within the quiescent niche, processes such as self-renewal, maintenance of quiescence or exit from the niche followed by proliferation and differentiation are highly controlled by growth factors and cytokines locally secreted by osteoblasts and stromal cells. Examples of such molecules are TGF-β, which is a negative regulator of the cell growth, Ang-1 responsible for the stem cell quiescence or WNTs and FGF-1, which promote stem cell expansion (upper right section). These factors can now dictate HSC fate by triggering specific signaling downstream modulators within the HSC, such as MYC, β-catenin, STATs, SMADs or C/EBPα. The possible intrinsic, that is, epigenetic regulators are represented in the lower part of the figure.
It is only recently that two groups of researchers independently revealed the spatial organization of the stem cells within the niche (14,15). Calvi et al. (14) demonstrated that activation of parathyroid hormone (PTH)-related protein receptors by feeding mice with PTH resulted in increased number of osteoblasts in the endosteum, thereby enlarging the stem HSC niche which allowed increase in HSC numbers. These osteoblasts expressed elevated levels of Jagged-1, the ligand for Notch receptors. Activation of Notch1 has been shown to result in enhanced HSC self-renewal, possibly by inducing a set of self-renewal genes including Hes1 (18) (Fig. 2). Conditional deletion of BMPR1 also leads to increased numbers of N-cadherin+CD45− osteoblastic (SNO) cells and subsequently to higher stem cell numbers (15). Thus, BMP signaling via the BMPR type IA complex controls the number of HSCs by regulating the niche size. N-cadherin is not only expressed on osteoblasts, but also on a subpopulation of murine LSK cells (15) where it interacts and forms an adherence complex with β-catenin (19). WNTs, as potent inducers of β-catenin signaling, have been implicated in the maintenance and expansion of murine HSCs (20,21). How N-cadherin–β-catenin interactions can affect HSC fate within the niche is currently unclear. It is possible that under certain circumstances metalloproteinases (MMPs), such as MMP9 or ADAM10, result in cleavage of membrane-bound N-cadherin, thereby increasing the cytoplasmic β-catenin pool that is now available to act in the canonical WNT pathway to induce expression of target genes, including CyclinD1 and c-MYC (22). Alternatively, the sole function of cadherins and integrins might be to physically associate HSC with osteoblasts or extracellular matrix (ECM) at the appropriate positions within the bone marrow niche (upper left part of Fig. 2). Integrins such as VLA-4 and VLA-5 are efficient activators of the small GTPase family members RAC1 and RAC2. Targeted deletion of both RAC1 and RAC2 results in mobilization of stem cells, indicating their importance in appropriate positioning of HSCs within the bone marrow microenvironment (23,24). In line with these observations, lack of VLA-4 expression restricts competitive repopulation activity and self-renewal potential of HCSs (25). Further support for the view that stem cells need to be physically associated with their bone marrow microenvironment arises from studies that demonstrated that in the absence of membrane-bound Kit Ligand (mKL) in the niche, HSCs are not maintained (26,27). Furthermore, it has been shown that MMP-9-mediated cleavage of mKL into soluble KL results in the translocation of HSCs from the quiescent endosteal niche towards vascular-enriched niches (discussed in more detail in the next section) favoring differentiation and HSC mobilization into the peripheral circulation (28). Recently, it was demonstrated that HSCs sense relatively high [Ca2+] levels via a seven transmembrane-spanning calcium-sensing receptor which is expressed on HSCs. This receptor is required to retain HSCs in close proximity to the endosteal surface of the bone, possibly by mediating the association of HSCs with collagen I (29). It has been reported recently that MUC1 can initiate calcium signaling through association with its ligand ICAM-1 (30), and as MUC1 can mediate transendothelial migration of breast cancer cells (31), these data suggest that MUC1-mediated cytoskeletal rearrangements might be involved in the interaction between HSCs and the niche as well.
The role of growth factors and cytokines in the stem cells niche
Once localized within the niche, locally secreted cytokines and growth factors can dictate stem cell fate by initiating specific signal transduction within the HSC (upper right section of Fig. 2). Transforming growth factor-β (TGF-β) is one of the few known negative regulators of HSCs. It maintains the stem cells in a slow cycling or quiescent state partly by blocking the cell surface expression of cytokine receptors like c-KIT, FLT3, MPL and IL-6R (32). The cell cycle arrest of human HSCs requires up-regulation of p57 which is a known tumor suppressor (33). Whether TGF-β is a negative regulator under physiological in vivo conditions is not clear yet, as HSCs with deleted TGF-β type I receptors have normal differentiation abilities and a normal cell-cycle distribution (34). In addition, the angiopoietin-1 (ANG-1) produced by stromal cells enhances the ability of HSCs to become quiescent through interaction with its tyrosine kinase receptor TIE2 (35). The mechanism of cell-cycle inhibition by TIE-2 still remains to be identified, but the cyclin-dependent kinase inhibitor p21 has been shown to be important for maintenance of HSCs quiescence (36). Interestingly, the gene encoding p21 is directly repressed by c-MYC (37), another key regulator in the stem cell niche. Resting HSCs are characterized by high p21 levels and an absence of c-MYC expression, whereas increasing levels of c-MYC activity have been linked to reduced p21 expression and a more active state of the HSC (38).
Quite different from the growth-inhibiting effects induced by TIE-2 or TGF-β, growth-promoting factors have also been identified. We have reported a role of fibroblast growth factor-1 (FGF-1) in stem cell self-renewal and expansion (39). Upon binding to its receptors, FGFR1–4, a variety of signal transduction pathways can be activated, including the MAPK pathway, STATs and PI-3K (40). Prolonged cultures of murine bone marrow cells supplemented with soluble FGF-1 resulted in robust expansion of HSCs (41) and these cells represent excellent targets for retrovirus-mediated gene delivery (42).
Intrinsic epigenetic modulators
Epigenetic modification of the chromatin structure underlies the differentiation of pluripotent HSCs into their committed progenies. Recent evidence indicates that members of the PcG protein complex play key roles in normal and leukemic hematopoiesis (lowest segment of Fig. 2). Notable examples are EZH2 and BMI-1 (43). EZH2 controls gene repression through recruitment of histone deacetylases followed by chromatin deacetylation and methylation of histone H3 at residue lysine 27 (44). In contrast, BMI-1 is recruited to methylated histone H3 lysine 27 and has a role in maintenance of the epigenetic memory (45,46). We have recently reported that EZH2 prevents HSC senescence which also links chromatin remodeling to aging (47). BMI-1 has been implicated in the maintenance of hematopoietic cells. Loss of function analysis reveals defects in HSCs renewal (48) and enforced expression leads to ex vivo expansion of mouse HSCs (49). BMI-1 regulates senescence and cell proliferation through p19ARF and p16INK4A (50). Genome-wide analysis of PcG complexes in Drosophila, mouse and human embryonic stem cells revealed that they repress developmental regulators, including the HOX gene cluster, and that their target genes are predominantly regulatory genes that control major differentiation pathways (51–54).
Mounting evidence suggests that HOX proteins fulfill a critical role in hematopoiesis. HOXB4-defficient mice exhibit mild proliferation defects in HSCs and have normal hematopoiesis (55). Retroviral overexpression of HOXB4 results in increased proliferation of murine stem cells. However, overexpression of HOXB4 in human CD34+ cells was reported to either increase proliferation (56) or promote myeloid differentiation (57). Molecular mechanism and target genes responsible for the HOXB4-induced expansion remain to be elucidated. HOXA9, HOXA7 and MEIS1 are expressed in CD34+ cells and are downregulated upon differentiation (58). Disruption of HOXA9 in mice leads to decreased numbers of progenitors and competitive defects of HSCs (59). In contrast, enforced expression of HOXA9 promoted proliferative expansion of both HSCs and progenitor cells (60).
LSCs AND THEIR MICROENVIRONMENT
Similarities between HSCs and LSCs
Whether LSCs also depend on the niche for self-renewal is currently unclear. Although there are clear differences between normal stem cells and LSCs, there are also striking similarities. A graphical presentation of the LSC niche and some of the important regulatory molecules are shown in Figure 3 and will be discussed in this section. Most of the available data suggest that leukemia is a stem cell disease, in which the original stem cell self-renewal mechanisms are preserved but in which the tight control is lost due to transformation events. This appears to be true for most of the leukemic ‘hits’ that have been identified, apart from a few exceptions including PML–RARα (61), MLL–ENL (62) and MOZ-TIF2 (63). Although these examples are capable of imposing self-renewal characteristics on committed progenitors, the majority of genetic defects are observed or need to be experimentally introduced in primitive HSCs in order to create a transplantable leukemia. Thus, it seems reasonable to assume that many molecular mechanisms that enable self-renewal are shared among normal stem cells and LSCs. Indeed, both normal stem cells and leukemic HSCs depend on SDF-1-mediated CXCR4 signaling for homing and mobilization (64). WNT-induced β-catenin signaling has been implicated in the maintenance and expansion of murine HSCs (20,21), whereas inhibition of β-catenin by overexpression of Axin severely impaired the self-renewal capacity of progenitors in CML (chronic myeloid leukemia) (65). The adhesion of normal CD34+ stem/progenitor cells to bone marrow stroma and fibronectin is mediated by the integrins VLA-4 and VLA-5 (66–68), and a similar role is fulfilled by integrins in leukemic cells (69–71). Similar to mouse studies, we have shown that RAC is also required for the interaction of human HSCs with bone marrow stroma and their self-renewal. Moreover, LSC self-renewal and expansion on MS5 stroma also depends on RAC activity (manuscript submitted for publication). Thus, many of the molecules that mediate the interaction between stem cells and the bone marrow niche are utilized by both normal stem cells and leukemic HSCs.
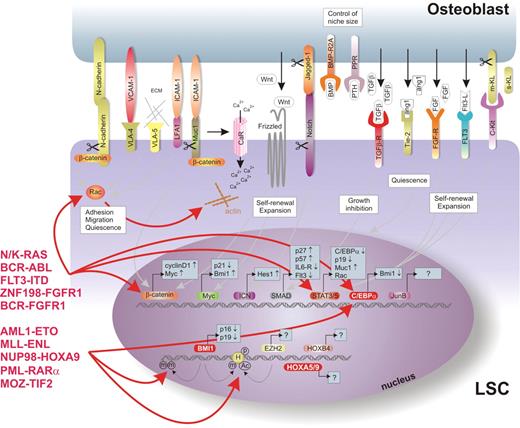
Signal transduction pathways in the LSC niche. A graphical representation of signaling pathways involved in the LSC fate in the niche. Whether LSCs are also depending on the niche for self-renewal is currently unclear. There are certain similarities between normal stem cells and LSCs, as well as striking differences. It seems reasonable to assume that many molecular mechanisms that allow self-renewal are shared among normal stem cells and LSCs. The differences in the signaling are most likely initiated by chromosomal translocations that are frequently observed in leukemias, such as BCR–ABL and AML1–ETO, or mutations in RAS or tyrosine kinase receptors such as FLT3 and c-KIT.
Do hematopoietic and LSCs share the same niche?
At diagnosis, most of the leukemic cells will be found in the BM or the blood, like the normal HSCs, and only in rare cases in non-hematopoietic organs. It is plausible that LSCs spread throughout the bone marrow and metastasize other sites there. Thus, they are able to escape the growth-inhibiting signals provided by the primary niche that maintain stem cell quiescence and prevent unlimited proliferation or self-renewal of normal HSCs. Recently, it has been shown that stem cells can home to ‘vascular niches’ (28) and one can speculate that exactly these niches can serve as a ‘home’ to the metastatic stem cells. Clearly, elucidating the localization of LSCs in the bone marrow and the identification of mechanisms by which LSCs escape the self-renewal cues of normal HSCs might provide a therapeutic window in which the LSC can specifically be targeted.
The dissimilarities between normal and leukemic cells that potentially allow us to manipulate and specifically eliminate the leukemia most likely find their origin in differences in signal transduction. These differences in signaling routes are in most cases initiated by chromosomal translocations that are frequently observed in leukemias, such as BCR–ABL and AML1–ETO, or mutations in RAS or tyrosine kinase receptors such as FLT3 and c-KIT (Fig. 3). As a consequence of these genetic lesions, often a constitutive downstream signaling is initiated. For example, it has been observed that NF-kB is highly activated in leukemic cells but not in HSCs (72), which is frequently mediated by the Ras pathway (73). The anti-apoptotic effects of the BCR–ABL oncogene can also be mediated via activation of NF-kB (74). Consequently, NF-kB inhibition can be a possible selective apoptotic therapy for patients with acute and chronic leukemias (72,75). Several parallel signaling pathways are activated by BCR–ABL, including the RAS, PI3-K/Akt, c-MYC and STAT5 pathways (76,77). Also, BCR–ABL is associated with f-actin and a variety of cytoskeletal proteins, particularly with focal-adhesion-associated adaptor proteins including Paxillin, Fak and Vinculin (78). Rho GTPases including RAC1 are activated by BCR–ABL and several reports have indicated that cells expressing BCR–ABL have an increased potential to associate with bone marrow stromal cells or ECM proteins such as fibronectin (79,80). Moreover, it was demonstrated that BCR–ABL-mediated leukemogenesis requires the activity of RAC, as homing to the bone marrow was impaired by expression of dominant negative RACN17, whereas the survival of mice transplanted with BCR–ABL and RACN17 double transduced cells was improved (79). By retroviral introduction of BCR–ABL into human CD34+ cells, we have observed that the interaction with bone marrow stromal cells is strongly increased which was associated with increased clonogenic activity (unpublished data), similarly as described by others (81,82). These data may imply that LSC self-renewal driven by BCR–ABL involves an enhanced interaction with the bone marrow microenvironment, but further studies are required to substantiate this hypothesis, particularly because data from primary human CML progenitor cells suggested that adhesion to stroma and ECM was reduced due to the expression of BCR–ABL (83). Furthermore, STAT5 signaling has been implicated downstream of BCR–ABL in leukemic transformation (84). More recently, it was demonstrated that STAT5 is required for the efficient induction and maintenance of BCR–ABL-induced CML in mice (85).
Other arguments for an improved LSC interaction with the niche coupled to elevated self-renewal properties came from studies in which FLT3-internal tandem duplications (FLT3-ITDs) or activating mutants of STAT5A were overexpressed in human cord blood-derived CD34+ cells. A constitutive activation of STAT5 is observed in the majority of AML cases, quite often due to FLT3-ITDs that are one of the most frequent mutations found in AML patients (86). Enforced activation of STAT5A in human CD34+ cells resulted in elevated self-renewal which was associated with an improved interaction with the microenvironment (87). Similar observations were made using murine HSCs (88). Also, expression of FLT3-ITDs in human CD34+ cells resulted in elevated clonogenic activity and CAFC frequencies (89), which depends at least in part on the activity of STAT5 (unpublished data). STAT5 signaling has also been implicated in various other chromosomal translocations that are observed in AML, including those resulting from fusions of the FGF receptor 1 with BCR and the zinc finger gene Znf198 (90–92). Particularly, cellular transformation of Znf198–FGFR1 fusions of BaF3 cells depends on STAT5 activity (93) (Fig. 3).
Recent understandings of leukemogenesis imply that at least two mutations complement each other to exert the AML phenotype (reviewed in 94). Typically, one mutation impairs the hematopoietic differentiation whereas the other promotes proliferation and/or survival. As examples, such ‘collaborative’ hits have been reported between activated FLT3 mutations and PML–RARα (95), MLL–SEPT6 (96), AML1–ETO (97), and most recently NUP98–HOXD13 and NUP98–HOXA10 fusions (98). In addition, it has been recently shown that NUP98–HOXA9 cooperates with BCR–ABL in mouse models of CML blast crisis (99,100). Whether the leukemic cell needs collaborative hits to improve its interactions with the extrinsic stem cell niche remains an open question. One possibility is that LSCs have much more active migration machinery when compared with normal HSCs that allows them to escape growth inhibition or quiescence promoting signals induced by osteoblasts and stromal cells in the niche. A number of membrane-associated ligands are normally present in the niche, and it has been shown that they can be cleaved by MMPs. The expression levels of MMPs such as MMP9 are often elevated in AML blasts (101). It is tempting to speculate that these MMPs might increase soluble concentrations of, for example, KL to enable leukemic self-renewal or expansion outside the niche. Furthermore, epigenetic modulation of the LSC by promoter methylation or histone modifications might result in a block in differentiation and elevated self-renewal, even in the absence of a microenvironment. Such examples could include BMI-1, which contains methyltransferase activity and has been shown to be involved in the regulation of self-renewal of HSCs (48,49,102), possibly by alleviating HSC senescence by downmodulation of p16INK4A and p19ARF (50). Also, the PcG protein EZH2 can directly methylate DNA (103) and prevents stem cell exhaustion (47). Whether and how chromosomal translocation products can affect the epigenetic state of the LSC is currently unclear. However, it is striking that, for example, CB CD34+ cells expressing AML1–ETO proliferate in liquid cultures in absence of stroma for over 7 months maintaining self-renewal of immature cells retain lymphoid and myeloid potential. This suggests that AML1–ETO affects the leukemic (stem) cell self-renewal in a microenvironment-independent manner (104).
CONCLUDING REMARKS
In conclusion, although a number of similarities exist between normal and leukemic stem cells in terms of molecular mechanisms that regulate self-renewal and interactions with the bone marrow microenvironment, clear differences exist as well. Future studies will be focussed on the purification procedures in order to be able to isolate LSCs to better homogeneity, on the localization of LSCs within the bone marrow compartment and on the differences between molecular mechanisms that LSCs utilize for self-renewal and niche interactions. These differences could be used as guidance for further studies to determine whether therapeutic windows exist in which small molecule inhibitors can specifically target the LSCs in a clinical setting.
ACKNOWLEDGEMENT
This work was supported by grants from the European Union (Eurythron EU FP6) and NWO-VENI (2004).
Conflict of Interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}