




Abstract
A truncated form of the Huntington's disease (HD) protein that contains the polyglutamine repeat, Httex1p, causes HD-like phenotypes in multiple model organisms. Molecular signatures of pathogenesis appear to involve distinct domains within this polypeptide. We studied the contribution of each domain, singly or in combination, to sub-cellular localization, aggregation and intracellular Ca 2+ ([Ca 2+ ] i ) dynamics in cells. We demonstrate that sub-cellular localization is most strongly influenced by the first 17 amino acids, with this sequence critically controlling Httex1p mitochondrial localization and also promoting association with the endoplasmic reticulum (ER) and Golgi. This domain also enhances the formation of visible aggregates and together with the expanded polyQ repeat acutely disrupts [Ca 2+ ] i levels in glutamate-challenged PC12 cells. Isolated cortical mitochondria incubated with Httex1p resulted in uncoupling and depolarization of these organelles, further supporting the idea that Httex1p-dependent mitochondrial dysfunction could be instrumental in promoting acute Ca 2+ dyshomeostasis. Interestingly, neither mitochondrial nor ER associations seem to be required to promote long-term [Ca 2+ ] i dyshomeostasis.
INTRODUCTION
Huntington's disease (HD) is a late-onset, autosomal dominant neurodegenerative disease characterized by motor abnormalities, cognitive deficits and neuropsychiatric symptoms. The disease results in widespread neuronal dysfunction and selective neurodegeneration in the CNS, particularly in the striatum. An expansion of a glutamine stretch within the Huntingtin (Htt) protein above approximately 40 repeats appears to confer a dominant toxic property that is deleterious to neurons as well as to decrease normal Htt activities ( 1 ). HD represents one of a growing number of polyglutamine (polyQ) repeat diseases that cause region-specific neuronal degeneration ( 2 ). Normal repeat Htt is primarily extranuclear; however, mutant Htt progressively accumulates in the nuclei of neurons in mouse models and human brain, ultimately forming visible nuclear inclusions ( 3–5 ). Cleavage of full-length Htt appears to be an obligate step in pathogenesis, since mutation of a critical caspase 6 site in full-length expanded repeat Htt eliminates pathology in YAC transgenic mice ( 6 ) and an N-terminal fragment of Htt can accumulate in nuclei ( 5 ). In addition to the repeats themselves, other Htt sequences may influence hallmark features of HD. The first 17 amino acids and the proline-rich region that each flank the polyQ stretch appear to influence sub-cellular localization, protein stability and alter the ability of the mutant protein to form aberrant protein–protein interactions including those leading to aggregation ( 7–11 ).
Nuclear accumulation of mutant Htt is strongly implicated in HD ( 12–15 ). Blocking this accumulation by fusing a nuclear export signal to mutant Htt rescues neurotoxicity induced by expanded Htt ( 12–14 ), and the onset and progression of HD pathogenesis in R6/2 mice is accelerated when exon 1 protein (Httex1p) is targeted to the nucleus using a nuclear localization signal (NLS) ( 12 ). However, as we previously showed, the first 17 amino acids can act as a cytoplasmic retention signal even when challenged with a strong NLS ( 11 ); therefore, a complex interaction between the protein domains, protein modifications and esence specific cellular processes must contribute to the nuclear localization that is observed. Although nuclear localization appears necessary for pathogenesis, extranuclear localization of Htt may also contribute to HD pathogenesis ( 12 ) through disruption of vesicle trafficking ( 16 , 17 ), BDNF transport ( 18 ), microtubule structure ( 19 ), NMDA receptor (NMDAR) and synaptic activity ( 20–23 ) and organelle morphology ( 24–26 ).
A functional consequence of the expression of expanded Htt is disruption of the homeostasis of intracellular free Ca 2+ ([Ca 2+ ] i ) ( 27 , 28 ). Synergistic systems may be involved, including an interference with Ca 2+ sequestration by mitochondria ( 29 ), to trigger [Ca 2+ ] i deregulation and apoptosis ( 25 ). Indeed, mitochondrial dysfunction has been implicated in HD pathogenesis beginning with early work showing that inhibitors of mitochondrial respiratory chain activities (3NP and malonate) recapitulate aspects of HD ( 30 , 31 ) and a direct interaction of Htt with mitochondria postulated to contribute to this dysfunction. An association between Htt and mitochondria has been shown for both truncated and full-length mutant Htt models ( 29 , 32–34 ). A functional contribution is suggested by studies showing that GST–Httex1p fusion proteins cause mitochondrial swelling and [Ca 2+ ] i disruption when incubated with isolated mitochondria ( 29 ) and that respiratory activity is reduced in the presence of mutant Htt ( 29 , 35 , 36 ), possibly through an association of Htt with an outer mitochondrial membrane protein. Here also, the amino acid domains flanking the expanded polyQ repeat may influence pathogenesis as a polyQ tract alone is less effective at inducing mitochondrial swelling and [Ca 2+ ] i disturbances than complete Httex1p.
Another hallmark feature of HD pathology is aggregation and several lines of evidence suggest that this process is also influenced by amino acids flanking the polyQ repeat. Antibodies directed against either the first 17 amino acid domain or the proline-rich region decrease the aggregation potential of mutant Htt exon 1 protein (Httex1p) fused to GFP, whereas antibodies directed against the polyQ tract increase aggregation potential ( 9 , 10 ). In contrast to the conclusion that the prolines may facilitate aggregation, addition of a proline-rich sequence C-terminal to an expanded polyQ peptide decreases aggregate formation and stability ( 37 ). Removal of the proline-rich region suggests that it plays a key role in aggregation, although there are differential effects dependent upon the nature of the epitope tag used ( 11 , 38 ).
Although mutant Htt can associate with mitochondria and trigger deleterious effects, the domain(s) of Httex1p that contribute to mitochondrial association and perturbations in [Ca 2+ ] i homeostasis have not been defined. Likewise, a systematic analysis of individual domains potentially involved in aggregation has not been performed in mammalian cells. Here, we investigate the influence of three key domains encoded by Httex1p upon sub-cellular localization, aggregation and [Ca 2+ ] i homeostasis. These domains include the first 17 amino acids, expanded or unexpanded polyQ repeats and the proline-rich region. We find that these Httex1p domains modulate the sub-cellular localization of Htt, influence aggregation potential and differentially modulate [Ca 2+ ] i disturbances in transfected cells.
RESULTS
The first 17 amino acids and the proline-rich region are involved in cytoplasmic localization of httex1p
We previously showed that the 17 amino acid domain of Htt, the sequence of which is highly conserved across species, has properties of a cytosolic retention signal ( 11 ). It can target GFP to the cytosol even in the presence of a fused NLS, via a mechanism that is distinct from CRM-1-dependent export. To systematically determine the contribution of the 17 amino acid domain, the proline-rich region and the polyQ domain of Httex1p to sub-cellular localization, plasmids encoding these domains alone or in combination were generated. Httex1p was used to represent the simplest Htt polypeptide that causes HD-like phenotypes in model organisms without additional amino acids present. Each polypeptide was epitope-tagged in frame to GFP on the C-terminus (Fig. 1 A). Immortalized striatal neurons (St12.7) and differentiated PC12 cells, both highly transfectable neuron-like cells, were transiently transfected with each construct and expression monitored by western analysis (Supplementary Material, Fig. S1A and B, respectively). Each protein is expressed, although in general, expanded repeat containing proteins show lower abundance of soluble protein, presumably due to stability of the expressed protein, aggregation and/or altered cellular degradation processes.
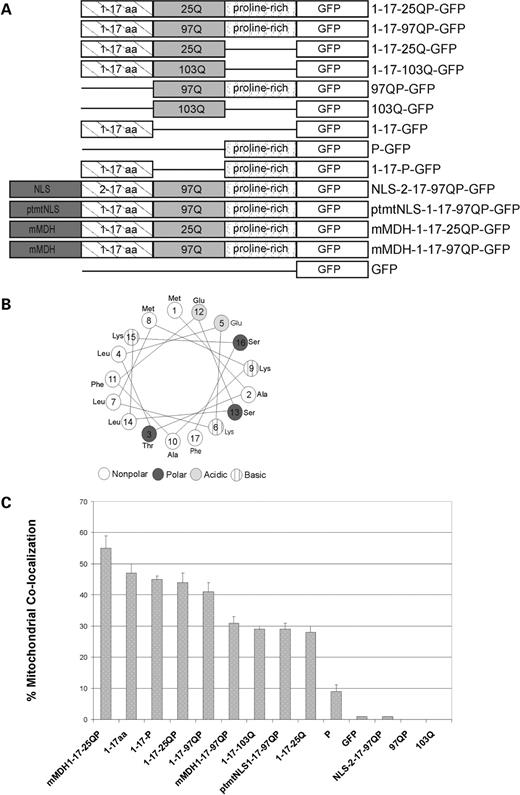
Httex1p constructs and % co-localization of Httex1p plasmids with mitochondria. ( A ) Schematic of Httex1p constructs. mtNLS, mutant NLS; mMDH, mitochondrial malate dehydrogenase mitochondrial targeting sequence. ( B ) The first 17 amino acids of Httex1p form an amphipathic α-helix. Using helical wheel software, the first 17 amino acids are predicted to form an amphipathic α-helix where hydrophobic and hydrophillic amino acids are positioned on opposite sides of the α-helix. ( C ) % co-localization of Httex1p polypeptides with mitochondria is shown. The first 17 amino acids are required for co-localization. Zeiss 510 software was used to analyze confocal microscope images of Httex1p polypeptides in St12.7 cells and Pearson's coefficient was determined per Htt construct and multiplied by 100 to obtain % co-localization. Standard error was calculated.
Sub-cellular distribution was evaluated 48 h post-transfection and representative images are shown in Figure 2 . Localization of Htt is visualized by GFP fluorescence (first image per panel). Complete Httex1p is primarily cytosolic in these cells (Fig. 2 A and B). When the proline-rich region is expressed in the absence of other Htt domains (P-GFP), it is primarily nuclear similar to GFP (Fig. 2 F and N), indicating that the proline-rich region does not independently influence cellular targeting of GFP. However, when the proline-rich region is removed from expanded Httex1p, the protein shows a more uniform distribution throughout the cell, suggesting that the proline-rich region enhances cytoplasmic localization of Htt in the context of Httex1p (compare 1-17-97QP-GFP, Fig. 2 B, with 1-17-103Q-GFP, Fig. 2 D). This diffuse distribution is also observed when the prolines are deleted from unexpanded Httex1p (1-17-25Q-GFP, Fig. 2 C).
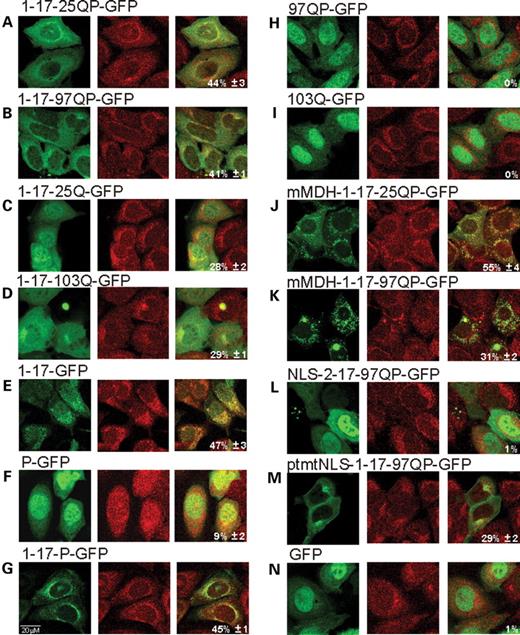
Httex1p polypeptides co-localize with mitochondria. Transient transfections of each Httex1p construct were performed in St12.7 cells and shown in ( A – N ). Forty-eight hours post-transfection, cells were stained with Mitotracker Red. Confocal microscopy was used to analyze cellular localization of Htt constructs and co-localization with mitochondria. Representative images are shown. The first panel is GFP fluorescence, second panel is Mitotracker Red and third panel is the merged image. % mitochondrial co-localization is annotated in the third panel.
The first 17 amino acids alone (1-17-GFP, Fig. 2 E) are cytosolic as described previously ( 11 ). When expressed together with the proline-rich region, a punctate cytoplasmic pattern is observed (eg. 1-17-P-GFP, Fig. 2 G). Consistent with the N-terminal region acting as a cytoplasmic retention signal, expanded Httex1p without the first 17 amino acids (97QP-GFP, Fig. 2 H) or expanded polyQs alone (103Q-GFP, Fig. 2 I) are primarily nuclear. To determine whether the cytosolic localization signal from the 17 amino acid domain overrides other targeting signals in the context of Httex1p, an NLS was fused to the N-terminus of Httex1p (NLS-2-17-97QP-GFP, Fig. 2 L) and found to target the protein primarily to the nucleus. One possibile explanation is that the NLS might simply block recognition of the first 17 amino acids; however, fusion of a non-functional NLS to the N-terminus of Httex1p (ptmtNLS-1-17-97QP-GFP, Fig. 2 M) is cytosolic, similar to Httex1p. When the NLS is fused to the C-terminus of the 17 amino acid domain (1-17-NLS-GFP), localization remains cytosolic ( 11 ). These results suggest that the cytoplasmic retention capability of the first 17 amino acids is functional and can be overcome by an NLS in the context of Httex1p; however, only when the NLS is placed N-terminal to the 17 amino acid sequence.
Httex1P co-localizes with mitochondria
The Htt protein, either endogenous or exogenous full-length or N-terminal fragments, can associate with mitochondria ( 29 , 33 , 34 ), and in particular can co-fractionate with the outer mitochondrial membrane ( 32 ). Since we observed localization to punctate cytoplasmic structures that resemble mitochondria (1-17-P-GFP), mitochondrial co-localization was assessed for Httex1p in both St12.7 and PC12 cells (Fig. 2 ) (data not shown). Quantitation of mitochondrial localization for multiple images is shown in Figure 1 C and is computed using a Pearson's correlation to represent the ratio of overlap between the number of mitochondrial pixels and Htt pixels. Both unexpanded and expanded Httex1p show the same relative level of mitochondrial association (Fig. 2 A and B); 1-17-25QP-GFP shows 44%±3 and 1-17-97QP-GFP shows 41%±3 mitochondrial localization (Figs 1 and 2 ), whereas a GFP control shows no mitochondrial localization (Figs 1 C and 2 N).
Complete Httex1p, unexpanded and expanded, uncouple the electron transport chain, increase respiration, decrease membrane potential and increase reactive oxygen species production in isolated cortical mitochondria
Expanded polyQ stretches purified as GST fusion proteins have a functional impact on isolated mitochondria ( 29 , 36 , 39 , 40 ), consistent with the idea that mutant Htt alters energy production in part through direct interactions with mitochondria ( 29 , 32 ). On the basis of the mitochondrial association observed for Httex1p, we sought to confirm the direct impairment of mitochondrial function and to extend these studies to determine the nature of the impairment. GST fusion proteins encoding unexpanded and expanded Httex1p (5 µg of GST-20QP or GST-51QP per mg of mitochondrial protein) or GST (control) were added to isolated cortical mitochondria and effects on respiration, mitochondria membrane potential and reactive oxygen species (ROS) production were monitored. The addition of 20QP or 51QP to isolated mitochondria utilizing complex-I, NADH-linked substrates (pyruvate/malate) and locked in state IV respiration (in the presence of oligomycin to inhibit ATP synthase), resulted in a significant increase in oxygen consumption, indicating increased proton leakage across the inner mitochondrial membrane (Fig. 3 A). This uncoupling effect was even more pronounced in mitochondria that were pre-incubated with Httex1p proteins and utilizing complex-II, FADH-linked substrates (succinate); however, in this case, only 51QP demonstrated significant effects on ATP phosphorylation (state III respiration) and state IV respiration (Fig. 3 B). The presence of either 20QP or 51QP significantly reduced mitochondrial membrane potential (Fig. 3 C) as shown previously ( 36 ) and, surprisingly, increased mitochondrial ROS production in mitochondria utilizing complex-I substrates that were locked in state IV respiration (Fig. 3 D). These results are consistent with studies showing that Httex1p polypeptides can have a direct effect upon mitochondrial function; however, point to a mechanism whereby the polyQ expansion results in an uncoupling of the proton gradient as well as increased ROS production, indicating a functional consequence of the observed association.
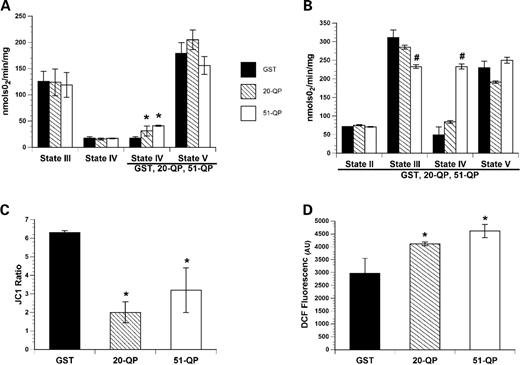
Httex1p proteins uncouple mitochondrial respiration, reduce mitochondrial membrane potential and increase mitochondrial ROS production in isolated cortical mitochondria. ( A ) GST fusion proteins encoding both unexpanded (20QP) and pathogenic (51QP) polyQ repeat lengths of Htt increased state IV (in the presence of oligomycin to inhibit mitochondrial ATP synthase) oxygen consumption in mitochondria utilizing complex-I, NADH-linked substates, pyruvate and malate. The Htt constructs were added to chamber following the addition of ADP (state III respiration) and oligomycin resulting in a significant increase in oxygen consumption compared with the GST addition alone. ( B ) To assess the effect of Httex1p proteins on complex-II, FADH-linked driven respiration, mitochondria were pre-incubated with GST, 20QP or 51QP for 1 min prior to the addition of the complex-II substrate succinate (state II). The presence of 51QP significantly reduced oxygen consumption in the presence of ADP (state III) and significantly increased state IV respiration. ( C ) The presence of either 20QP or 51QP significantly reduced mitochondrial membrane potential which was measured using the ratiometric indicator 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolycarbocyanine iodide (JC-1) in the presence of complexes-I and II substrates. Mitochondrial ROS production was assessed using the indicator DCF. ( D ) 20QP or 51QP significantly increased mitochondrial ROS production when utilized complexes-I and II oxidative substrates. Bars represent group means, SEM ( n = 3–5/group); asterisk indicates P < 0.01 compared with GST controls and hash indicates P <0.01 compared with control and 20QP.
The first 17 amino acids are required for mitochondrial association
To further investigate the contribution of the protein domains flanking the polyQ stretch to the observed mitochondrial association, each Httex1p domain, either singly or in combination, was assessed. Since the 17 amino acid domain appears to be responsible for the cytosolic localization of Httex1p, we tested whether it contributes to the observed mitochondrial association. Mitochondrial targeting sequences are composed of 20–60 amino acids abundant in positively charged amino acids, hydroxylated residues and few, if any, negative charges. These sequences are predicted to form amphipathic α-helices (helical wheel) in membranes or membrane-like environments ( 41 ). When evaluated for its potential to form a helical wheel ( http://cti.itc.Virginia.EDU/~cmg/Demo/wheel/wheelApp.html ), this domain indeed fits within the predicted structure (Fig. 1 B). Consistent with this, when the first 17 amino acids are expressed alone, 47%±3 mitochondrial co-localization is observed (Figs 1 C and 2 E). When this domain is deleted, Htt polypeptides 97QP-GFP or 103Q-GFP do not co-localize to mitochondria (Figs 1 C and 2 H and I, respectively), demonstrating that the first 17 amino acids are required for the association of Httex1p with mitochondria.
Although the proline-rich region alone (P-GFP) marginally associates with mitochondria (9%±2; Figs 1 C and 2 F), the prolines do not significantly enhance the association of the first 17 amino acids in the absence of the polyQ stretch (1-17-P-GFP; 45%±1) (Figs 1 C and 2 G). However, the prolines do contribute to this association in the presence of either normal or expanded polyQ repeats: the association with mitochondria is reduced from 44%±3 (1-17-25QP-GFP) to 28%±2 (1-17-25Q-GFP) when the prolines are deleted (Figs 1 C and 2 A and B).
To compare the mitochondrial association of Httex1p with the same polypeptide targeted directly to the mitochondrial matrix, the mitochondrial targeting sequence from rat malate dehydrogenase (MDH) ( 42 ) was fused in frame to the N-terminus of unexpanded (mMDH-1-17-25QP-GFP) and expanded (mMDH-1-17-97QP-GFP) Httex1p. An increased mitochondrial co-staining is observed for mMDH-1-17-25QP-GFP (Fig. 2 J, 55%±4) compared to Httex1p with 25Q (1-17-25QP-GFP) (Fig. 2 A, 44%±3). Furthermore, mMDH-1-17-25QP-GFP more strongly associates with mitochondria than mMDH-1-17-97QP-GFP (Fig. 2 K, 31%±2), presumably reflecting aberrant folding and aggregation of the expanded repeat polypeptide (described before for Fig. 9 ), which reduces the ability to target the protein to the mitochondrial matrix.
As expected, fusing an NLS to the N-terminus of mutant Htt (NLS-2-17-97QP-GFP) completely abolished the ability of Htt to associate with mitochondria (Figs. 1 C and 2 L). Taken together, these results suggest that: (i) the first 17 amino acids are required for an association of Httex1p with mitochondria and (ii) the proline-rich region enhances mitochondrial localization in the context of Httex1p (see Fig. 1 C for overview).
Httex1p polypeptides can also co-localize with endoplasmic reticulum and Golgi
We investigated whether Httex1p polypeptides can associate with other cytoplasmic organelles, e.g. endoplasmic reticulum (ER) (Fig. 4 ) and Golgi (Fig. 5 ). Using ER tracker Blue-White DPX for visualization, we found that both complete unexpanded Httex1p (Fig. 4 A, 48%±2) and expanded Httex1p (Fig. 4 B, 36%±2) can co-localize with ER; however, unexpanded Httex1p shows a stronger association, suggesting that the expansion may reduce this interaction. In the absence of a polyQ repeat, the two protein domains flanking the polyQ repeat (1-17-P-GFP) retain the ability to associate with ER (Fig. 4 C, 44%±2). Similar to data for mitochondria, the 17 amino acid domain is required for this association (97QP-GFP shows no co-localization with ER, Fig. 4 E). In contrast to mitochondria, the prolines are required for a strong interaction in the absence of other domain the 17 amino acid domain only marginally associates (Fig. 4 D, 15%±1).
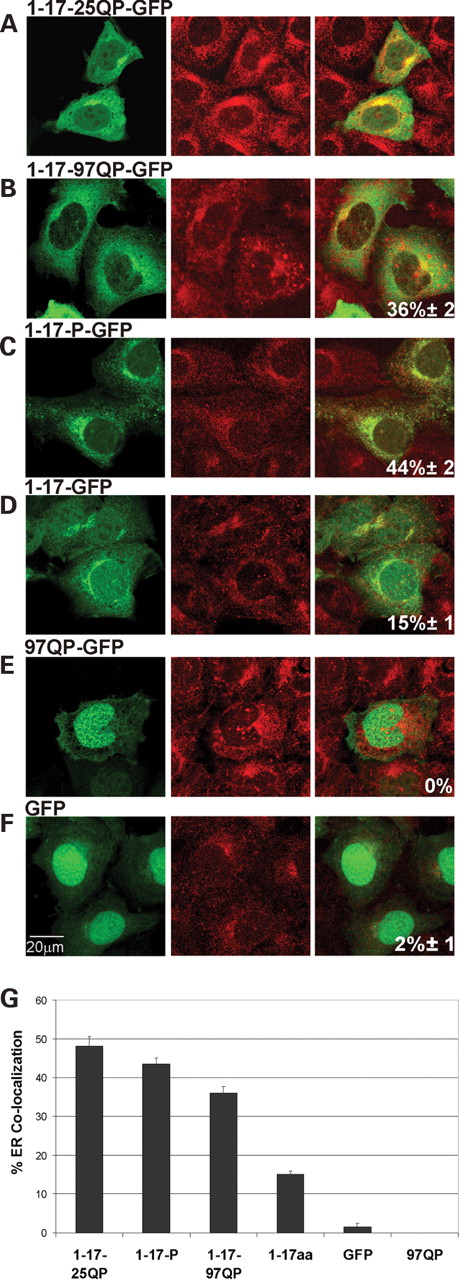
Httex1p polypeptides differentially associate with ER. ( A – F ) Httex1p constructs were transiently transfected into St12.7 cells and 48 h post-transfection, cells were stained with ER tracker Blue-White DPX to visualize ER. Confocal microscopy was performed to analyze Htt cellular localization and ER association. ( G ) Compilation of % co-localization of Httex1p plasmids with ER. Zeiss 510 software analysis was performed and Pearson's coefficient determined as described above.
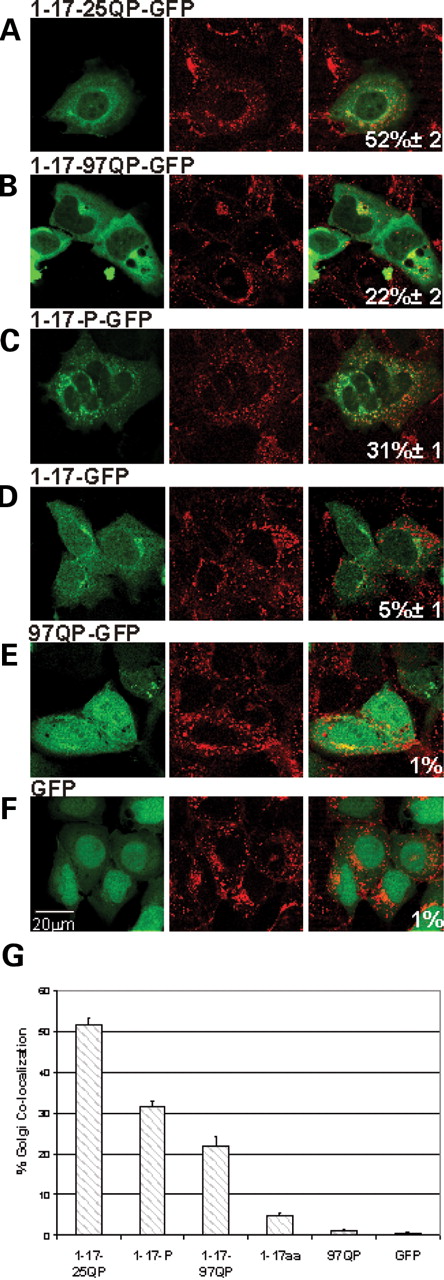
Httex1p polypeptides differentially associate with Golgi. ( A – F ) Httex1p constructs were transiently transfected into St12.7 cells and Golgi visualized using wheat germ agglutinin Texas Red-X and Pearson's correlation calculated as described for mitochondrial and ER co-localization. ( B ) Compilation of % co-localization of Httex1p plasmids with Golgi.
Interactions between larger Htt polypeptides and Golgi and other phosphoinositol phosphates have been shown previously ( 43 , 44 ). To assess whether Httex1p can associate with Golgi, wheat germ agglutinin conjugate Texas Red-X was used for visualization. In general, a similar profile is observed for ER and Golgi interactions: (i) a combination of the first 17 amino acids, the prolines and an unexpanded repeat provide maximal association (Fig. 5 A, 52%±2), (ii) the first 17 amino acids and the prolines are both required to promote an interaction of greater than 20% (Fig. 5 G) and (iii) expanded repeats reduce the interaction (Fig. 5 A and B, 52% compared with 25%).
Httex1p domains differentially contribute to membrane-bound organelle localization
A comparison of the relative overlap in staining of Httex1p polypeptides with mitochondria, Golgi and ER from independent experiments is shown in Figure 6 . Unexpanded Httex1p associates with all three cytoplasmic organelles; however, it does not favor any single organelle. The 17 amino acid domain alone also shows an association with all three organelles; however, it strongly favors an association with the mitochondrial membrane (47%±3 for mitochondria as opposed to 15%±1 for ER and 5%±1 for Golgi).
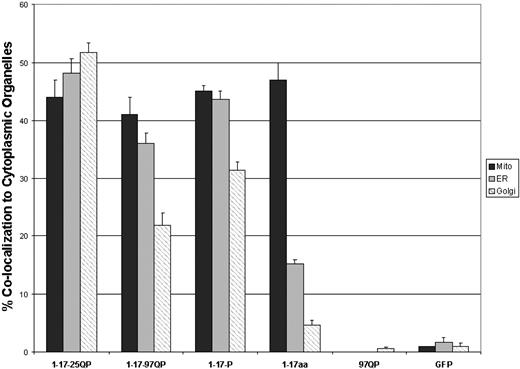
Schematic comparison of Httex1p polypeptide association with ER, Golgi and mitochondria. % co-localization for each cytoplasmic organelle is presented.
When the first 17 amino acids are expressed with the proline-rich region (1-17-P-GFP), the prolines enhance the association with ER and Golgi, pointing to potential interactions of the proline-rich region of Htt with membrane proteins. This is in contrast to the association with mitochondria, which is solely dependent upon the presence of the first 17 amino acids.
An expansion of the polyQ tract significantly reduces the visible association with ER and Golgi. As wild-type Htt is required for normal organelle morphology and function (e.g. mitochondria, ER and Golgi) ( 24 ), a decreased association due to the repeat expansion may also reduce necessary functions and contribute to the disruption of organelle morphology and function observed in HD.
Expanded Httex1p disrupts [Ca 2+ ] i homeostasis following both chronic and acute stress
Alterations in [Ca 2+ ] i levels play a critical role in promoting neuronal death in many neurological conditions including HD ( 27 ). The issue of [Ca 2+ ] i dyshomeostasis is particularly relevant in the context of the excitotoxic overdrive that takes place in HD. Several studies indicate that HD is most likely associated with excessive glutamate release, impaired glial glutamate re-uptake and overactivation of both NR1A/NR2B NMDA and mGluR5 receptors that are highly expressed in striatal medium spiny neurons (MSNs) ( 22 , 26 , 45 ). Thus, we investigated how Httex1p domains contribute to [Ca 2+ ] i deregulation triggered by toxic activation of glutamate receptors in a neuronal-like cell line.
Differentiated PC12 cells were transiently transfected with Htt-encoding plasmids and real-time changes in [Ca 2+ ] i levels monitored with a microfluorimetric assay. Httex1p constructs tested in these experiments represent those that when expressed show either maximal or little to no mitochondrial association and, with the exception of complete Httex1p (1-17-25QP-GFP), contain expanded repeats. Specific targeting of Httex1p to mitochondria was also tested (mMDH-1-17-97QP-GFP). Transfected PC12 cells loaded with the Ca 2+ -sensitive probe fura-2 AM were observed for 5 min, before a 20 min exposure to 500 µ m glutamate in a physiological buffer containing 10 m m CaCl 2 . After agonist washout, recovery was monitored for an additional 20 min. Comparison was made between transfected cells (identified by GFP fluorescence at the beginning of the experiment) and non-transfected cells (used as control). Two main perturbations in [Ca 2+ ] i handling were investigated: (i) alterations in baseline [Ca 2+ ] i levels due to prolonged exposure to expressed mutant Htt (chronic stress), and (ii) altered [Ca 2+ ] i levels upon glutamate exposure (acute stress).
Comparison among all constructs showed that in only one case, with complete expanded Httex1p (1-17-97QP-GFP) expression, did Htt promote both higher baseline [Ca 2+ ] i levels (chronic deregulation) and significantly higher [Ca 2+ ] i rises during glutamate receptor activation (acute deregulation) (Figs 7 B and 8 ; Supplementary Material, Table S3). The observed [Ca 2+ ] i deregulation may be the result of Htt interference on several homeostatic systems: Ca 2+ pumps and exchangers at the plasma membrane level, mitochondria and the ER. Since expanded Httex1p co-localizes with both mitochondria and the ER (Figs 2 B and 4 B, respectively), it is likely that some of the observed dyshomeostatic effects can be attributed to disruption of one or several of these regulatory systems.
![Time course of glutamate-triggered [Ca 2+ ] i rises by Httex1p plasmids. To monitor [Ca 2+ ] i responses in PC12 cells transfected with the indicated Htt plasmids, cells were loaded with the Ca 2+ -sensitive fluorescent probe fura-2 and [Ca 2+ ] i rises monitored before, during and after a 20 min exposure to 500 m m glutamate (in the presence of 10 m m Ca 2+ ). Traces show mean [Ca 2+ ] i rises expressed as fura-2 340/380 ratios (mean±SEM; red, transfected cells; black, untransfected cells). Note that upon glutamate exposure, only 1-17-97QP-GFP PC12 transfected cells undergo actue disruption of Ca 2+ homeostasis using integral analysis of cytosolic glutamate-driven [Ca 2+ ] i loads (see also supplementary Material table S3). In cells transfected with 97QP-GFP (Figure 7C), due to a climbing baseline before glutamate exposure, initial evaluation of glutamate-driven [Ca 2+ ] i changes gives the erroneous impression that these cells also undergo acute [Ca 2− ] i deregulation. However, when overall [Ca 2+ ] i load is investigated by integral analysis of fura-2 ratio changes occurring from T:5 (the time point immediately before the addition of glutamate) and T:25 (the time point immediately after glutamate washout), no statistically difference is observed when comparing 97QP-GFP transfected and untrasfected cells (see Supplementary Materials Table S3).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/16/1/10.1093_hmg_ddl440/1/m_ddl44007.jpeg?Expires=1716536825&Signature=4Sv15r1VceE78Msk9vQq2U3xnhxPTKC9kh30nk8ArMTWnwYn85RpwXbpxMo~W20gljynhXMvZOFTESoprJI9LXorNIjyJNHTLCNRS42ge56bC5qbdk863Ze~gkvrpUc7GIWRwRIzwamIL4TB4FPZF8wJ0cKX80AvdOZvU~nYfkgojfIpS~FkoMhM~EnFITe1NMeZAvs07SbeYxlUd2sgSCRoW4n-BFfk40FTrWWsAsemNvl4wsJp3uU4c1Mepc7kxSiE4C7Yy02QvwHM0tP-OTaLfOu8eDiZwnFcO-U4SOUxP3JEhco2vHsXfeO~wzuO6pmDkkWNK6Z3XbcJSs3OdQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Time course of glutamate-triggered [Ca 2+ ] i rises by Httex1p plasmids. To monitor [Ca 2+ ] i responses in PC12 cells transfected with the indicated Htt plasmids, cells were loaded with the Ca 2+ -sensitive fluorescent probe fura-2 and [Ca 2+ ] i rises monitored before, during and after a 20 min exposure to 500 m m glutamate (in the presence of 10 m m Ca 2+ ). Traces show mean [Ca 2+ ] i rises expressed as fura-2 340/380 ratios (mean±SEM; red, transfected cells; black, untransfected cells). Note that upon glutamate exposure, only 1-17-97QP-GFP PC12 transfected cells undergo actue disruption of Ca 2+ homeostasis using integral analysis of cytosolic glutamate-driven [Ca 2+ ] i loads (see also supplementary Material table S3). In cells transfected with 97QP-GFP (Figure 7C), due to a climbing baseline before glutamate exposure, initial evaluation of glutamate-driven [Ca 2+ ] i changes gives the erroneous impression that these cells also undergo acute [Ca 2− ] i deregulation. However, when overall [Ca 2+ ] i load is investigated by integral analysis of fura-2 ratio changes occurring from T:5 (the time point immediately before the addition of glutamate) and T:25 (the time point immediately after glutamate washout), no statistically difference is observed when comparing 97QP-GFP transfected and untrasfected cells (see Supplementary Materials Table S3).
![Baseline [Ca 2+ ] i levels after Htt transfection. Effects of different Htt polypeptides on Ca 2+ homeostasis were evaluated in PC12 cells 48 h post-transfection. As with Figure 7 , cells were loaded with fura-2 and baseline [Ca 2+ ] i levels analyzed. Bars show fura-2 340/380 ratios (mean±SEM). Values of transfected cells are expressed as percentage of the average ratio of untransfected cells (asterisk indicates statistical difference with P < 0.05).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/16/1/10.1093_hmg_ddl440/1/m_ddl44008.jpeg?Expires=1716536825&Signature=mSE9VjWWpkB4SpsyCnK45HxbbO6CXhnT4AHoNhu8D7JJXScIlFct4a0Hx7RTrc8oLOdeN8Oos51kjZCGrPME1ZO10afGS2dmRLSsGFg3ogweGfFTAsCj-wuCGoIfW98CnJsGPDk2N4L~ROvSo~MeSOtZHBnAexJbHHTcWOxonLVi3PT8IGj9WJvBHPDcCJx~KzZNHi86JupoHUStpajqd8EDzgs9rse4isAG4O7LZmsqVzbS20Uf~nwTZM2uEgSDJVJzoWxEDUrp-zrMEpOWkWzxvcMJDObd7lnyeT-s6R5FCZCLD7~YKeclR5VmDJJU0VzXFIRJgflM9zBZQ0QI7Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Baseline [Ca 2+ ] i levels after Htt transfection. Effects of different Htt polypeptides on Ca 2+ homeostasis were evaluated in PC12 cells 48 h post-transfection. As with Figure 7 , cells were loaded with fura-2 and baseline [Ca 2+ ] i levels analyzed. Bars show fura-2 340/380 ratios (mean±SEM). Values of transfected cells are expressed as percentage of the average ratio of untransfected cells (asterisk indicates statistical difference with P < 0.05).
PC12 cells expressing unexpanded Httex1p do not show perturbed basal [Ca 2+ ] i levels (Figs 7 A and 8 ), indicating that the polyQ repeat expansion is necessary to promote [Ca 2+ ] i deregulation. Consistent with the requirement for the first 17 amino acids in organelle targeting, disruption of Ca 2+ signaling is altered when this domain is removed. PC12 cells transfected with 97QP-GFP and 103Q-GFP constructs show increased basal [Ca 2+ ] i levels (Fig. 8 ), but have no effect on glutamate-driven [Ca 2+ ] i dyshomeostasis (Figs 7 D and F; Supplementary Material, Table S3), suggesting that acute effects may require an association with mitochondria and/or the ER. Neither of these polypeptides associate significantly with either the ER or mitochondria (Figs 2 and 6 ) (data not shown); however, they show strong segregation in the nucleus, similar to GFP alone. Therefore, the observed alteration in basal [Ca 2+ ] i levels this case may be due to their ability to promote aberrant nuclear events that interfere with [Ca 2+ ] i homeostasis.
Protein domains flanking the polyQ stretch do not alter [Ca 2+ ] i homeostasis in the absence of an expanded repeat
To investigate whether cytosolic organelle association is sufficient for different Htt polypeptides to chronically alter [Ca 2+ ] i levels, the protein domains flanking the polyQ stretch, the first 17 amino acids and the proline-rich region, expressed either singly (1-17-GFP and P-GFP) or in combination (1-17-P-GFP) or targeted to the nucleus (NLS-2-17-97QP-GFP) or mitochondria (mMDH-1-17-97QP-GFP), were tested. We find that in the absence of an expanded polyQ repeat, no disruption of [Ca 2+ ] i levels is observed following expression of the polypeptide, regardless of its ability to localize to mitochondria or ER (Fig. 8 ). Interestingly, direct targeting of Httex1p to the mitochondrial matrix (mMDH-1-17-97QP-GFP) eliminates the deregulatory action of the expanded polyQ region on [Ca 2+ ] i homeostasis (Figs 7 H and 8 ). Given the fact that both 97QP-GFP and 103Q-GFP are able to alter basal [Ca 2+ ] i levels due to chronic Htt expression, it seems likely that the addition of the NLS or mMDH sequence N-terminal to the first 17 amino acids alters the interaction properties of the first 17 amino acids and the expanded polyQ stretch responsible for disruption of Ca 2+ regulatory systems.
The first 17 amino acids increase the ability of Httex1p to form visible aggregates
It has been hypothesized that aggregate formation may interfere with organelle function and intracellular trafficking ( 29 , 46 , 47 ); therefore, we determined whether visible aggregation might contribute to disruption of [Ca 2+ ] i homeostasis. Since aggregation is dependent upon the presence of a repeat expansion, expanded repeat containing polypeptides that show comparable expression levels were used and visible aggregation/inclusion formation monitored in transfected St12.7 cells. In general, the extent of aggregation correlates with the length of the polyQ repeat, protein context and promoter strength ( 14 , 48–50 ). In these studies, the polyQ repeat length is controlled between 97 and 103Qs and levels of expression were shown to be similar by western analysis (Supplementary Material, Fig. S1). As shown in a representative experiment (Fig. 9 ), either deletion of the 17 amino acid domain or addition of amino acid sequences N-terminal to Httex1p reduces aggregation (on average 2-fold). It has previously been shown that targeting of mutant Htt to mitochondria and ER reduces Htt aggregation ( 51 ); however, it is not clear whether it is indeed the targeting of the protein to these organelles per se or whether masking the first 17 amino acids alone (e.g. ptmtNLS-1-17-97QP-GFP) is sufficient to reduce aggregation.
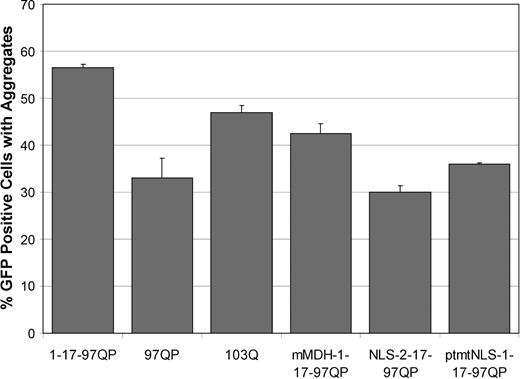
Aggregation analysis of Httex1p constructs. Effects of different Htt polypeptides on aggregation were evaluated following transient transfection of aggregation competent Httex1p plasmids into St12.7 cells. Each construct used showed comparable expression levels of expanded repeat proteins (Supplementary Material, Fig. S1). Each trial was performed in duplicate in three independent experiments. Images of six fields per construct were taken (at least 300 GFP-positive cells) and the total number of GFP positive cells and GFP cells containing visible aggregates were counted. % aggregation was calculated using the total amount of GFP cells containing aggregates divided by the total number of GFP positive cells. The presence of the first 17 amino acids increased, whereas the proline-rich region appeared to decrease, the ability of Httex1p to aggregate when Htt is fused to GFP.
Taken together, aggregation analyses indicate that although domains flanking the polyQ domain contribute to the aggregation potential of the expanded repeat polypeptides, the level of visible aggregation per se does not contribute to [Ca 2+ ] i dyshomeostasis, either positively or negatively. For instance, a similar degree of aggregation (moderate) is observed for 97QP-GFP, 103Q-GFP and mMDH-1-17-97QP-GFP; however, a difference in their ability to influence [Ca 2+ ] i handling is observed. 97QP-GFP and 103Q-GFP impair Ca 2+ signaling during chronic stress, and mMDH-1-17-97QP-GFP does not cause any Ca 2+ handling deficits. In addition, the difference in aggregation between 1-17-97QP-GFP and 103Q-GFP is minimal (~7%); however, only 1-17-97QP-GFP impairs Ca 2+ handling upon glutamate exposure. It will be of interest to determine whether oligomeric species or other forms of aggregated Htt not monitored in the present assays might be involved in [Ca 2+ ] i dyshomeostasis.
DISCUSSION
Sub-cellular localization of wild-type and mutant Htt appears to have a significant impact on HD pathogenesis. Here we systematically assess the contribution of three domains of Htt encoded by exon 1 to sub-cellular localization and cellular processes. We find that the 17 amino acid domain is involved in the normal association with cytosolic membrane-bound organelles: the 17 amino acid domain is required and sufficient for mitochondrial association and is required for co-localization with Golgi and the ER. The proline-rich region is also required for maximal association with the ER and Golgi and, in the presence of a polyQ repeat, enhances mitochondrial co-localization. In the absence of other domains, the first 17 amino acids preferentially target GFP to mitochondria over ER and Golgi and show mitochondrial association that is similar to an unexpanded Httex1p polypeptide targeted to the mitochondrial matrix. These data suggest that the first 17 amino acids, which can theoretically form an amphipathic α-helix, may represent a possible mitochondrial targeting signal, consistent with earlier postulated roles for Htt ( 52 ). Results from our study indicate that different domains of Httex1p facilitate sub-cellular localization and can target and critically interfere with the functioning of specific intracellular compartments, including mitochondria, ER and the nucleus. A truncated form of Htt protein was used, capable of causing disease in mice that recapitulates key aspects of HD and includes transcriptional changes observed in human patient brain ( 53 ), in order to focus upon these distinct domains.
Mutant Htt interference with mitochondrial function and Ca 2+ homeostasis
Mutated Htt plays an important role in the perturbation of Ca 2+ signaling potentially by acting, through synergistic mechanisms: enhansing the activation of NMDAR by interfering with the receptor trafficking via the PSD95–NR1A/NR2B complex ( 54 ), binding to IP3 sensitive receptors on the ER (InsP3R1) to promote from Ca 2+ mobilization through the metabotropic receptor-driven activation of IP3 ( 26 , 27 ) and interfering with mitochondrial Ca 2+ uptake ( 29 ). Htt-driven mitochondrial dysfunction also seems to play a key role in HD (reviewed in 55 ). Mitochondria from HD lymphoblasts have increased susceptibility to various apoptotic stressors ( 56 ) and this susceptibility is associated with repeat length and abnormal mitochondrial membrane potential ( 29 ). A link between Ca 2+ dyshomeostasis and mitochondrial dysfunction has been described in excitoxically challenged neurons from YAC mutant full-length transgenic mice and from HD knock-in mice ( 27 , 28 , 57 ).
Previous studies also suggest that pathogenic proteins can directly disrupt Ca 2+ signaling in at least three ways: (i) increased Ca 2+ influx through activation of cell surface receptors, (ii) mitochondrial Ca 2+ overload by promoting influx through direct pore formation into the outer mitochondrial membrane or by increasing mitochondrial permeability through permeability transition pore (PTP) opening and (iii) induction of oxidative damage that leads to both mitochondrial and cytosolic Ca 2+ dyshomeostasis ( 58 ). Each of these processes may be involved in the Ca 2+ dyshomeostasis observed in the presence of expanded polyQ.
Our data confirm and expand previous findings indicating a critical role exerted by mutant Htt in perturbing mitochondrial function. In our experiments, incubation of mitochondria with GST fusion protein expressing expanded Httex1p suggests that Htt can disrupt mitochondrial function, impair respiration, depolarize mitochondria and increase ROS production in vitro . These effects can be possibly explained by Htt ‘poking holes’ in the outer mitochondrial membrane. This hypothesis seems to be substantiated by the fact that both 20QP or 51QP: (i) increase state IV respiration in the presence of complex-I substrates and (ii) alter the mitochondrial membrane potential, indicating that Httex1p polypeptides may either be acting as proton channels across the inner mitochondrial membrane or may disrupt the integrity of the inner membrane such that protons are able to cross back into the matrix. The increase in ROS production that we measured without any obvious reduction in maximum electron transport system (ETS) activity is surprising since reducing membrane potential and increasing oxygen consumption should reduce ROS production ( 59–61 ). These data are supported, however, by previous reports showing that mitochondrial respiration and ATP production are impaired in knock-in Hdh Q111 striatal cells even in the absence of deficits of respiratory complex activity ( 35 ). Whereas a pathologic-length polyQ protein (Q62 glutamines fused to GST) directly inhibits ADP-dependent mitochondrial respiration (state III), the levels and activities of electron transport chain complexes, ATP synthase and the adenine nucleotide translocase remain unaltered ( 40 ). In the latter study, an increase in ROS production was also observed ( 40 ). One possible explanation for this disparity could be an alteration in mitochondrial pH due to the alteration in proton conductance. Another could be that the disruption of the inner membrane by Httex1p increases the access of the ROS indicator 2′-7′-dichlorodihydro-fluorescein (DCF) to ROS; however, this is unlikely since the form of DCF we utilized should freely cross both mitochondrial membranes. The significant effects of 51QP on complex-II-driven respiration with regard to ATP phosphorylation (state III respiration) as well as state IV respiration are also intriguing since 20QP did not significantly alter respiration in this paradigm. Since the involvement of complex-II with HD is well established ( 55 ), these data infer that Httex1p proteins differentially interact with components of the ETS in addition to disrupting inner membrane proton conductance.
Regarding the deranging effects of mutant Htt on neuronal [Ca 2+ ] i homeostasis, the scenario offered by our experiments seems complex. Single cell Ca 2+ imaging data in differentiated PC12 cells indicate that only Htt polypeptides that are able to associate with mitochondria and ER, for example, contain an exposed 17 amino acid domain and an expanded polyQ repeat, cause acute perturbation of [Ca 2+ ] i levels upon glutamate receptor overactivation. However, our data also indicate that the ability of mutant Httex1p to associate with either mitochondria or ER is not sufficient to account for disturbances in the ability of neurons to buffer Ca 2+ over extended periods of time. These findings are consistent with studies in cultured MSNs from YAC expanded repeat transgenic mice exposed to various apoptotic stimuli, including compounds that inhibit mitochondrial function or promote [Ca 2+ ] i deregulation ( 62 ). In these studies, neurons expressing expanded polyQ repeats were found to be more vulnerable to the toxic effects of [Ca 2+ ] i overload when produced by NMDAR overactivation as opposed to direct inhibition of mitochondrial function, again suggesting that mutant Htt can set in motion a complex set of toxic mechanisms (e.g. altered function of plasma membrane Ca 2+ channels, cytosolic organelles and nuclear-encoded events) that affect multiple systems for maximal pathogenesis.
Httex1p association with cytosolic organelles
Mutant Htt shows progressive accumulation in the nucleus in HD and this localization appears critical for disease. As a consequence of this nuclear accumulation, transcriptional regulatory mechanisms may be altered and gene expression changes are an early pathogenic event in both transgenic mouse models and in human HD brain ( 53 , 63 , 64 ). An indication of dysregulated transcription is the reduction of HD phenotypes in cells, flies and mice by histone deacetylase (HDAC) inhibitors ( 65–67 ). A very intriguing scenario is offered by the fact that interference with transcription factor activity can also affect mitochondrial function and most likely [Ca 2+ ] i homeostasis. Several recent studies indicate that transcription factors such as p53, NF-B, IB and Elk-1 co-localize with mitochondria, target the activity of PTP and impact cell viability ( 68–73 ), and a set of key studies show that function of the transcriptional co-activator, peroxisome proliferator-activated receptor co-activator-1 alpha (PGC1α), involved in mitochondrial biogenesis, is impaired by mutant Htt ( 74 , 75 ). Relevant to these studies, HDAC inhibitor activity also influences cell signaling pathways, protein stability and production of ROS ( 76 ) and ameliorates mutant Htt-mediated mitochondrial-dependent Ca 2+ handling defects ( 77 ).
In light of these nuclear events, it may seem counterintuitive that the 17 amino acid domain targets Httex1p to cytosolic organelles; however, this may be most important for physiological Htt functions that are disrupted upon repeat expansion. An association with sub-cellular organelles may be involved in the normal function of wild-type Htt since deletion of the Hdh gene in mouse embryonic stem cells results in an abnormal staining pattern and distribution of the normally perinuclear clustering of ER, Golgi and mitochondria ( 24 ), and siRNA-mediated inhibition of wild-type Htt expression causes a specific increase in the aberrant configuration of the ER network in N2a cells ( 78 ). From the data presented here, it is likely that the first 17 amino acids are involved in the interactions of normal Htt with cytosolic organelles. In striatal cells, full-length normal repeat Htt co-localizes with Golgi ( 43 ), and binding to the plasma membrane and membranes enriched in phosphoinositol phosphates is governed by a domain between amino acids 172 and 372 (44). These results demonstrate that domains in addition to the first 17 amino acids influence organelle interactions. A direct effect is supported by the association between vesicles of mouse brain lipids and Htt; whereby this interaction caused bilayer perturbation that was dependent upon the presence of the proline-rich region ( 79 ).
Aggregation
Since each of the Htt polypeptides capable of altering [Ca 2+ ] i homeostasis are also capable of forming visible aggregates, we investigated whether the presence of these inclusions correlate with the inability to buffer Ca 2+ . In other studies, the presence of cytosolic Htt, following expression of full-length mutant Htt, caused impaired mitochondrial movement specifically at sites of aggregates in primary cortical neurons ( 46 ). This blocked mitochondrial movement was observed before neurons displayed increased glutamate excitotoxicity, altered mitochondrial Ca 2+ handling or altered mitochondrial morphology ( 80 ). In our study, we find that the first 17 amino acids enhance the propensity of Htt to aggregate, which may contribute to these aggregation effects. It has been shown that mitochondrial targeting of or masking of this domain with additional N-terminal residues comparably reduces the levels of visible aggregation ( 51 ). In contrast, the presence of the proline-rich region appears to reduce the aggregation potential of Httex1p at the time point tested when fused to GFP. These data are in agreement with studies showing that flanking amino acids influence polyQ toxicity: Httex1p polypeptides containing the proline-rich region aggregate more slowly in yeast ( 81 ) and an oligoproline region attached to the C-terminus of a polyQ polypeptide decreases the rate of aggregate formation ( 82 ). However, the tendency to form inclusions is not required for the observed [Ca 2+ ] i disturbances. The process of Htt aggregation is complex and involves a multi-step process, involving monomers, soluble and insoluble oligomers and fibrils, and ultimately leading to inclusion body formation ( 83–85 ). Therefore, we speculate that toxic oligomeric species may accumulate around mitochondrial and ER membranes and influence the ability of Htt to dysregulate [Ca 2+ ] i homeostasis. Supporting this model, a recent study showed that soluble amyloid oligomers can permeabilize membranes ( 86 ) and therefore may initiate downstream pathologic processes such as [Ca 2+ ] i dyshomeostasis, production of ROS and mitochondrial dysfunction. Furthermore, in HD models, oligomeric fragments were detected in post-synaptic membrane fractions from brains of R6/2 transgenic HD mice at 8 weeks of age, at which time mice are symptomatic ( 79 ). The contribution of toxic oligomeric species to Ca 2+ dyshomeostasis and the requirement for the first 17 amino acids in these effects will be investigated further.
Summary
Our study provides an integrated approach to dissect the impact of Httex1p domains upon HD phenotypes. Specific domains play important roles in modulating sub-cellular localization and aggregation of Htt, as well as contributing to the ability of Htt to disrupt [Ca 2+ ] i homeostasis in cells, and we provide evidence of the critical nature of the first 17 amino acids in modulating these effects together with the presence of the expanded repeat. The influence of the first 17 amino acids on larger Htt proteolytic cleavage products and upon full length protein will be determined in future studies. Our results lend support to a complex scenario in which polyQ repeats promote an interplay between nuclear events and potential aberrant membrane associations or direct disruption of mitochondrial functioning, leading to enhanced [Ca 2+ ] i dyshomeostasis. Unraveling this interplay may help in the development of future therapeutic intervention.
MATERIALS AND METHODS
Generation of Httex1p plasmids
Complete Httex1p or truncated Httex1p (without the proline-rich region) plasmids encoding either a normal range or expanded polyQ tract with alternating CAG/CAA amino acid repeats were fused in frame to GFP at the C-terminus (1-17-97QP-GFP, 1-17-103Q-GFP, 1-17-25QP-GFP and 1-17-25Q-GFP) in pcDNA3.1 (Invitrogen) as described previously ( 87 , 88 ). The following plasmids were also generated and cloning is described in Supplementary Material: 1-17-P-GFP (first 17 amino acids, proline-rich region fused to GFP), P-GFP, (M-L-Q-proline-rich region fused to GPF), 1-17-GFP (first 17 amino acids fused to GFP), 97QP-GFP (M-A-S-F-97Qs, proline-rich region fused to GFP), 103Q-GFP (same as 97QP-GFP without proline-rich region), mMDH-1-17-97(25)QP-GFP (mitochondrial malate dehydrogenase mitochondrial targeting sequence fused to exon 1 with 97 or 25Qs), NLS-2-17-97QP-GFP (SV40 nuclear localization sequence fused to exon1 with 97 or 25Qs) and ptmtNLS-1-17-97QP-GFP (point mutant, non-functional NLS as described in Benn et al . ( 12 ) fused to exon 1 with 97 or 25Qs). In some cases, additional amino acids were introduced or retained due to cloning.
All Httex1p constructs were verified by sequencing and transient transfection into PC12 and St12.7 mammalian cells to observe expression (Supplementary Material, Fig. S2) and cellular localization using light microscopy (Zeiss Axiovert 25).
Aggregation analysis of Htt plasmids
Immortalized striatal neurons (St12.7) ( 89 ) and rat adrenal pheochromocytoma (PC12) were grown in six-well plates and transiently transfected with Httex1p plasmids (1 µg) using Lipofectamine 2000 (Invitrogen). St12.7 cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) and 10% fetal bovine serum (FBS) at 33°C and PC12 cells were grown in DMEM with 10% horse serum (HS) and 5% FBS at 37°C (all reagents from Invitrogen). PC12 cells were plated on collagen (BD Biosciences)-coated dishes and were differentiated 24 h post-transfection using 50 ng/ml NGF (Harlan Bioproducts, Indianapolis, IN, USA) in DMEM with 1% HS. Each plasmid was transfected in duplicate per experiment and three independent experiments were performed. 48 h post-transfection, cells were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) pH 7.4, stained with 4’,6-diamidino-2-phenylindole (DAPI) and mounted on microscope slides using Vectashield mounting medium for fluorescence H-1000 (Vector Laboratories). Using Auxiovision Release 4.2 software, images of each Httex1p plasmid were captured at 32 × on Zeiss Axiovert 25 microscope. Images were taken from six random fields per construct to have an approximate cell count of at least 300 and two measurements were made: the number of transfected cells (GFP) and the number of GFP cells containing visible aggregates. The % GFP-positive cells with aggregates was calculated by using the number of GFP cells with aggregates divided by the total number of transfected cells multiplied by 100.
Co-localization experiments of Htt plasmids and mitochondria, ER and Golgi
Transient transfections of each Httex1p plasmid were performed in both St12.7 and PC12 cells as described above in six-well dishes containing cover slips. For PC12 cells, the cover slips were first coated with poly- l -lysine (Sigma) before placing into collagen-coated six-well dishes. Twenty-four hours post-transfection, PC12 cells were differentiated as described above. Forty-eight hours post-transfection, cells were incubated with their corresponding media containing 300 n m of Mitotracker Red CM-H 2 XROS (Molecular Probes) for 1 h at 37°C. Cells were then washed first with their corresponding media followed by 1 × cold PBS and then fixed in 4% paraformaldehyde for 30 min. Cells were washed two times with PBS, stained with DAPI (Sigma) for 5 min, washed again with PBS and finally mounted on microscope slides as described above. Confocal microscopy (Zeiss LSM 510 Meta) was performed on slides to determine co-localization of Httex1p plasmids with mitochondria. Zeiss 510 image software analysis was performed to determine % localization with mitochondria. Individual cells (two to three per field) were analyzed from at least three different images per Htt construct. Pearson's correlation index, an overlap coefficient that is commonly used to measure the amount of overlap between two image pairs ( 90 ), was obtained for each construct and multiplied by 100 to give % mitochondrial co-localization.
Similar approaches were used to quantitate ER and Golgi staining. For ER staining, cells were washed with 2 ml of cold PBS 48 h post-transfection, fixed in 4% paraformaldehyde and then incubated with 1 µ m ER-tracker Blue-White DPX (Molecular Probes) in PBS for 30 min at 37°C. For Golgi staining, cells were washed with 2 ml of cold PBS, fixed in 4% paraformaldehyde and permeablized with cold methanol at −20°C for 10 min. Cells were then washed two times in 2 ml PBS and blocked with 1% BSA in PBS for 30 min at 37°C. After blocking with BSA, cells were washed again two times with PBS and incubated with 2 µg/ml wheat germ agglutinin conjugate Texas Red-X (Molecular Probes) in PBS for 30 min at 37°C. Cells were then treated as described above for mitochondria and % co-localization determined.
In vitro analysis of GST Httex1p proteins on isolated cortical mitochondria
The effects of Httex1p proteins on mitochondrial respiration and ROS production were monitored using purified cortical mitochondria. Cortical mitochondria were isolated from the cortex of adult, male Sprague-Dawley rats as previously described with slight modifications ( 59 , 91 , 92 ). The animals were decapitated and the brains carefully removed and placed in ice-cold isolation buffer (215 m m mannitol, 75 m m sucrose, 0.1% BSA, 20 m m HEPES, 1 m m EGTA and pH adjusted to 7.2 with KOH). All subsequent steps were conducted at 4°C. Total mitochondria (synaptic and non-synaptic fractions) were isolated from the cortex of adult, male Sprague-Dawley by differential centrifugation or utilizing a discontinuous Percoll gradient. Mitochondria isolated by differential centrifugation were used in assessment of mitochondria membrane potential and ROS production. Mitochondria isolated using Percoll gradients were used for all the oxymetric assessments. Synaptic mitochondria were released from synaptoneurosomes using nitrogen cell disruption as previously described ( 59 , 60 , 92 ). Further purification utilized a discontinuous Percoll gradient and differential centrifugation ( 93 ). The final mitochondrial pellet was resuspended in EGTA-free isolation buffer at a concentration of ~10 mg/ml and stored on ice. The protein concentration was determined using the BCA protein assay kit by measuring absorbance at 560 nm with a Biotek Synergy HT plate reader (Winooski, VT, USA).
GST fusion proteins encoding both normal (GST-20QP) and pathogenic (GST-51QP) polyQ repeat lengths of Htt were expressed in Escherichia coli cells and purified using glutathione–Sepharose beads as described previously ( 87 ). Proteins were eluted off the glutathione beads using 15 m m glutathione (Sigma) in Buffer A (20 m m HEPES pH 7.4, 125 m m KCl, 10% glycerol and 100 m m PMSF). Bradford assays and SDS–PAGE analysis (compared with BSA) were performed to determine protein concentration. Mitochondrial oxygen consumption was measured using a Clark-type electrode in a continuously stirred, sealed chamber at 37°C as previously described ( 59 , 60 , 92 , 94 ). Isolated mitochondrial protein (25–50 mg) was suspended in respiration buffer (215 m m mannitol, 75 m m sucrose, 0.1% BSA, 20 m m HEPES, 2 m m MgCl, 2.5 m m KH 2 PO 4 at pH 7.2) in a final chamber volume of 0.25 ml. Respiratory control ratio was calculated as the ratio of oxygen consumption in the presence (state III) or absence (state IV) of ADP using 5 m m pyruvate and 2.5 m m malate (complex-I-driven respiration) or 10 m m succinate (complex-II-driven respiration) as substrates. The effects of Htt proteins on mitochondrial respiration were assessed either following pre-incubation with the Htt proteins for complex-II-driven respiration or the Htt proteins were added to the chamber following initiation of state IV respiration by the addition of oligomycin to inhibit the flow of protons through the ATP synthase for complex-I-driven respiration. In all experiments, mitochondrial respiration results were expressed as nmols O 2 /min/mg of mitochondrial protein in the chamber.
Mitochondrial ROS production was assessed using the ROS indicator DCF as described previously ( 59 , 91 , 94 ). Mitochondrial membrane potential was measured as previously described ( 95 ) using the dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolycarbocyanine iodide (JC-1) (Molecular Probes), which exists as a green fluorescent monomer at low membrane potential, but reversibly forms red fluorescent ‘J-aggregates’ at polarized mitochondrial potentials. Briefly, for both assays, 50 µg of isolated mitochondria were incubated in a total volume of 100 ml respiration buffer at 37°C in the presence of pyruvate (5 m m ), malate (2.5 m m ), succinate (10 m m ) and oligomycin (1 µ m ). Htt proteins or GST alone was present throughout the experiment. Controls included the addition of FCCP (1 m m ) to depolarize the mitochondria and inhibit membrane potential-dependent ROS production (minimum ROS production) and antimycin A (1 m m ) to maximize ROS production by inhibition of the ETS ( 59 , 60 , 92 ). In each experiment, non-specific DCF oxidation was accounted by subtracting the fluorescence intensity measured in control wells in which no mitochondria were added. Data for ROS measurements were expressed as raw fluorescence units. Mitochondrial membrane potentials were expressed as the ratio of red aggregates to green monomers.
Ca 2+ imaging studies on differentiated PC12 cells transfected with Httex1p constructs
PC12 cells were plated on coverslips, differentiated for 4 days using NGF (50 ng/ml, International Clinical Services GmbH), and transfected with Lipofectamine 2000 (Invitrogen) using Httex1p constructs. Fluorescence imaging was performed using a Nikon TE 2000 (Nikon) inverted microscope equipped with a xenon lamp, filter wheel (Lambda 10, Sutter Instruments) at 40 × epifluorescence (N.A. 1.3) oil immersion objective. Images were acquired with a 12-bit digital CCD camera (Orca 100, Hamamatsu) and analyzed (after background subtraction from a cell-free region of the dish) with Metafluor 6.0 software (Universal Imaging). PC12 cells were loaded with the Ca 2+ -sensitive probe, fura-2 AM (3 μ m +0.2% pluronic acid) at room temperature for 30 min, washed in HCSS (120 m m NaCl, 5.4 m m KCl, 0.8 m m MgCl 2 , 20 m m HEPES, 15 m m glucose, 1.8 m m CaCl 2 , 10 m m NaOH pH 7.4) and kept in the dark for additional 30 min. Fura-2 excitation is at 340 and 380 nm, with emission at 510 nm. All experiments were performed in the presence of the cell-permeable heavy metal chelator TPEN (1 μ m ) in order to avoid contamination of the fura-2 signal by endogenous metals like Zn 2+ . Transfected cells were identified by the green fluorescence of GFP with the imaging set-up and non-transfected cells were chosen as the control. PC12 cells were also exposed to a 10 min glutamate challenge and analysis of the resultant [Ca 2+ ] i changes were expressed as variations of fura-2 ratio values. Two different evaluations were performed: changes in fura-2 ratio values before exposure to glutamate (average of baseline ratios) and integral analysis of fura-2 ratio changes during (t-5:25; acute glutamate exposure) and after (t-25:45; washout) glutamate challenge.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We would like to thank Katalin Illes, Ya-Zhen Zhu, Francesca Faricelli, Margherita Capasso, Valerio Frazzini and Zifu Wang for technical assistance and advice. This work was supported by the Hereditary Disease Foundation (to L.M.T. and J.L.M.), a Huntington's Disease Society of America Coalition for the Cure grant (to L.M.T.), the High Q Foundation (to L.M.T. and J.L.M.), NIH awards HD36081 (J.L.M.), NS 42157-04, PRIN 2004 (S.L.S.), RBNE03PX83-006 FIRB 2003 (S.L.S.) and NS045283 (J.L.M. and L.M.T.). We gratefully acknowledge the DNA Array Core Facility and Optical Biology Shared Resource of Cancer Centre Support Grant CA-62203 from the University of California, Irvine.
Conflict of Interest statement. None of the authors have financial interests or connections, direct or indirect, or other situations that might raise the question of bias in the work reported or the conclusions, implications or opinions stated—including pertinent commercial or other sources of funding for the individual author(s) or for the associated department(s) or organization(s), personal relationships or direct academic competition.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}