




Abstract
Many paradigms for our understanding of cancer etiology have been shaped in mammalian model systems. However, it has become evident that both genetic and epigenetic components actively influence the progression and severity of cancers. The complexity of epigenetic mechanisms in mammals has invigorated the use of non-mammalian model organisms in several research areas. Key contributions from this approach include (1) the in-depth characterization of epigenetic mechanisms and their interactions, resulting in an improved understanding of epigenetic pathways, (2) the establishment and refinement of techniques for genome-wide epigenetic profiling and (3) the discovery of novel epigenetic modifiers with potentially druggable enzymatic activities. Recent findings in all three areas will improve our understanding of epigenetic misregulation in cancer and facilitate the translation of basic research concepts into clinical applications.
The term ‘epigenetics’ generally is used in reference to meiotically and mitotically heritable molecular events involving a wide variety of protein complexes and regulatory mechanisms that do not involve changes in the DNA sequence. Understanding the pathways leading to the initiation, establishment, maintenance and plasticity of epigenetic states is crucial not only to recapitulate the events associated with cellular differentiation and homeostasis but also those ultimately leading to cellular transformation. Understanding epigenetic regulation thereby opens new ways for the diagnosis and treatment of cancer. In this regard, non-mammalian model systems, such as fungi ( Schizosaccharomyces pombe , Saccharomyces cereviseae , Neurospora crassa ), insects ( Drosophila melanogaster ) and plants ( Arabidopsis thaliana ) have provided the molecular details on how epigenetic marks are established, propagated and maintained.
Originally, epigenetic states of eukaryotic DNA have been discussed in the context of two structurally distinct domains, euchromatin and heterochromatin. Both chromatin structures represent functionally different states that interdependently regulate gene expression and chromosome behavior. In the classic view, euchromatic domains represent the accessible (nuclease-hypersensitive) and transcriptionally active parts of a genome whereas heterochromatic domains such as centromeres and telomeres were viewed to be less accessible (nuclease-resistant) and transcriptionally inactive. This simplistic view has been revised leading to our current understanding that the epigenetic state of a given genomic region is determined by the local balance between activities that promote or inhibit heterochromatin and euchromatin formation ( 1 ).
It is suggestive that the developmental ground state of the epigenome at the beginning of the individual life cycle correlates with a rather open chromatin configuration ( 2 ). Therefore, heterochromatin formation appears to be a key factor in the epigenetic regulation of gene expression and chromosome behavior during development, tissue differentiation and tumorigenesis ( 3 , 4 ). A substantial amount of research has been focused on elucidating the pathways regulating heterochromatin ( 5 ). To this day, three molecular mechanisms have been discovered that are intricately related in initiating and maintaining epigenetic states in general and heterochromatic states in particular: DNA methylation, histone modifications and RNA-associated silencing.
MECHANISMS OF EPIGENETIC GENE REGULATION
The best-described epigenetic mark is (cytosine-5) DNA methylation. This modification is being established by DNA methyltransferases (DNMTs), and interpreted by methyl CpG-binding domain (MBD) proteins. Eukaryotic cytosine methyltransferases contain 10 sequence motifs that are also present within bacterial (cytosine-5) methyltransferases suggesting that the eukaryotic DNA methylation system developed from the archetypal bacterial DNA restriction-modification systems ( 6 ). Some eukaryotes either have no cytosine methyltransferase coding sequence ( S . cerevisiae ) or appear to have recently lost the DNA methylation system ( C . elegans ). Other organisms, such as A . thaliana , have more than 10 cytosine methyltransferase homologues ( 7 ). The common functional denominator of the DNA methylation system in eukaryotes seems to be the silencing of genes and repetitive DNA such as viruses and transposable elements (see below).
The amino termini of histones H3 and H4 and the amino and carboxyl termini of histones H2A, H2B and H1, are subject to a variety of post-translational modifications, such as acetylation, methylation, phosphorylation and ubiquitination. The potential epigenetic functions of additional histone modifications (sumoylation, glycosylation, ADP ribosylation, biotinylation and carbonylation) remain to be analyzed in future studies ( 8 ). The modifications that have been studied most extensively are acetylation and methylation of lysine residues in histones H3 and H4. These marks are set by highly conserved histone-modifying proteins including histone acetyltransferases and histone methyltransferases, and can be removed by histone de-acetylases and recently discovered histone de-methylases ( 9 ). Increased acetylation, for example, is indicative of transcriptional activity, whereas hypo-acetylation correlates with the transcriptionally repressed state. Other modifications, like di- and trimethylation of histone H3 lysine 9 have been found to be a common mark for silent chromatin.
An important role for non-coding RNAs, miRNAs and the RNA interference pathway in the formation of heterochromatic domains has also been described (Fig. 1 ). For example, large non-coding RNAs can serve as scaffolds for the nucleation and spreading of heterochromatic domains during epigenetic processes ( 10–12 ). In addition, microRNAs have been shown to interact with nascent mRNA to alter chromatin of the corresponding DNA in Arabidopsis ( 13 ). The first demonstration that RNA can guide DNA methylation (RNA dependent DNA methylation, RdDM) was made in transgenic tobacco plants, where viroid-containing transgenes became methylated after infection, even though there is no DNA phase in the viroid replication cycle ( 14 ). These observations suggested for the first time the existence of RNA-mediated control of DNA methylation. A later study in S . pombe revealed that small RNAs in the RNAi pathway can transcriptionally silence genes and are required for centromeric heterochromatin at repetitive DNA ( 15 ). Deletion of components of the RNAi machinery, including the RNaseIII-like enzyme Dicer, the PAZ/Piwi family protein Argonaute 1 and the RNA-dependent RNA polymerase rdp1 caused the accumulation of complementary transcripts from centromeric heterochromatic repeats, the transcriptional de-repression of transgenes integrated at the centromere, loss of histone H3 lysine 9 methylation and impairment of centromere function. These results unequivocally proved the strict dependence of heterochromatin formation and targeting of H3 lysine 9 methylation on the RNAi pathway.
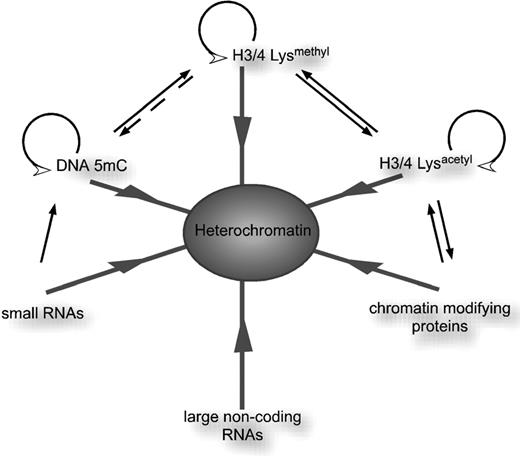
Epigenetic modifications and mechanisms leading to heterochromatin formation. Three different covalent modifications are important to distinguish the heterochromatic from the euchromatic state. Hypermethylation and hypoacetylation at lysine residues on core histones H3 and H4 as well as DNA methylation on cytosines contribute to the formation of heterochromatin. Each of the epigenetic modifications can be directly propagated and may influence acquisition of the other two. Targeting of the molecular machineries catalyzing these modifications can be accomplished through various RNAs as well as chromatin modifying protein complexes. Abbreviations: H3/4-Lys methyl , histone H3 or H4 methylated on lysine residues; H3/4-Lys acetyl , histone H3 or H4 acetylated on lysine residues; DNA 5mC, DNA methylation.
Comprehensive and genome-wide studies have begun to elucidate the biochemical pathways that collaborate to initiate and maintain heterochromatin ( 16 , 17 ). The canonical assembly process seems to occur in a stepwise manner involving a self-perpetuating cycle of histone modifications (deacetylation, histone H3 lysine 9 methylation) that recruit silencing complexes and spread along the chromatin fiber by self-oligomerization. Histone modifying enzymes contain specific motifs, such as the chromo and bromo domains, to recognize histone methylation and acetylation, respectively. This allows the targeting of histone modification activities, followed by the spreading of the epigenetic marks. The initial targeting of this process can involve sequence-specific DNA binding proteins ( 5 , 18 , 19 ) and/or the transcription of small RNAs originating from target loci ( 15 ).
Importantly, evidence from fungi, flies and plants demonstrates an interdependence of chromatin-based modifications and DNA methylation (Fig. 1 ). DNA methylation influences the extent of histone H3 lysine 9 methylation, and most H3 lysine 9 methylation is lost at centromeric and repeated heterochromatic loci in Arabidopsis DNMT mutants ( 20–22 ). Conversely, histone methylation can influence cytosine methylation in several non-mammalian model organisms, suggesting the existence of feedback loops between these modifications that propagate the silenced state ( 23–25 ). Additional loci that are required for DNA methylation and gene silencing, such as the HISTONE DEACETYLASE 6 ( HDA6 ) gene ( 26 , 27 ) and the DECREASE IN DNA METHYLATION 1 ( DDM1 ) gene ( 28 ) have been isolated in genetic screens. Model systems such as Arabidopsis will be crucially important to understand how DNMTs cooperate with histone modifying enzymes and chromatin-remodeling proteins to maintain gene silencing.
MAPPING THE EPIGENOME
The important role of aberrant DNA methylation in cancer development is widely accepted and underlined by the large number of published reports on epigenetically silenced tumor suppressor genes in various cancer types ( 4 , 29 ). Even though the knowledge of methylation and expression status of important regulatory genes is assumed to have great diagnostic and prognostic potential ( 30 ), the overall picture of the genome-wide methylation profile of mammalian cells is still rather diffuse. The characterization of the human methylome (i.e. collecting information about genome-wide methylation profiles) in various healthy and tumorous tissues represents a major challenge for the human epigenome project. In this context, non-mammalian model organisms play an important role in the search for fundamental patterns of genomic DNA methylation. In addition, the smaller genome size of these organisms ( A. thaliana : 125 Mb, N. crassa : 40 Mb, D. melanogaster : 180 Mb) facilitates comprehensive, genome-wide analyses.
In the filamentous fungus N. crassa , DNA methylation is catalyzed by a single DNMT, DIM-2, which is not essential for organismal viability ( 29 ). This stands in contrast to mammalian organisms that utilize four DNMTs and that require DNA methylation for embryonic development ( 31 ). Nevertheless, the Neurospora genome methylation level (2–3%) is comparable to mammals ( 32 ), and the availability of a DNMT mutant, combined with the comparatively low genome complexity, has provided an opportunity to study the biological function of DNA methylation. The characterization of Neurospora DNA methylation patterns was greatly facilitated by methyl-binding-domain agarose chromatography ( 33 ). This method is based on an affinity column consisting of the methyl-CpG binding domain of the MeCP2 protein attached to a solid matrix. The degree of methylated DNA enrichment is proportional to the density of methylated CpGs within the sequence. Hundreds of clones were further investigated by DNA sequencing and Southern blotting, using methylation sensitive restriction enzymes and candidate sequences were validated in the dim-2 DNMT mutant ( 34 ). Most of the methylated sequences were found to represent repeats and relics of transposons (Fig. 2 ), which had been subjected to repeat-induced-silencing, a genome-defence mechanism in fungi ( 34 ). Additionally, methylation of the centromeric region of N. crassa chromosomes was also detected. From these data, it was concluded that the predominant function of DNA methylation in Neurospora is the protection of the genome from transposon proliferation. Of note, a similar function of DNA methylation has also been proposed for mammals, based on the finding that the IAP retroelement is strongly upregulated in mouse mutants lacking the DNA methyltransferase Dnmt1 ( 35 ).
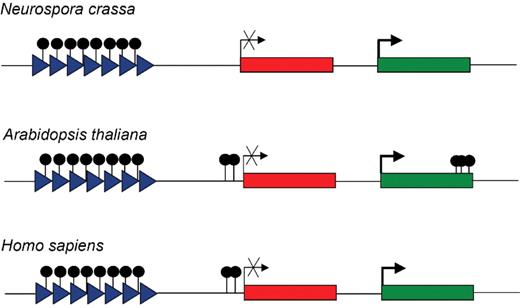
Distribution of DNA methylation in fungi, plants and humans. In N. crassa , DNA methylation is mainly found in transposon sequences and repetitive elements (triangles). These elements are also densely methylated in A. thaliana . However, additional DNA methylation is observed in genes. Promoter methylation is found in tissue-specific genes and correlates with lower expression levels. In contrast, methylation in the gene body and in the 3'-end is associated with constitutive gene expression. The human genome displays dense methylation in repeats and transposon sequences, but also in genes that are subject to epigenetic silencing. The latter effect becomes more pronounced in cancer cells.
An important role for epigenetic mechanisms in the silencing of transposons has also been described in Arabidopsis . This conclusion was based on an analysis of histone methylation and DNA methylation patterns of the interstitial heterochromatic knob on chromosome 4 and a comparison to mRNA expression profiles ( 36 ). The DNA methylation profile was obtained by comparison of total genomic DNA with the ‘methylation-depleted’ fraction that was generated by methylation-dependent restriction digest with the enzyme McrBC and subsequent size fractionation. As expected, high levels of histone H3 lysine 9 trimethylation and DNA methylation were found in the heterochromatic knob, whereas histone H3 lysine 4 methylation was low. The repressive H3K9 and DNA methylation marks were not distributed evenly, but enriched in transposable elements (Fig. 2 ). In contrast, most of the 106 known genes within the knob were found to be unmethylated. The few heavily methylated genes were usually found in close proximity to a transposon insertion. Mutation of DDM1 , a SWI2/SNF2 protein involved in chromatin remodeling and the regulation of DNA methylation ( 28 ) led to a reduction of DNA methylation, a complete loss of H3K9 methylation and an increased transposon expression. Together, these results provide convincing evidence that heterochromatin formation in Arabidopsis is strongly influenced by transposons and repetitive elements that trigger repressive epigenetic modifications, such as H3K9 trimethylation and DNA methylation.
The most recent milestone in the field of epigenome mapping also used Arabidopsis as a model system ( 37 ). After selection by a methylcytosine-specific antibody or a methyl-DNA binding protein, methylated DNA sequences were hybridized to a whole genome-tiling array at 35 bp resolution. The data represent the first comprehensive map of DNA methylation of an entire genome. In agreement with a previous report ( 36 ), dense methylation was found in transposons and other repetitive heterochromatic sequences. The same tiling arrays were used for expression profiling, thereby allowing a coupled analysis of DNA methylation and its impact on the regulation of gene expression. Surprisingly, DNA methylation was also detected in expressed genes: about 5% of all expressed genes were methylated within their promoter regions and one-third of all expressed genes showed a high level of DNA methylation in their transcribed region (Fig. 2 ). While promoter-methylated genes tended to be regulated in a tissue-specific manner and appeared to have a low expression level, body-methylated genes tended to be constitutively expressed at a comparatively higher level (Fig. 2 ). To differentiate between the role of CG methylation (catalyzed by MET1) and non CG-methylation (catalyzed by DRM1, DRM2, CMT3), a MET1 null mutant and a DRM1 DRM2 CMT3 triple mutant were also analyzed. In the MET1 mutant, increased reactivation of pseudogene and transposon expression and overexpression of promoter-methylated genes were observed. In the DRM1 DRM2 CMT3 triple mutant, the most significant increase in expression was reported in genes within a euchromatic environment, and a correlation between the loss of DNA methylation and overexpression was observed. This underlines an important role for non-CG methylation in the regulation of gene expression. It is possible that the maintenance-methylation activity of MET1 is particularly suited for the stable silencing of transposons, while the more dynamic nature of DRM and CMT3 activities might be advantageous for the expression control of endogenous genes ( 37 ).
IDENTIFICATION OF NEW TARGETS FOR ONCOLOGY DRUG DEVELOPMENT
Non-mammalian model organisms have also played an important role in the discovery of novel epigenetic modifier proteins. This is mostly due to the fact that these organisms allow for systematic and genome-wide mutagenesis screens that have been instrumental for the identification of novel genes involved in epigenetic regulation. In the fruit fly D. melanogaster , two different screening approaches have established the groundwork for a systematic characterization of two fundamental epigenetic networks. The analysis of an eye phenotype that is caused by a pigmentation gene being translocated to the proximity of pericentromeric chromatin uncovered a class of genes termed modifiers of position-effect variegation ( 38 ). Similarly, the analysis of mutations inducing homeotic transformations, i.e. changes in body segment identity, has defined the Polycomb and trithorax group of genes ( 39 ). The molecular analysis of these genes has shown that they encode key regulators of chromatin ( 40 ). Many genes identified in Drosophila screens have been highly conserved in evolution and their human homologues have been associated with various aspects of cancer. In addition, several of these genes encode proteins with enzymatic activities, some of which represent attractive targets for pharmaceutical intervention.
There are several modifiers of position effect variegation that have been shown to function as chromatin modifying enzymes. The most widely known of these is the Su(var) 3–9 gene, which encodes a histone H3 lysine 9 (H3K9) methyltransferase ( 41 ). H3K9 can be mono-, di- and trimethylated and the latter two modifications are catalyzed by the SET domain of Su(var) 3–9 ( 42 , 43 ). H3K9 di- and trimethylation are prominent marks of heterochromatin and have been widely conserved during evolution ( 44 ). Similarly, Su(var) 3–9 genes also show a strong conservation in many species, including mammals. A double knockout of the two mouse Su(var) 3–9 homologues, Suv39h1 and Suv39h2 caused chromosomal abnormalities and an increased tumor incidence, which suggested an important function of these enzymes in cancer biology through the maintenance of genome stability ( 45 ).
A detailed analysis of Su(var) 3–9 dependent gene silencing in Drosophila also identified the JIL-1 kinase as an important regulator of chromatin states ( 43 ). JIL-1 kinase catalyzes histone H3 serine 10 (H3S10) phosphorylation ( 46 ) and antagonizes the effects of Su(var) 3–9 mediated H3K9 methylation ( 43 , 47 ). Another H3S10 kinase, the human Aurora B kinase, has recently been shown to antagonize the effects of H3K9 methylation by causing displacement of the trimethyl-H3K9-binding heterochromatin protein HP1 ( 48 , 49 ). Aurora B kinase represents a key regulator of mitosis that has been shown to be overexpressed in various types of human cancers ( 50 ). Thus, H3S10 kinases like JIL-1 and Aurora B represent interesting candidate targets for oncology drug development. In a similar context, histone demethylases ( 9 ) should also warrant further validation as candidate drug targets. Ectopic expression of the H3K9 demethylase GASC1, for example, causes loss of H3K9 di- and trimethylation ( 51 ), which has been associated with genome instability ( 45 ). GASC1 is significantly overexpressed in prostate tumors and lymph node metastases and experimental reduction of GASC1 levels in human cancer cell lines causes a strong reduction in cell proliferation ( 51 ). Thus, the enzyme has several characteristics that establish it as a candidate target for drug development.
There is also increasing evidence that altered Polycomb regulation plays an important role in human tumorigenesis. The first known mammalian homologue of Drosophila Polycomb genes, Bmi-1 , was identified as a MYC-interacting proto-oncogene that promoted lymphomagenesis in mice ( 52 ). While Bmi-1 does not have an enzymatic activity of its own, it stimulates the histone H2A lysine 119 ubiquitinylation activity of the E3 ligases Ring1a and Ring1b ( 53 ). The H2A lysine 119 ubiquitinylation activity of Ring proteins and its connection with epigenetic gene silencing had initially been discovered in Drosophila ( 54 ). Targeting this activity by specific small-molecule inhibitors might be a promising approach to inhibit the oncogenic potential of Polycomb proteins in human cancers.
Another key enzyme in Polycomb silencing is the Enhancer of zeste gene that was also initially discovered in Drosophila . A human homologue, termed EZH2 , was found to be strongly overexpressed in metastatic prostate cancer ( 55 ). siRNA knockdown of EZH2 in human prostate cancer cell lines induced cell cycle arrest and inhibited cellular proliferation, which functionally implicated the gene in the progression of prostate cancer ( 55 ). Later work in Drosophila showed that E(z) proteins catalyze methylation of histone H3 lysine 27, an epigenetic mark associated with gene silencing ( 56–58 ). This finding also explained the observation that EZH2 overexpression was associated with large-scale gene silencing in prostate cancer ( 55 ). The global gene-silencing defect of prostate cancer cells may be reversible upon administration of EZH2 inhibitors that have yet to be developed.
CONCLUDING REMARKS AND OUTLOOK
Non-mammalian models have played an essential role in developing concepts and technologies for cancer epigenetics and will continue to shape this field in several areas. For example, the highly complex epigenetic systems in Arabidopsis , combined with their exceptional experimental accessibility, will be crucial for further refining our mechanistic understanding of epigenetic regulation and, consequentially, epigenetic deregulation in cancer. Also, the high-resolution mapping of methylated DNA fragments by immunoprecipitation, followed by hybridization to tiling arrays should now be applied to human cancer samples and to corresponding control tissues. This will allow detailed insights into the genes and pathways affected by epigenetic mutations in cancer patients. Lastly, novel epigenetic modifier proteins that have been identified through systematic genome wide mutagenesis screens in model systems should be evaluated as potential targets for anticancer drug development.
ACKNOWLEDGEMENTS
The authors acknowledge support from Deutsche Forschungsgemeinschaft, Priority Programme Epigenetics.
Conflict of Interest statement . None declared.

{kind=link}
{kind=link}