




Abstract
SOX9 is a temporal and tissue-specific transcription factor involved in male sexual development and bone formation. Haploinsufficiency of SOX9 is known to cause campomelic dysplasia (CD). CD cases without SOX9 coding region mutations have been described in association with translocations that have breakpoints mapping as far as 932 kb upstream from the gene. These rearrangements suggest that position effects acting from a great distance regulate SOX9 gene expression. Studies of one such case (900 kb upstream to SOX9) have led to the delineation of a potential 2.1 kb cis-acting regulatory element 1.1 Mb upstream of SOX9, termed SOX9cre1. We investigated the role of this putative regulator in SOX9 expression. SOX9cre1 increases the activity of a minimal SOX9 promoter in reporter constructs in a dose-dependent and tissue-specific manner, consistent with an enhancer role. In silico studies identify a putative binding site within SOX9cre1 for GLI1, a downstream mediator of sonic hedgehog (SHH). Furthermore, the stimulation of primary human chondrocyte cells in culture with SHH increases endogenous SOX9 expression 3-fold. Electrophoresis mobility shift assay (EMSA) studies that demonstrate physical interactions between the GLI1 transcription factor and a putative binding site within SOX9cre1, as well as experiments in which reporter constructs are co-transfected with GLI1, suggest a direct interaction between GLI1 and SOX9cre1. GLI1-SOX9cre1 interactions are verified in chromatin immunoprecipitation experiments. These data support a direct molecular link between the Hh signaling pathway and SOX9 regulation, wherein SHH stimulates SOX9 through its mediator GLI1, and are consistent with a mechanism of SOX9 regulation through distal chromatin interactions.
INTRODUCTION
SOX9 is a tissue-specific and temporally regulated transcription factor required for different developmental pathways. This 56 kDa protein is involved in both male sexual determination downstream of SRY as well as chondrocyte differentiation and maturation. The involvement of SOX9 in these distinct pathways is further revealed by SOX9 mutations or haploinsufficiency, each causing a unique dominant condition termed campomelic dysplasia (CD), that includes both skeletal abnormalities and sex reversal in two-thirds of XY individuals (1–4). CD is a rare and usually severe condition when resulting from a mutation in SOX9, often causing death during infancy. Common features of CD include bowing of the long bones in the legs (campomelia), hypoplastic scapulae, a bell-shaped and under-developed thorax, Robin sequence (micrognathia, cleft lip/palate and retroglossia), 11 pairs of ribs and other bony abnormalities (5,6). SOX9 is apparently responsible for activating critical collagen genes necessary for chondrogenesis and bone formation (7–10). SOX9 loss of function mutations also lead to sex reversal in two-thirds of male patients, whereas overexpression of SOX9 enables Sertoli and Leydig cell differentiation and potentially causes female-to-male sex reversal (4,11,12). SOX9 is expressed in select tissues, including the male testes (Sertoli cells), chondrocytes, pancreas, fetal brain and relatively few others; it appears in low abundance and is active only at specific times (1). The exact function of SOX9 in tissues other than testes and bone remains to be elucidated.
There have been reports of CD cases without mutations in coding sequences but instead associated with translocation or genomic deletions with rearrangement breakpoints occurring up to 1 Mb upstream of SOX9 in 17q24.3 (1,3,4,13–18). SOX9 is located within a vast ‘gene desert’, thus translocation breakpoints are not likely disrupting other nearby and potentially related genes, although there have been reports of non-coding RNAs in the region (13). The CD-associated translocation cases, along with in silico studies of both mice and pufferfish syntenic genomic regions, suggest that position effects may be altering SOX9 expression potentially by isolating or removing distal cis-acting regulatory elements (18–21). Reduced gene expression may result in either hypomorphic alleles or complete haploinsufficiency. Such putative regulatory elements may be located further than 400 kb upstream to SOX9, as YAC constructs containing this large region failed to completely recapitulate proper SOX9 expression in transgenic mice (18). Although patients with mutations in, and translocations close to, SOX9 have severe phenotypes, most CD translocation cases with breakpoints mapping distantly upstream have a milder phenotype (13,15,16). It has been postulated that the rearrangement breakpoints in these cases fall into distal (∼800–1000 kb) and proximal (∼1–400 kb) ‘clusters’ that may correlate with mild and severe phenotypes, respectively (22). We reported a CD case with a breakpoint mapping to the distal cluster and provided evidence that genomic regions as far as 1 Mb away from the SOX9 coding sequence may be in physical proximity to SOX9 in interphase cells (16). Using in silico methods based on evolutionary conservation, we postulated cis-acting regulators of SOX9 and designated them as SOX9 cis-regulatory elements 1 and 2 (SOX9cre1 and SOX9cre2), located at 66,519,309 and 66,531,252 respectively, on the March 2006 genome assembly.
The regulation of SOX9 is incompletely understood; nevertheless, it has been proposed that SOX9 is regulated at least partially by the hedgehog (Hh) family of intercellular ligands. Sonic hedgehog (SHH) has been shown to transiently activate SOX9 expression in non-SOX9-expressing chick pre-somitic tissue and can activate its expression more completely in the presence of bone morphogenic protein (BMP) and the transcription factor Nkx3.2 (BAPX1 in humans) (23–26). Shh overexpression upregulates Sox9 in mice as evidenced by reporter assays incorporating a 6.8-kb Sox9 promoter region both in vivo and in vitro (24). Shh has been proposed to act as an activator of Sox9 in mesenchymal condensations early in limb development, whereas later during differentiation, in chondrocytes and Sertoli cells, respectively, its homologues Indian hedgehog and Desert hedgehog regulate cell maturation (27–29). These intercellular ligands exert their downstream effects on gene expression through the Gli family of transcription factors (30,31). Recently, SHH has been implicated in promoting chondrogenic differentiation of bone-marrow-derived human mesenchymal stem cells in vitro (32). However, a role for regulation of SHH in SOX9 expression remains tentative as there is currently a paucity of direct molecular evidence linking this signaling pathway to SOX9 expression.
In this study, we provide evidence that SOX9cre1 acts as a tissue-specific enhancer for SOX9. Furthermore, we show that SHH stimulation itself can activate SOX9 expression in primary human chondrocyte (PHC) cultures, suggesting that SHH is indeed involved in human SOX9 expression. We also document that SOX9cre1 enhancement is subject to modification by signals from the Hh signaling pathway and that it maintains a physical interaction with the GLI1 transcription factor, a major mediator of the Hh signaling pathway. These data provide direct molecular evidence for interactions between the SHH pathway and distal SOX9 elements in gene activation. Our experiments further elucidate the complex regulatory mechanisms required to maintain a proper ‘window of gene expression’ for dosage-sensitive transcription factors such as SOX9.
RESULTS
SOX9cre1 enhances SOX9 reporter activity in vitro in a tissue-specific manner
We identified potential SOX9 distal regulatory elements using three convergent approaches: (i) in silico predictions, including 3-way RP scores (33), (ii) the proximity to regions flanking the syntenic Ods mouse deletion (21) and (iii) localization further upstream than reported translocation breakpoints that conveyed a CD phenotype through position effects (16). These elements, SOX9cre1 and SOX9cre2, appeared to be in close physical proximity to SOX9 in interphase nuclei, despite a linear genomic location of 1.1 Mb centromeric to the gene, suggesting that these elements may potentially act in a contact model of regulation (16,34). To examine empirically if these elements had the ability to regulate SOX9 expression in vitro, we constructed a series of reporter constructs using an SEAP reporter plasmid containing a minimal SOX9 promoter (Table 1). This 298-bp promoter was chosen based on homology of the human genomic sequence to the known functional elements in the mouse Sox9 promoter (35). The purpose of the small promoter was to minimize SOX9 expression due to the promoter alone so as to enable better quantification of the potential-regulatory ability of SOX9cre1 and SOX9cre2.
A list of reporter constructs used in reporter and co-transfection assays
| Plasmid | SOX9 promoter | Putative regulatory sequence | |
|---|---|---|---|
| SOX9cre1 | SOX9cre2 | ||
| pGBW101 | + | ||
| pGBW101-1 | + | + | |
| pGBW101-2 | + | + | |
| pGBW101-1,2 | + | + | + |
| pSEAP2-BASIC | |||
| Plasmid | SOX9 promoter | Putative regulatory sequence | |
|---|---|---|---|
| SOX9cre1 | SOX9cre2 | ||
| pGBW101 | + | ||
| pGBW101-1 | + | + | |
| pGBW101-2 | + | + | |
| pGBW101-1,2 | + | + | + |
| pSEAP2-BASIC | |||
A list of reporter constructs used in reporter and co-transfection assays
| Plasmid | SOX9 promoter | Putative regulatory sequence | |
|---|---|---|---|
| SOX9cre1 | SOX9cre2 | ||
| pGBW101 | + | ||
| pGBW101-1 | + | + | |
| pGBW101-2 | + | + | |
| pGBW101-1,2 | + | + | + |
| pSEAP2-BASIC | |||
| Plasmid | SOX9 promoter | Putative regulatory sequence | |
|---|---|---|---|
| SOX9cre1 | SOX9cre2 | ||
| pGBW101 | + | ||
| pGBW101-1 | + | + | |
| pGBW101-2 | + | + | |
| pGBW101-1,2 | + | + | + |
| pSEAP2-BASIC | |||
We observed that SOX9cre1, but not SOX9cre2, enhances SOX9 expression in vitro approximately 2-fold (Fig. 1A). The presence of the two putative regulatory elements together in pGBW101-1,2 may provide a synergistic effect to SOX9cre1 alone (P = 0.05; t-test).
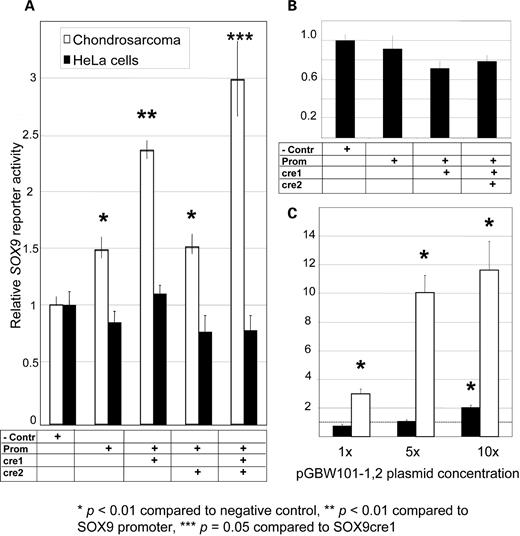
Reporter assays supporting SOX9cre1 as a tissue-specific enhancer of SOX9 in Chondrosarcoma (SW1353) cells. Y-axes represent relative SOX9 reporter activity. (A) Results of 65-h transfection of reporters in SW1353 cells (white) and HeLa cells (black) with 10.7 billion constructs listed in Table 1(∼1.78 × 10−14 mol or 80 ng of the largest construct) as well as a β–galactosidase transfection control. Results were normalized by this internal control and then normalized again by the negative control (pSEAP2-BASIC) setting a baseline expression level at 1. These data show that SOX9cre1, but not SOX9cre2, acts to enhance the minimal SOX9 promoter’s activity (P = 1.9 × 10−5). This enhancement was not observed in HeLa cell experiments, suggesting that the SOX9cre1 element and the SOX9 promoter act in a tissue-specific manner. The combination of both SOX9cre1 and SOX9cre2 yielded higher reporter activity than SOX9cre1 alone (P = 0.05) (n = 6 for all samples and n = 9 for controls). (B) HeLa cell experiments were reproduced in a shorter experiment (20 h) because of the decreased cell-cycle time in this cell line. Again, there was no observable increase in reporter activity in the presence of the promoter or SOX9cre1. (C) Both cell lines were sensitive to the amount of reporter transfected. Adding 5- and 10-fold amounts of reporter constructs dramatically increased reporter activity in SW1353 cells. HeLa cells also saw a statistically significant increase in reporter activity with a 10-fold increase in plasmid concentration, in effect losing the tissue-specific enhancement. The dashed line represents the baseline expression of the negative control (n = 3 for 5 × and 10 × samples).
Importantly, SOX9cre1 does not enhance the activity of the SOX9 minimal promoter in HeLa cells, regardless of the assay times post transfection (Fig. 1A and B). Transfections in chondrosarcoma cells (SW1353 cells) that are derived from SOX9-expressing tissues and have baseline SOX9 expression (36) resulted in a statistically significant increase of reporter activity from the SOX9 minimal promoter in the presence of SOX9cre1, whereas non-SOX9-expressing HeLa cells did not show any significant reporter activity above that of the negative controls (Fig. 1A). Although HeLa cells have a shorter doubling time, the results were consistent at 20 or 65 h of assay time post-transfection (Fig. 1A and B). As anticipated, the assays were sensitive to the amount of constructs transfected; assays performed in SW1353 cells revealed a change in reporter activity from 3-fold (of control levels) to 12-fold when increasing the amount of input vector 10-fold (100 × 109 plasmids) (Fig. 1C). Although not as dramatic, HeLa cells also expressed a statistically significant increase (2-fold) in reporter activity with the pGBW101-1,2 construct in the presence of a 10-fold increase of reporter vectors (P = 4.8 × 10−4 with t-test, Fig. 1C). Although the apparent tissue-specific enhancement of SOX9 was consistent with low dosages of transfected constructs (as no significant increase of reporter activity could be demonstrated in HeLa cells), high levels of transfected constructs result in increased reporter activity in either cell type. The observation of dosage sensitivity to reporter concentrations in the HeLa background is consistent with a potential repressive element in SOX9 expression.
SHH stimulates SOX9 expression in PHC cultures and pre-chondrocyte human ATDC5 cell lines
The Shh intercellular ligand has been implicated in Sox9 expression in chick pre-somitic and somatic mesoderm explants as well as in transgenic mouse embryos and cultured CH310T1/2 cells via Shh overexpression (24,25). Shh is thought to affect Sox9 expression in early stages of limb bud development, when mesenchymal tissues are beginning to differentiate into pre-chondrocytic cells. To examine if human tissues are responsive to SHH stimulation, we obtained PHC cells and cultured them in the presence or absence of SHH. Although primary chondrocyte cultures quickly de-differentiate into fibroblast-like cells, we postulated that they may have the potential to remain responsive to SHH stimulation.
Stimulating PHC cells with 500 ng/ml SHH in the presence of BMP-2 led to a 3-fold increase in SOX9 expression (Fig. 2A), levels similar to those observed in mouse models in Shh overexpression studies (24). Stimulation with SHH also induced a 3-fold increase in GLI1 expression, a known target of SHH signaling and thus acting as a positive control. BMP-2 alone had no effect on SOX9 or GLI1 levels (Fig. 2B). Studies of human ATDC5 cells, which can be stimulated to express chondrocyte markers (37), also revealed a statistically significant increase in SOX9 expression (nearly 2-fold), and a very pronounced GLI1 response to SHH (Fig. 2C), even though this cell line already expresses SOX9 in abundance. The human chondrosarcoma cell line SW1353 did not demonstrate any measurable response to SHH stimulation, perhaps because this is a tumor cell line no longer responsive to external cell-signaling stimuli (Fig. 2D). HeLa cells do not express SOX9 in the presence or absence of SHH, although there is a 1.5-fold increase of GLI1 expression (P = 0.05, t-test) with the addition of 500 ng/ml of SHH (data not shown). The lack of SOX9 expression in the presence of SHH stimulation despite evidence that HeLa cells are SHH-responsive suggests that other factors are necessary for SOX9 regulation but absent in this cell type. These may be positive or negative factors, and likely alter cellular responses to SHH in a tissue-specific manner, as observed by the different responses to stimulations among different cell types.
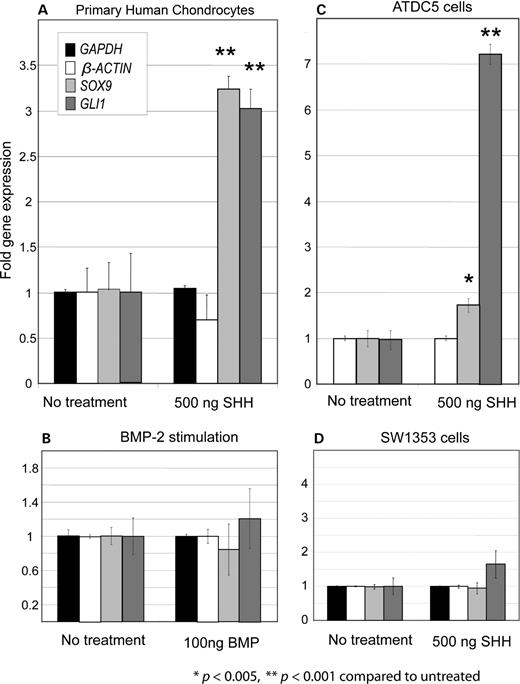
Twenty-four-hour SHH stimulations in human cells revealed its involvement in SOX9 expression. Fold gene expression is measured in the Y-axis. (A) Stimulating PHC cells with 500 ng/ml of SHH and 100 ng/ml of BMP-2 led to a 3-fold increase in endogenous SOX9 expression in RT–PCR assays. The same increase was seen in GLI1 levels, a known SHH target. All gene expression profiles were normalized by GAPDH levels. β–ACTIN expression was not significantly affected by SHH stimulation (n = 9 for GAPDH and GLI1, n = 18 for β–ACTIN and SOX9). (B) BMP-2 stimulation alone (100 ng/ml) had no effect on SOX9 expression (n = 9). (C) ATDC5 cells, which can be stimulated to produce chondrocyte differentiation markers, display a statistically significant response to SHH stimulation. SOX9 levels increase nearly 2-fold, and GLI1 levels increase dramatically, over 7-fold (n = 18 for GLI1 and SOX9, n = 27 for control). This cell line expresses a high level of SOX9 endogenously when confluent (data not shown). (D) Chondrosarcoma cells (SW1353) did not respond to SHH stimulation, possibly because it is a tumor line and not responsive to external stimuli (n = 15 for SOX9 and n = 9 for all other samples).
SOX9cre1 contains a potential GLI1 binding site
The Gli family of transcription factors is well characterized as a mediator of the Hh signaling pathway. Hh ligands interact with the integral membrane receptor Patched-1, resulting in the liberation of the membrane-bound smoothened, whose function allows the activation of Gli transcription factors and their entry into the nucleus (reviewed in 31). The most potent intercellular ligand of this pathway, Shh (27), has been implicated previously in Sox9 expression by cell stimulation and gene overexpression, but there has been no direct link established through its downstream mediators.
We sought a direct relationship between the Shh pathway, SOX9 expression and the putative cis-regulatory sequence in humans by screening for the known 9 bp GLI1 binding sites (38–40) in the 2.1 kb SOX9cre1 and 388 bp SOX9cre2 sequences. Although we did not find an exact match to the originally described GACCACCCA putative site (38), we identified a sequence, GGCCACCCA, in SOX9cre1 that matches 8/9 bp (Fig. 3A andB). This sequence has previously been predicted to act as a strong substrate for GLI1 binding (41). No other putative GLI1 binding sites were identified in SOX9cre1, SOX9cre2 or the minimal SOX9 promoter. This putative regulatory element is located at chr17:66,521,188. It is located within an ∼130 bp of genomic sequence that is conserved in mouse (81%), rat (80%) and chicken (78%). This segment contains sequence similarities to chromosomes 1, 4 and X, as well as a transcript that codes for an mRNA binding protein, SERPINE-1, from chromosome 7. Both mouse and rat sequences have a one-nucleotide substitution (A to G) in the putative binding site. Although SOX9cre1 has a high degree of homology with other species, the region surrounding the putative site displays an interesting pattern of conservation. The syntenic region of a human sequence including 590 bp surrounding the putative site (66,520,972–66,521,561) is analogous to 6.4 kb in the mouse, 6.7 kb in the rat and 8.2 kb in the chicken genomes. Most of the intervening, non-homologous sequence is repetitive. These intervening sequences are also absent in other primate species, including the chimp and rhesus monkey.
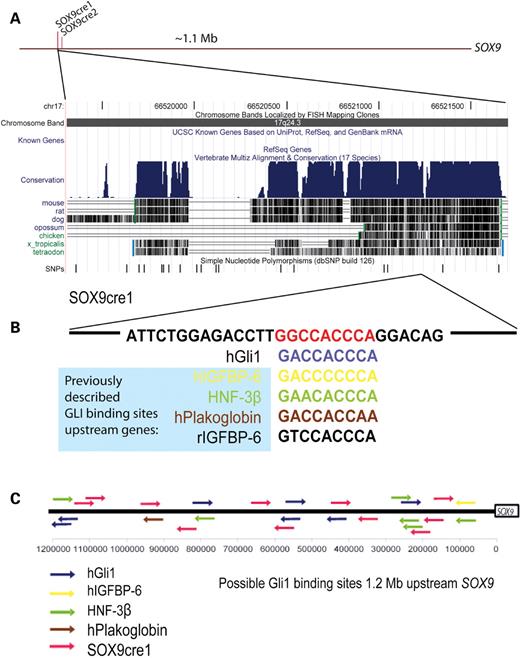
In silico search for Shh signal mediator binding sites. (A) Relative location of SOX9cre1 and SOX9cre2 upstream of SOX9, and a view of the multi-species conservation of SOX9cre1 from the UCSC genome browser, March 2006 assembly. (B) SOX9cre1 contains a putative GLI1 binding site, GGCCACCCA, which matches eight of 9 bp with the hGLI1 sequence, and is located in the distal portion of SOX9cre1. This is compared to other putative sites found in other GLI1 targets (38–40). (C) A search of the 1.2-Mb genomic region upstream of SOX9 yielded 26 possible GLI1 binding sites. Eleven of these contain the SOX9cre1 putative site.
Other transcription factors thought to be involved in SOX9 expression include Smads 1, 5 and 8 (of the BMP pathway) and Nkx3.2/BAPX1 (25,42); however, these proteins have either ambiguous or common DNA binding sites, making them less suitable for a computational-based search in a large genomic region, or they are thought to regulate transcription in a means other than by direct DNA binding (43,44).
We screened a region which spans 1.2 Mb upstream of SOX9 for potential GLI1 binding sites, using as input sequences those shown previously to bind GLI1 in electrophoresis mobility shift assay (EMSA) experiments (Fig. 3C) (38–40). These included hGli1(A1) (GACCACCCA), hIGFBP-6 (GACCCCCCA), HNF-3β (GAACACCCA), hPlakoglobin (GACCACCAA) and the SOX9cre1 sequence (GGCCACCCA). There were 26 possible binding sites in this 1.2 Mb region. The SOX9cre1 putative site occurs 11 times in this 1.2 Mb genomic interval, whereas 4.5 such sites would be predicted by chance alone. Although it is unlikely that GLI1 binds all of these sites in vivo, the presence of known binding motifs and clustering suggest that there may be multiple GLI1 binding sites upstream of SOX9.
SHH regulates SOX9 expression through GLI1 in SW1353 cells
To determine if the increase in endogenous SOX9 expression levels in the presence of SHH in human cells is because of its mediation through the GLI1 transcription factor, we transfected SW1353 cells with small amounts of a GLI1 expression cassette using a T1 promoter. As reported earlier, this cartilage tumor cell line is not responsive to intercellular signals. However, transfecting these cells with 100 ng/ml of the GLI1 construct increased endogenous SOX9 expression levels 2.6-fold as measured by quantitative reverse transcription–polymerase chain reaction (RT–PCR) experiments (Fig. 4A). The observation that GLI1 can partially recapitulate SHH cell-signaling effects, even in cells that have lost the ability to respond to exogenous intercellular signals, is consistent with the hypothesis that SHH mediates its SOX9 inducing effects through GLI1. We also attempted to force GLI1 expression in HeLa cells using up to 10 times the amount of expression cassette, but were unable to identify any SOX9 expression. These data suggest that the other elements required for SOX9 regulation that are not present in HeLa cells may be acting downstream of GLI1.
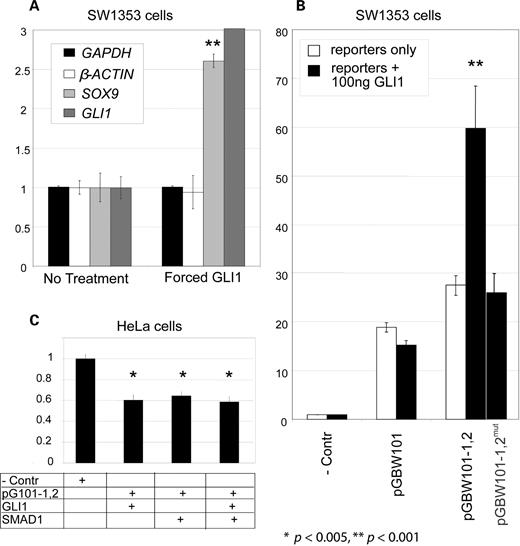
The SHH signal mediator, GLI1, likely effects SOX9 expression through SOX9cre1. (A) Transfection of 100 ng of a GLI1 expression cassette in SW1353 cells shows a statistically significant increase in endogenous SOX9 levels in RT–PCR experiments, suggesting that SHH affects SOX9 expression through this GLI1. Fold gene expression is measured in the Y-axis. All gene expression profiles are normalized to GAPDH levels. GLI levels increase 28-fold in the presence of the GLI1 overexpression cassette (n = 9). (B) GLI1 increases SOX9cre1 enhancement of the SOX9 minimal promoter (pGBW101) when reporter constructs are co-transfected with the GLI1 transcription factor expression cassette. Relative reporter activity to the negative control is displayed in the Y-axis. Results were normalized to β-gal and appropriate negative controls. The addition of 100 ng GLI1 construct to 13 × 109 plasmids (2.16 × 10−14 mol) of the pGBW101-1,2 reporter increases reporter activity more than 2-fold. GLI1 co-expression had no significant effect on 13 × 109 plasmids of the minimal SOX9 promoter reporter, suggesting that the increase in enhancement acts through SOX9cre1. Furthermore, removal of the putative GLI1 site from pGBW101-1,2 abolishes its ability to respond to GLI co-transfection. (C) The presence of GLI1 and SMAD1 did not increase reporter activity in HeLa cell cultures. Reporters containing the SOX9 promoter and SOX9cre1 and SOX9cre2 even showed a significant decrease in reporter activity when co-transfected with GLI1 and/or SMAD1 of the BMP pathway in expression cassettes.
GLI1 modifies SOX9cre1 enhancement of SOX9
SOX9cre1 contains a possible GLI1 binding site, and Hh signaling is implicated in SOX9 expression. Therefore, we examined whether these downstream signals from the Hh pathway can mediate their cell-signaling effects on SOX9 at least in part through SOX9cre1. Introduction of an expression cassette containing GLI1 along with SOX9cre1-containing reporter constructs (listed in Table 1) enabled the measurement of the effect of GLI1 on SOX9cre1. Co-transfection of a GLI1-expressing construct in SW1353 cells with the reporter vectors containing SOX9cre1 led to a doubling of reporter activity compared with the reporter alone (Fig. 4B). The same GLI1 expression cassette, co-transfected into cells with the vector pGBW101 containing the SOX9 minimal promoter, had no significant positive effect on reporter activity (Fig. 4B). This indicates that the reporter response to GLI1 is SOX9cre1-dependent, suggesting that the GLI1 acts to regulate SOX9 expression through the SOX9cre1-regulatory element. To provide evidence that the putative GLI1 binding site in SOX9cre1 mediates SHH/GLI1 signaling, we constructed a mutant reporter vector (pGBW101,1,2mut) using site-directed mutagenesis to eliminate this binding site. The mutant vector had no measurable response to GLI stimulation. Co-expression of low levels of SHH- and BMP-related transcription factors and reporters had no apparent effect in HeLa cells (Fig. 4C).
Direct binding of GLI1 to the putative binding site on SOX9cre1
Tissue culture experiments have demonstrated that GLI1 enhances SOX9 reporter activity in a SOX9cre1-dependent manner, and computational predictions suggest a putative GLI1 binding site within SOX9cre1. EMSAs were used to examine direct interactions between GLI1 and SOX9cre1 (Fig. 5A–C). Interactions between GLI1 and the putative binding site GGCCACCCA within SOX9cre1 were tested along with controls (Fig. 5A). GLI1, and not a control protein (SMAD1), specifically bound a 30-bp double-stranded DNA oligonucleotides from SOX9cre1 that contains the putative binding site. This binding was quenched with 25-fold excess of unlabeled oligonucleotides, but was retained when the excess oligonucleotides contained a mutated binding site. Serial dilutions of cold and mutant cold competitors document that the signal loss is specific to the competitor with the putative binding site (Fig. 5B). These data indicate not only that GLI1 directly interacts with SOX9cre1, but it does so at the novel putative binding site.
![Physical binding of GLI1 to SOX9cre1 in EMSA experiments. (A) An EMSA displays the binding of the GLI1 transcription factor to the putative element in SOX9cre1. Probes with the IGFBP-6 putative site are used as a positive control, and the SMAD1 protein is used as a negative control. Binding is abolished with excess cold competitor, but retained with mutated cold competitor, signifying that the binding occurs at the putative site. (B) Serial dilutions of competitors display a dose-dependant quenching of the signal with cold competitor. (C) The signal band is dose-dependent on the quantity of GLI1 added. Increases in probe concentration only increase background. (D) Construction of the GLI1 transcription factor in vitro is verified by autoradiography with [3S] methionine and by western blot with GLI1 antibodies. GLI1 was created using erythrocyte extracts, and the negative controls consisted of the vector without GLI1.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/16/10/10.1093/hmg/ddm061/2/m_ddm06105.jpeg?Expires=1716599418&Signature=Ougdo5JMD~tFju6jgUflEZkMUkCUznFl~SQIkJEL~-BFM~LrWUA7ThOmOlQKpGms2Qxx~E2EEp8cl~EBYdE3HYmzAaxISlvyKlbxisegzh6hQO34MIIY0IhmNWyp~C6t9u8HNegx9y~7fvEiFs1zO-DjHG6bFEI6mYgQgZ8rsrueA9INxf2qF9oPTYBgHP2PceIKkaBRPxryDYVCC9tgS~5q31WVLRxXqFizQuycFvR-N019ZxicGGdbs2ygqhaE5aEhalCuiUSyUIdtWSW3IphDLZ9PlGmcoJg9fFmT9ry1FMG2SPo8ZaqLGyu85WzcPkRGw4x-F1-SaU8g~J8kEA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Physical binding of GLI1 to SOX9cre1 in EMSA experiments. (A) An EMSA displays the binding of the GLI1 transcription factor to the putative element in SOX9cre1. Probes with the IGFBP-6 putative site are used as a positive control, and the SMAD1 protein is used as a negative control. Binding is abolished with excess cold competitor, but retained with mutated cold competitor, signifying that the binding occurs at the putative site. (B) Serial dilutions of competitors display a dose-dependant quenching of the signal with cold competitor. (C) The signal band is dose-dependent on the quantity of GLI1 added. Increases in probe concentration only increase background. (D) Construction of the GLI1 transcription factor in vitro is verified by autoradiography with [3S] methionine and by western blot with GLI1 antibodies. GLI1 was created using erythrocyte extracts, and the negative controls consisted of the vector without GLI1.
Chromatin immunoprecipitation of SOX9cre1 GLI1 binding site
In vitro experiments such as reporter assays and EMSAs demonstrate SOX9cre1’s ability to enhance SOX9 reporter activity and bind the GLI1 transcription factor. To further document the function of SOX9cre1, we employed chromatin immunoprecipitation (chIP) to evaluate protein–DNA interactions with this putative cis-acting regulatory sequence in PHC cells. Antibodies against human GLI1 and acetylated histone H3 (Acetyl-H3) were used in the immunoprecipitations. Acetyl-H3 is known to be bound to DNA during active chromatin remodeling and its presence is thought to mark active transcription sites (45). chIP quality was measured with GAPDH controls (Fig. 6A). Our data indicate that after 20 min of SHH stimulation, Acetyl-H3 is bound to SOX9cre1 over 5-fold more than untreated controls, suggesting that SOX9cre1 is involved in active transcription in response to SHH stimulation (Fig. 6B). The maximum DNA enrichment occurs 20 min after SHH stimulation, as seen in other chIP stimulation studies (46). GLI1 is bound to SOX9cre1 near the putative binding site nearly 4-fold more than non-treatment controls at 20-min SHH stimulation (Fig. 6C). These data indicate not only that GLI1 and Acetyl-H3 both bind SOX9cre1, but also that this binding is a direct response to SHH stimulation in PHC cells. Similar chIP experiments on SHH-stimulated HeLa cells failed to enrich for SOX9cre1 sequence above untreated controls (data not shown). These data support previous observations of the tissue-specific activity of SOX9cre1.
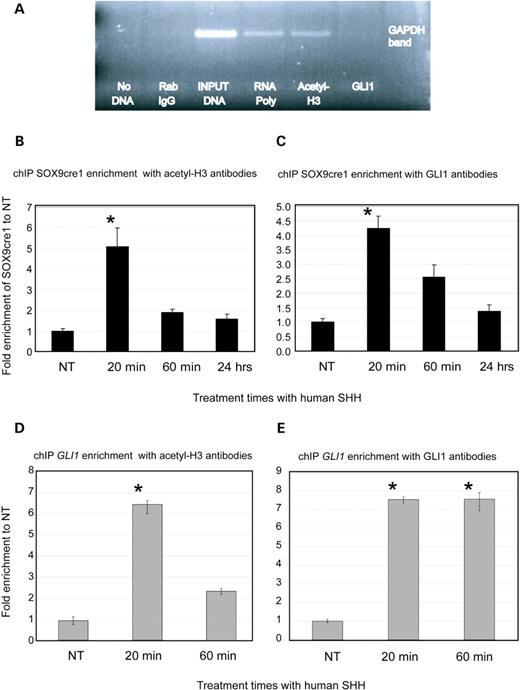
chIP experiments in PHC cells demonstrating GLI1/SOX9cre1 interactions. (A) A positive control experiment of chIP DNA using primers for GAPDH. ChIP enriches DNA sequences which bind to the protein–antibody complexes of interest. PCR is then used to screen this DNA library for the presence of any specific sequence. GAPDH, which is actively expressed in most cells, is enriched in chIP using RNA polymerase and Acetly-H3 antibodies . Normal rabbit IgG serves as a non-specific negative control. Because GLI1 is not involved in GAPDH expression, chIP with GLI1 antibodies does not enrich GAPDH DNA. The input positive control is chromatin DNA that is pre-cleared of repetitive sequences, but is not immunoprecipitated. (B) Results of chIP experiments screening for the enrichment of SOX9cre1 near the GLI1 putative binding site with Acetyl-H3 antibodies. ChIP was conducted after SHH stimulation for different periods of time. qPCR was used to quantify the results. Absolute measurements were obtained with serial dilutions of the input control. For each time point, data were normalized by the non-specific rabbit IgG control at the same time interval to ensure equal loading. All data are reported as fold increases in enrichment compared with the non-SHH-stimulated control. Our data indicate that after 20 min of stimulation with SHH, there is maximum binding of Acetyl-H3 to the SOX9cre1 element, although some binding was still evident at 24 h. This suggests that SOX9cre1 is involved in active transcription. (C) chIP of SOX9cre1 with GLI1 antibodies. These data also indicate maximal protein–DNA interactions at 20 min after SHH stimulation, and confirm that GLI1 binds SOX9cre1 under SHH stimulation in the native cell. (D) chIP of GLI1 with acetyl-H3 antibodies. GLI1 is a direct target of SHH signaling, and acetyl-H3 binding suggest active transcription of GLI1 after SHH stimulation, serving as a positive control for the chIP and the SHH stimulation. (E) chIP of GLI1 with GLI antibody. This suggests that GLI1 may be involved in regulating its own expression.
chIP experiments in PHC cells were also used to assess the GLI and Acetyl-H3-binding capacities of the 10 most proximally described putative GLI sites identified by pattern matching in Fig. 3C in response to SHH stimulation (data not shown). Three of these elements were occupied by Acetyl-H3. These were located at chr17:67,555,504, 67,359, 951 and 67,284,958 (P < 0.01, t-test). Two of the elements were enriched with GLI1 antibodies significantly above controls (approximately 2-fold), and were located at 67,545,317 and 67,359,951 (P < 0.05). The element located at 67,359,951 is bound by both Acetyl-H3 and GLI1, suggesting that it may be involved in gene expression secondary to SHH stimulation. Interestingly, this sequence is not conserved among species. Further experiments, such as reporter assays, are necessary to elucidate any potential role this sequence may have in SOX9 expression in humans.
DISCUSSION
SOX9, like other dosage-sensitive tissue-specific and temporally specific transcription factors, must be strictly regulated to achieve proper expression levels. SOX9 alterations and/or reductions in expression caused by gene mutations usually results in infant lethality, but decreased SOX9 dosage because of extragenic effects can also have profound phenotypic consequences (15). The observation that distal genomic regions as far as 1.1 Mb appear to affect the expression of SOX9 may allude to the extensive regulatory networks in the gene desert of 17q24.3 necessary to achieve proper gene expression levels. An alternate viewpoint might emphasize that this desert is more like a ‘sea’ of cis-acting regulatory elements, and its disruption results in position effects. Although the distal SOX9cre1 regulator alone may have only a weak enhancing effect on SOX9 reporter activity, this could still be significant because very small changes in SOX9 levels may have phenotypic consequences in a majority of cases. Furthermore, the ‘sea’ of regulation upstream (and possibly downstream) of SOX9 is likely comprised of many such regulatory sites that may work in concert to drive gene expression levels that impact cellular differentiation and development. Recently, several suspected regulatory regions located within 290 kb of the mouse Sox9 were shown to control gene expression in different tissues, including the neural crest, inner ear and pancreas (47). Although none of these evolutionary conserved regulatory elements (dubbed E1–E7) drove expression in chondrocytes or Sertoli cells, these findings suggest that the extended regulatory control region upstream of SOX9 is capable of controlling gene expression in a tissue-specific and temporally specific manner. Thus, these elements may have a critical role to play in the development of gene expression throughout evolution.
This type of regulation is not unique to SOX9. SHH regulates other key developmental pathways that may also have similar regulatory schemes, and was itself demonstrated to be regulated in a similar fashion as a 1-Mb upstream enhancer of SHH has been reported (48). Additionally, transcription factors such as Pax6 in the mouse and POU3F4 are apparently subject to analogous distal regulators (205 and 900 kb, respectively) and are also affected by position effects (49–51). Recently, DAX1, another dosage-sensitive gene shown to result in partial sex reversal when its expression is altered, has been shown to be susceptible to similar position effects (Smyk et al., manuscript in preparation). It appears that genes responsible for controlling key developmental pathways may be regulated by extensive networks that ensure proper expression levels.
Our data indicate that SHH may be directly involved in SOX9 regulation in human chondrocytes and chondrosarcomas. Cell culture responses were tissue-specific, even in the presence of mediators of the Hh and BMP pathways. HeLa cells failed to show increased reporter activity in the presence of the minimal SOX9 promoter or SOX9cre1; in fact, there was a significant decrease in reporter activity when compared with the negative control (p-BASIC). It is possible that HeLa cells were unable to recapitulate SOX9 expression in the presence of GLI1 and SMAD1 because other tissue-specific factors are necessary for proper gene expression, or because there are repressive elements that bind to these regions that cannot be removed. Such elements may prevent unwanted gene expression. It has been demonstrated that Nkx3.2/BAPX1 is a Shh- and BMP-activated transcriptional repressor important in chondrocyte differentiation and that it may be required for Sox9 expression, as the two genes form a positive auto-regulatory loop (25). Nkx3.2 has been shown recently to directly bind and repress the activity of Runx2, another transcription factor that is necessary for chondrocyte maturation, which opposes Sox9 function (26). It is possible that non-SOX9 expressing tissues have negative regulators similar to Runx2 that bind to the minimal SOX9 promoter and/or regulatory elements that must be overcome for SOX9 expression, but these tissues lack inhibitors of these negative regulators.
Chromatin structure and organization may also be an important determinant in SOX9 expression. According to contact models of gene regulation, distal cis-acting regulatory regions may be brought in proximity to the gene of interest by transcription factors and activators and by looping out the intermediate DNA, creating an active chromatin hub or similar structure (52,53). Previously, we documented that distal elements appear to be brought close to SOX9 in living cells (16). Our reporter constructs enable us to bring putative regulatory sequences and SOX9 in close proximity as they appear in our co-localization experiments, because these artificial constructs do not contain the 1.1 Mb present in the genomic sequence. Perhaps the prevailing chromatin structure created by these protein–DNA interactions can explain why translocations seem to cluster to two regions, approximately 300 and 900 kb upstream SOX9. Such clustering may reflect bias of ascertainment because of conveyed phenotypes rather than genomic ‘hotspots’ for chromosomal translocations.
The precise regulation of SOX9 within a window of gene expression is critical as can be witnessed by phenotypes with mild perturbations of surrounding regions and is likely similar to the regulation of other developmental regulatory genes under the control of SHH, such as PAX1 or BAPX1 (as well as SHH itself). Comprehending the regulation in the region of 17q24.3 is critical to the understanding of essential developmental control genes and warrants further investigation. In this sense, genomic approaches, such as ChIP-chip, could potentially provide further insight into the complex regulatory mechanisms involved in such a large genomic region.
MATERIALS AND METHODS
Generation of reporter constructs and expression cassettes
A 298-bp SOX9 minimal promoter was designed using BLAST2 to compare the known functional components of the mouse promoter (35) with human sequences. This sequence was PCR-amplified with the primers 5′-GCATTCCGAGAGTACGACAA-3′ and 5′-CCAGCCCAGGGTCTCTTTA-3′ from the human BAC RP11-1116I6, cloned into the TOPO-TA vector (Invitrogen, Carlsbad, CA), sequence verified for orientation and was digested with EcoRI and ligated into the pSEAP2-Basic reporter vector (BD Biosciences, Mountain View, CA) to construct the SOX9 promoter/SEAP reporter pGBW101.
The SOX9cre1 sequence was amplified with the primers 5′-CTCCTTTCTACAAGGAAAGTCAC-3′and 5′-TCTGTGTAGCGAGAGAAGTTCA-3′, whereas SOX9cre2 was amplified with the primers 5′-GTCCTCTGTCCCTCCTTTCC-3′ and 5′-AACTCCAGCAGTTTGCCACT-3′ by PCR using the BAC clone RP11-831L20 as a template. These putative regulatory sequences were then TOPO-TA cloned, sequence verified and subcloned into either the pcDNA3.1(+) or pcDNA3.1(−) (Invitrogen) vectors, respectively. The SOX9cre1/pcDNA3.1(+) construct was digested with XhoI and NheI and subcloned into pGBW101 to construct the Cre1/Prom/SEAP reporter pGBW101-1. The SOX9cre2/pcDNA3.1(−) construct was digested with XhoI and HindIII and subcloned into pGBW101 and pGBW101-1 vectors to construct pGBW101-2 and pGBW101-1,2 containing either SOX9cre2 or both SOX9cre1 and SOX9cre2, respectively (Table 1). Final constructs were sequence verified. Plasmid pGBW101-1,2mut was constructed using two rounds of site-directed mutagenesis, with the following primers along with their complementary reverse sequences: 5′-CTGGAGACCTTGCTAGCCCAGGACAGTGGTGGCAGGG (first round) and 5′- CTGGAGACCTTGCTAGGCTGGGACAGTGGTGGCAGGG (second round). Mutagenesis reactions were conducted with reagents and protocols from the QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). Final constructs were sequence verified.
GLI1 cDNA was amplified with PCR primers 5′-TATATATAGGTCTAGACCCAGACAGAGTGTCCCCAC-3′ and 5′-CCCCCCCCCCGCGGCCGCTTTTTTTTTTTTTTTTTT-3′ using Pfu polymerase and a GLI1-containing clone (Accession No. BC013000) (Open Biosystems, Huntsville, AL) as a template. This product was then digested with XbaI and NotI. The resulting fragment was gel-purified and cloned into the pcDNA3.1(−) expression vector. SMAD1 cDNA was cloned into the same expression vector using analogous methods with the SMAD1 clone (Accession No. BC001878) as a template. All plasmid constructs were DNA sequence verified.
Reporter and co-transfection assays
HeLa and chondrosarcoma cells (SW1353) (ATCC, Manassas, VA) were transfected with the above reporter constructs and a negative control that consisted of the pSEAP-Basic vector alone. Each experiment was conducted in triplicate, and this series was repeated at least twice (N = 6 total or more) for each set of conditions. For reporter assays, cells in each chamber were transfected with 10.7 × 109 plasmid copies regardless of size, with an equal amount of a β-gal expression vector driven by a cytomegalovirus promoter, which served as a transfection control. The plasmid number was kept constant (1.78 × 10−14 mol) to ensure equimolar transfections because of varying plasmid sizes. Transfections were allowed to proceed undisturbed for 18–65 h at 37°C in 15% fetal bovine serum (FBS) media, at which point the cell media containing the SEAP chemiluminescent protein was harvested, and cells were lysed to measure β-gal activity. Co-transfections with GLI1 were done in 1% FBS media to enhance GLI expression and interaction with reporters in SW1353 cells. Reporter and β-gal levels were measured using a tube luminometer. All transfected plasmids were treated with 0.5 µl of a transfection reagent, lipofectamine 2000 (Invitrogen) for 20 min, before being added to cells.
Cell lines
SW1353 cells were cultured in a 37°C incubator without carbon dioxide, in L-15 media with 15% FBS. HeLa cells were cultured under normal conditions [Dulbecco's modified Eagle medium (DMEM) with 10% FBS, 20% CO2]. ATDC5 cells were cultured in a mixed media (50% DMEM, 50% F-12) with 10% FBS in a 5% CO2 chamber (37). PHCs, obtained fresh from a lab volunteer after informed consent via incision of the ear, were cultured in a modified DMEM media (1% of 200 mml-glutamine, 1% of 100 mm pyruvate, 1% of 100 mml-cysteine) with 10% FBS in a 5% CO2 incubator. The cartilage was treated with pronase and collagenase to release the cells into the culture media. Cells were passaged less than three times.
SHH stimulations: quantitative PCR
ATDC5, PHC and SW1353 cells were stimulated with the N-terminus recombinant human SHH protein (R&D systems, Minneapolis, MN). Confluent cells were placed in 1% FBS DMEM media for 24 h prior to treatment. Cells were treated with 500 ng of SHH and 100 ng of BMP-2 (R&D Systems) in 1-ml wells. Experiments were performed in triplicate, and each batch experiment was repeated at least twice. The stimulations were allowed to proceed for 24 h. Cells were then lysed with TRIzol reagent (Gibco BRL, Gaithersburg, MD) and RNA was extracted according to protocols included with the kit. RNA was treated with DNAse1 (Ambion, Austin, TX) and cDNA was created using a Taqman Reverse Transcriptase Kit (Applied Biosystems, Foster City, CA).
Quantitative PCR (qPCR) experiments were conducted using the ABI 7000 sequence detection system. All RT–PCR reactions were performed in triplicate. Gene expression levels were normalized to GAPDH or β-ACTIN controls for treated and untreated samples. Treated samples were then normalized to non-treated controls to obtain the relative fold increase in expression. Primers used include: β-ACTIN forward 5′-GCCAACCGCGAGAAGATGAAC, reverse 5′-CTCCTTAATGTCACGCACGATTTC; GLI1 forward 5′-CGGGGTCTCAAACTGCCCAGCTT, reverse 5′-GGCTGGGTCACTGGCCCTC (54); SOX9 and GAPDH primers were obtained pre-fabricated from ABI and were used with FAM-labeled probes. SYBR Green (ABI) was used for detection of non-FAM products.
Electrophoresis mobility shift assays
GLI1 full-length protein was synthesized in vitro from the GLI1 expression vector and the TNT quick coupled transcription/translation system (Promega, Madison, WI). The expressed protein was analyzed for size and specificity using autoradiography in an sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) with [35S]methionine and western blotting with anti-GLI1 antibodies (Chemicon, Temecula, CA) (Fig. 5A). This protein was hybridized with 32P end-labeled double-stranded oligonucleotides that consisted of equimolar synthetic complementary single-strand oligonucleotides that were heated to 95°C for 5 min and allowed to anneal at room temperature overnight. These included a positive control for a known binding site upstream from the human IGFBP-6 gene (40), sense 5′-GACTCCCTGACCATGACCCCCCACAGGCTGCTG-3′ and antisense 5′-GACTCAGCAGCCTGTGGGGGGTCATGGTCAGGG-3′; and oligonucleotides from SOX9cre1 which included the anticipated computationally predicted GLI1 binding site, sense 5′-GACTATTCTGGAGACCTTGGCCACCCAGGACAG-3′ and antisense 5′-GACTCTGTCCTGGGTGGCCAAGGTCTCCAGAAT-3′; and the SOX9cre1 sequence with a mutated binding site, sense 5′-ATTCTGGAGACCTTGGAAAAAAAGGACAG-3′ and antisense 5′-CTGTCCTTTTTTTCCAAGGTCTCCAGAAT-3′. A total of 40 fmol of oligonucleotide was mixed with protein in 10 µl of hybridization buffer for each sample and placed at 4°C for 30 min. The buffer consisted of 25 mm Tris–HCl, pH 8.0, 50 mm KCl, 5 mm MgCl, 1 mm dithiothreitol, 10% glycerol and 1 µg of poly (dI-dC). The samples were then warmed to room temperature and subjected to electrophoresis on a 4% acrylamide gel for 90 min at 100 V. Phosphoimaging and autoradiography enabled visualization of the experimental results.
Chromatin immunoprecipitation
PHC cells were cultured until confluent with appropriate media and then placed in a 1% FBS media for 24 h. These cells (∼6 × 106) were then stimulated with 1 µg/ml of SHH and 100 ng/ml of BMP-2 (or no treatment) for varying lengths of time. Protein–chromatin interactions were captured with 1% formalin. Cells were lysed in an SDS lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris–HCl, pH 8.1) and sonicated for 60 s with a Bronson Sonifier 450 sonicator at 30% power and 70% duration. Immunoprecipitations were performed using the EZ ChIP kit (Millipore, Billerica, MA) and protocols, as well as the following rabbit polyclonal antibodies: anti-GLI1 (Chemicon), anti-acetylated histone H3 (Millipore), anti-RNA polymerase (Millipore) and normal Rabbit IgG (Millipore). Results were evaluated for specificity by PCR with primers for the GAPDH gene, 5′-TACTAGCGGTTTTACGGGCG and 5′-TCGAACAGGAGGAGCAGAGAGCGA. Quantitative data for SOX9cre1 were obtained using qPCR with primers near the GLI1 binding site, 5′-CCAGCTCTGGCTTACCTGGAT and 5′-TTGCAGACTCTCCTCTTTAAAGCTG. Primers used for GLI1 detection and amplification were as previously mentioned. Absolute quantifications were obtained with input control serial dilutions (pre-cleared chromatin not immunoprecipitated), and data were normalized by the non-specific negative control, normal rabbit IgG.
ELECTRONIC DATABASE INFORMATION
UCSC Genome Browser, http://genome.ucsc.edu/.
ACKNOWLEDGEMENTS
We thank Dr Brendan Lee for providing us with the ATDC5 cell line and β-ACTIN primers, and Drs Jeffery Rosen and Chad Shaw for their helpful discussions. This work was supported in part by the National Institutes of Child Health and Development (NIH) (POI HD38420). G.A.B.-W. was supported by an NIH Medical Scientist Training Grant. This research was supported in part by Grant PBZ/KBN (122/P05/01-7) form the Polish Ministry of Scientific Reserch and Information Technology to PS.
Conflict of Interest statement. The authors have no conflicts of interest to declare.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}