





Abstract
The pre-ovulatory surge of gonadotrophins triggers a marked and obligatory increase in follicular prostaglandin synthesis prior to ovulation, and the cyclooxygenase (COX) enzyme is a key rate-limiting step in the biosynthesis of prostaglandins. In the early 1990s, the pre-ovulatory rise in follicular prostaglandin synthesis was shown to result from the selective induction of a novel COX isoform, now referred to as COX-2. Differences in the time-course of COX-2 induction in species with a short versus a long ovulatory process suggest that the enzyme could be a molecular determinant that sets the alarm of the mammalian ovulatory clock. Some of the fine molecular mechanisms involved in the transcriptional activation of the COX-2 gene in granulosa cells have also been elucidated. The binding of trans-activating upstream stimulatory factors (USF) to a consensus E-box cis-element in the proximal region of the promoter was shown to play a predominant role in COX-2 transcription. Studies showed that COX-2 expression could also serve as a valuable marker for follicular commitment to ovulation during hyperstimulatory cycles. This paper presents a comprehensive review of the events that led to the characterization of COX-2 in pre-ovulatory follicles, updates current concepts on the control of COX-2 expression in pre-ovulatory follicles, and addresses the consequences of COX-2 inhibition to women fertility and potential implications of COX-2 expression in ovarian cancer.
Introduction
The ovulatory process involves a complex series of biochemical and biophysical events that ultimately lead to the rupture of the pre-ovulatory follicle and the release of the maternal germ cell. The process has all the signs of an acute, self-controlled inflammatory reaction, including hyperaemia, edema, leukocyte extravasation, and induction of proteolytic and collagenolytic activities (Espey, 1980; Espey and Lipner, 1994). Prostaglandins, which play a central role in inflammation, have been recognized as key mediators of ovulation for >30 years and, in retrospect, the history of this relationship can arbitrarily be divided into two major periods. During the first period, which spans from the early 1970s to the mid-1980s, radioimmunoassays and inhibitors of prostaglandin synthesis served as important experimental tools to establish the critical role of prostaglandins in follicular rupture, and results from these studies have been the subject of a number of reviews (Armstrong, 1981; Murdoch et al., 1993; Priddy and Killick, 1993; Tsafriri et al., 1993; Espey and Lipner, 1994). The second period, which spans from the late 1980s up to now, has taken advantage of the development of molecular biology techniques to unravel the regulation and role of key enzymes responsible for prostaglandin synthesis during ovulation. Undoubtedly, the discovery that the cyclooxygenase (COX) enzyme induced by the LH surge in granulosa cells prior to ovulation was a novel isoform (now known as COX-2) represented an important landmark in the field. This review presents a historical account of events that led to the characterization of COX-2 in pre-ovulatory follicles, and updates current concepts on the molecular control of follicular COX-2 expression during natural and hyperstimulatory cycles. The consequences of COX-2 inhibition to women's fertility, as well as the potential implication of COX-2 expression in ovarian cancer, are also presented. To begin, a brief overview of prostaglandin biosynthesis and COX enzymes is proposed; readers interested in a more comprehensive characterization are referred to excellent recent reviews (Garavito and Mulichak, 2003; Murakami and Kudo, 2004).
Prostaglandin biosynthesis
Prostaglandins are important mediators of a variety of biological and pathological processes, and have been implicated in a number of female reproductive functions, including ovulation, fertilization, luteolysis, implantation and parturition (Sirois et al., 2000; Matsumoto et al., 2001a; Lim and Dey, 2002; Olson, 2003; Goff, 2004). Prostaglandins, along with prostacyclins (PGI) and thromboxanes (TX), are often referred to as prostanoids, which belong to a large family of biomolecules containing a 20-carbon backbone structure, the eicosanoids (Smith and Marnett, 1991; Funk, 2001). All prostanoids are derived from fatty acids stored in cellular membranes, and their biosynthesis can be divided into three phases: (i) release of the arachidonic acid from membrane phospholipids by the action of phospholipase A2 enzymes; (ii) conversion of arachidonic acid into an unstable prostaglandin intermediate, PGH2, by cyclooxygenase enzymes; and (iii) conversion of PGH2 into various biologically active prostanoids by cell-specific terminal synthases (Figure 1) (Smith and Marnett, 1991; Murakami and Kudo, 2004). Once released, prostanoids act on target cells via specific seven-transmembrane, G protein-coupled receptors or in some cases through interactions with nuclear receptors (Breyer et al., 2001; Wright et al., 2001; Lim and Dey, 2002; Helliwell et al., 2004).
Cyclooxygenases: structure and function
The true rate-limiting step in prostanoid biosynthesis is believed to be under the control of the COX enzyme, also known as prostaglandin synthase, prostaglandin endoperoxide synthase or prostaglandin G/H synthase (abbreviated PGS, PGTS or PGHS). Because of its key role in generating potent inflammatory mediators, the enzyme is the primary target for a large group of non-steroidal anti-inflammatory drugs (NSAID) that includes aspirin, indomethacin, ibuprofen, celecoxib and rofecoxib (Vane, 2000; FitzGerald, 2003). The enzyme exhibits two consecutive catalytic activities, a cyclooxygenase activity responsible for the bisoxygenation of arachidonic acid to PGG2, and a peroxidase activity involved in the reduction of PGG2 to PGH2 (Figure 1) (Garavito and Mulichak, 2003). To date, three COX enzymes with similar activities but derived from two distinct genes have been characterized.
Cyclooxygenase-1
COX-1 is generally recognized as a constitutive enzyme involved in the biosynthesis of prostaglandins necessary for various homeostatic functions (Smith et al., 2000). The primary structure of COX-1 was first characterized in sheep and subsequently in a number of species (Merlie et al., 1988; DeWitt and Smith, 1988; Smith et al., 2000). The COX-1 gene is ∼22 kilobases (kb) in length, contains 11 exons, maps to human chromosome 9q32-q33.3, and is transcribed as a 2.8 kb mRNA (Tanabe and Tohnai, 2002). The COX-1 transcript encodes a 600–602 amino acid protein (depending on species) that exists as a homodimer and as a haem-binding, integral membrane protein preferentially, but not exclusively, associated with the endoplasmic reticulum. The promoter region of the COX-1 gene does not possess a canonical TATA or CATT box but is GC rich, in keeping with features of a housekeeping gene (Tanabe and Tohnai, 2002).
Cyclooxygenase-2
COX-2 is normally undetectable in most tissues but can be induced by a variety of stimuli, and is often referred to as the inducible isoform involved in inflammatory and pathological processes (Smith et al., 2000). COX-2 cDNA's were first isolated as immediate–early genes expressed in virally transformed chicken embryo fibroblasts (Xie et al., 1991) and in mitogen-stimulated Swiss 3T3 cells (Kujubu et al., 1991), but the presence of a COX isoform distinct from COX-1 had been suspected prior to its cloning (Rosen et al., 1989). The human COX-2 gene is more compact than the COX-1 gene, being ∼8.3 kb in length and containing 10 exons, and maps to a distinct chromosome (chromosome 1q25.2-q25.3) (Tanabe and Tohnai, 2002). However, the COX-2 transcript is larger than COX-1, being ∼4.4 kb in length, primarily because of a relatively long 3′-untranslated region that also contains numerous AUUUA motifs involved in mRNA instability (Smith et al., 2000). The COX-2 transcript encodes a 604 amino acid protein that preferentially, but not exclusively, localizes to the perinuclear envelope, and that is highly conserved across species (Figure 2). Likewise, all important structural and catalytic domains involved in COX function are conserved in COX-1 and COX-2 (Figures 1 and 2) (Garavito and Mulichak, 2003). The promoter region of the COX-2 gene is quite distinct from that of COX-1, as it contains a TATA box and several cis-elements (NF-kB, Sp1, AP-2, C/EBP, CRE-ATF, E-box) often associated with early response genes (Tanabe and Tohnai, 2002; Murakami and Kudo, 2004).
Cyclooxygenase-3
A third COX isoform, named COX-3, has recently been characterized in dogs (Chandrasekharan et al., 2002). Canine COX-3 is not derived from a distinct gene but corresponds to an alternatively spliced COX-1 variant in which intron 1 (90 nulceotides) is retained and codes for an in-frame insertion of 30 amino acids. The presence of a putative COX-3 isoform has also been detected in mice and rats (Kis et al., 2003; Shaftel et al., 2003), but the expression of a functional COX-3 in humans is still debated since intron 1 in this species is 94 nucleotides long, which creates a frameshift that results in a protein sequence not related to COX-1 (Dinchuk et al., 2003; Schwab et al., 2003b; Simmons, 2003). The enzyme is sensitive to acetaminophen and highly expressed in the central nervous system, suggesting that inhibiting COX-3 may represent an important mechanism for controlling the synthesis of prostanoids mediating pain and fever (Schwab et al., 2003a).
Prostaglandins and the ovulatory process
A potential relationship between prostaglandin biosynthesis and ovulation first emerged during the early 1970s (reviewed in Armstrong, 1981; Espey and Lipner, 1994). This was an exciting period in the prostaglandin field as John Vane (1971) had just demonstrated that the inhibition of prostaglandin synthesis was the underlying mechanism of action of aspirin and other related NSAID. Indomethacin, another prostaglandin synthesis inhibitor, rapidly became a drug of choice to determine the putative role of prostaglandins in various biological processes.
The indomethacin-dependent inhibition of ovulation was initially reported in rats and rabbits (Armstrong and Grinwich, 1972; Orczyk and Behrman, 1972), but was subsequently observed in numerous species, including pigs, sheep, cows and humans (reviewed in Armstrong, 1981; Sirois et al., 2000). In these species, as well as in others, the LH surge causes a marked increase in concentrations of PGE2 and PGF2α in ovarian follicles just prior to ovulation, with granulosa cells generally thought to be the primary site of prostaglandin synthesis. The ability of indomethacin to act directly at the ovarian level and block the pre-ovulatory rise in follicular prostaglandins was quickly favoured as the underlying mechanism responsible for causing anovulation (Armstrong, 1981; Murdoch et al., 1993). The relationship between prostaglandins and ovulation was further strengthened by studies in which the administration of antisera to prostaglandins was shown to block follicular rupture in mice and rabbits (Armstrong et al., 1974; Lau et al., 1974), and by investigations in which prostaglandins were able to restore ovulation in indomethacin-treated animals (Tsafriri et al., 1973; Diaz-Infante et al., 1974; Wallach et al., 1975a; Downey and Ainsworth, 1980). However, it should be noted that a number of other studies in the 1970s and 1980s provided opposite results and questioned the obligatory role of prostaglandins during ovulation (reviewed in Murdoch et al., 1993; Espey and Lipner, 1994). The issue would be ultimately resolved in the 1990s by genetic studies in mice (see below).
From follicular prostaglandin synthase to COX-2
Different alternatives were proposed as potential biochemical mechanisms responsible for the increase in follicular prostaglandins mediated by the LH surge, including an increase in arachidonic acid release and/or a rise in prostaglandin synthase activity. While the work of Clark et al. (1978) provided initial indirect evidence for the regulation of prostaglandin synthase activity, the ultimate demonstration of enzyme regulation came 10 years later.
Prostaglandin endoperoxide synthase
The collective work of two groups provided in 1987 the first direct evidence that prostaglandin endoperoxide synthase, referred to as PG synthase or PGS in their studies, was up-regulated in follicles prior to ovulation (Curry et al., 1987; Hedin et al., 1987; Huslig et al., 1987). It is interesting to realize that this discovery was made at a time when only one COX had been characterized, and the success of their work unknowingly depended on the ability of the antibodies that were raised against ovine PGS purified from seminal vesicles (now known as COX-1) to cross-react with the PGS isoform induced in pre-ovulatory follicles (now known as COX-2). Using intact and hypophysectomized immature rats primed with estradiol and/or gonadotrophins as in vivo models of pre-ovulatory follicular development, Hedin et al. (1987) established by immunoblots and immunofluorescence that PGS was selectively and transiently induced in granulosa cells during the ovulatory process (Figure 3A). The induction was also stage-dependent, as low or negligible expression was observed in small antral follicles and in pre-ovulatory follicles isolated prior to hCG treatment. A marked increase in PG synthase protein was also documented by enzyme immunoassays in ovaries of immature, pregnant mare serum gonadotrophin-primed rats isolated 8 h after an ovulatory dose of hCG (Huslig et al., 1987). The physiological occurrence of this biochemical event was ultimately confirmed in pre-ovulatory follicles of adult rats exhibiting normal estrous cycles (Curry et al., 1990).
The mechanisms responsible for gonadotrophin-dependent induction of PGS in pre-ovulatory follicles were further investigated using various models in vitro, such as short-term incubations of isolated pre-ovulatory follicles and cultures of granulosa cells (Wong et al., 1989). The pattern of PGS induction observed in vivo following hCG treatment was recapitulated in vitro by LH, FSH and forskolin. This agonist-dependent induction of PGS was not blocked after inhibition of PGS enzymatic activity with indomethacin, nor was it altered by addition of prostaglandins. However, it was abolished when transcriptional (α-amanitin) or translational (cycloheximide) inhibitors were added to the culture media, consistent with results of previous studies that showed that LH- or cAMP-stimulated production of follicular PGE2in vitro was dependent on transcription and translation (Clark et al., 1976; Zor et al., 1977). Attempts to relate the marked induction of PGS protein to a similar regulation in PGS transcript by Northern blots proved unsuccessful at the time, and understandably surprised the investigators (Wong et al., 1989). It would later become evident that the cDNA probe (now known as COX-1) available in these early studies was unable to hybridize with the isoform induced in pre-ovulatory follicles. However, this finding was perceived as an important discrepancy and raised initial suspicions as to the precise nature of the PGS isoform induced in granulosa cells.
Distinct PGS molecular weight variants
The production of anti-PGS antibodies with distinct specificities led to the identification of two molecular weight variants differentially expressed in follicles and other tissues (Figure 3B) (Wong and Richards, 1991). One antibody recognized a 72 000 mol. wt isoform (PGS72) rapidly induced by LH in granulosa cells of pre-ovulatory follicles but not expressed in other ovarian and non-ovarian tissues tested. In contrast, another antibody identified the presence of a 69 000 mol. wt isoform (PGS69) constitutively expressed in small antral and pre-ovulatory follicles (primarily in thecal cells), unregulated by LH, and present in various non-ovarian tissues. While it remains not entirely clear how antibodies with such distinct specificities could be generated from a unique source of antigen [PGS purified from ovine seminal vesicles (again presumably COX-1)], this serendipitous finding provided the first evidence for the presence of two PGS variants in the rat ovary. Likewise, it helped to reconcile results of previous immunohistological studies identifying PGS expression in thecal cells (Curry et al., 1987, 1990).
Purification of a novel isoform of prostaglandin endoperoxide synthase: COX-2
To resolve rising concerns as to the precise nature of the PGS isoform induced by LH/hCG in granulosa cells of rat pre-ovulatory follicles, referred to as rPGSi for inducible form of rat PGS, the purification and characterization of the enzyme appeared as the most appropriate approach (Sirois and Richards, 1992). A granulosa cell extract was prepared from rat pre-ovulatory follicles isolated 6 h after an ovulatory dose of hCG, a time when the expression of the enzyme previously proved to be maximal (Hedin et al., 1987). The rPGSi enzyme was purified from the extract by a combination of anionic exchange chromatography (Mevkh et al., 1985) and size fractionation. Amino-terminal microsequencing analysis revealed that the first 26 residues of the rat protein was quite different (only 58–61% identity) from the corresponding region of the ovine and mouse PGS (DeWitt and Smith, 1988; Merlie et al., 1988; DeWitt et al., 1990), but highly similar (96% identity) to the deduced amino acid sequence of a new mouse PGS-related cDNA clone that had just been characterized (Figure 3C) (Kujubu et al., 1991). Thus, the purification and characterization of a new PGS protein in rat granulosa cells (Sirois and Richards, 1992), along with the cloning of a novel PGS-related clone in chicken (Xie et al., 1991) and mouse fibroblasts (Kujubu et al., 1991), contributed collectively to the identification of the second PGS enzyme. The isoform that was first characterized would be thereafter referred to as COX-1, and the novel isoform as COX-2. The availability of a COX-2 cDNA probe allowed the regulation of the transcript during the ovulatory process to be revisited, and results demonstrated that it was indeed markedly induced in granulosa cells by LH/hCG (Sirois et al., 1992).
Species-specific time-course of COX-2 induction
Studies in rats revealed that the induction of COX-2 was very rapid, 2–4 h after hCG treatment, and preceded ovulation by ∼10 h (Figure 3D) (Sirois et al., 1992). It became of interest to determine whether the selective induction of COX-2 in granulosa cells was a molecular process conserved in other species. The bovine system was the first one tested, and results established that COX-2 was also induced in granulosa cells of pre-ovulatory follicles during hCG- or GnRH-induced ovulation (Sirois, 1994; Tsai et al., 1996), as well as following a natural endogenous LH surge (Liu et al., 1997). However, a striking difference was observed in the time-course of COX-2 induction in bovine follicles, as the induction was not rapid as in the rat but occurred only ∼18 h after hCG treatment (Figure 3D) (Sirois, 1994). Interestingly, the interval of time from COX-2 induction to follicular rupture, ∼10 h, was remarkably conserved in the bovine system (Figure 3D). The rapid versus delayed induction of COX-2 in species with a short (rat: 12–14 h post-hCG) versus a long (cattle: 28–30 h post-hCG) ovulatory process led to the hypothesis that the timing of COX-2 induction was involved in determining the species-specific length of the ovulatory process. This hypothesis was subsequently tested using the equine model, a species with a longer ovulatory process (39–42 h post-hCG). Results revealed that COX-2 induction was further delayed in this species and occurred only 30 h post-hCG, with follicular rupture again taking place ∼10 h later (Figure 3D) (Sirois and Doré, 1997; Boerboom and Sirois, 1998). Collectively, these results supported the concept that COX-2 could serve as a molecular determinant that sets the alarm of the mammalian ovulatory clock (Richards, 1997). Results from investigations in mice also agreed with the present model, as a rapid induction of COX-2 was also observed after hCG treatment (Joyce et al., 2001; Segi et al., 2003).
Studies on the regulation of COX-2 in human pre-ovulatory follicles have been limited. However, the transcript has been detected in granulosa cells and the protein measured in pre-ovulatory follicular fluid collected from women enrolled in an IVF programme (Narko et al., 1997; Tokuyama et al., 2001, 2003). Also, the pattern of COX-2 expression has been studied in the rhesus monkey, a primate model with an ovulatory process of ∼40 h (Duffy and Stouffer, 2001). A rise in COX-2 protein expression and in intrafollicular prostaglandin levels occurred 24 and 36 h post-hCG, respectively, which is consistent with results in other species. However, other aspects of COX-2 expression in primate follicles appeared unique, including the relatively early detection of COX-2 mRNA (12 h post-hCG) by RT–PCR and the localization of the protein in both granulosa and theca cell layers.
Control of follicular COX-2 expression
While the pre-ovulatory gonadotrophin surge is recognized as the primary trigger of follicular COX-2 induction prior to ovulation, numerous agonists acting through distinct signalling pathways have been shown to regulate the expression of the enzyme in ovarian cells. Furthermore, unravelling the fine molecular mechanisms involved in the transcriptional activation of the COX-2 gene by gonadotrophins has also been the focus of a number of investigations, and noticeable progress has been made in this field.
Agonist-dependent regulation of COX-2 mRNA and protein
The use of hCG to mimic the endogenous LH surge should be regarded as the first model shown to induce follicular COX-2 prior to ovulation (Hedin et al., 1987; Huslig et al., 1987). Several subsequent experimental paradigms in vivo and in vitro revealed that LH and FSH, as well as other direct activators of the cyclic adenosine monophosphate-dependent protein kinase (PKA) pathway, were equally able to up-regulate COX-2 expression in pre-ovulatory granulosa cells (Wong et al., 1989; Wong and Richards, 1991; Liu et al., 1999). However, the LH surge-mediated induction of the enzyme was also shown to involve other signaling pathways, including calcium-dependent protein kinase (PKC) and tyrosine kinase-dependent pathways (Wong and Richards, 1992; Morris and Richards, 1993, 1995).
Apart from gonadotrophins, the list of molecules capable of regulating COX-2 in ovarian cells seems to be continuously expanding. The pattern of COX-2 induction by GnRH in rat pre-ovulatory follicles or granulosa cells in vitro was very similar to that observed with gonadotrophins (Wong and Richards, 1992). Transforming growth factor α (TGF-α) was also shown to induce COX-2 in hen pre-ovulatory granulosa cells (Li et al., 1996), whereas interleukin-1β had a similar effect in human granulosa-luteal cells and rat granulosa cells (Narko et al., 1997; Ando et al., 1998; Saito et al., 2001). Likewise, stimulation of mouse granulosa cells with recombinant growth differentiation factor 9 (GDF-9), a factor exclusively produced by the oocyte, was shown to up-regulate COX-2, suggesting that oocyte-derived factors may contribute to increased prostaglandin synthesis during the ovulatory process (Elvin et al., 1999). Interestingly, the regulation of COX-2 appears to switch from a predominant PKA-dependent pathway in pre-ovulatory follicles to a predominant PKC-dependent pathway in the corpus luteum (Wu and Wiltbank, 2001, 2002). PGF2α and reactive oxygen species become strong inducers of COX-2 in luteal cells (Nakamura and Sakamoto, 2001; Tsai and Wiltbank, 2001).
In contrast to the impressive list of COX-2 agonists in ovarian cells, only a limited number of putative COX-2 repressors has been identified. TGF-β significantly suppressed basal and TGF-α-induced COX-2 mRNA in hen granulosa cells (Li et al., 1996), whereas the work by Hedin and Eriksson (1997) revealed that progesterone was able to decrease LH-stimulated COX-2 expression in rat pre-ovulatory follicles in vitro.
Regulation of the COX-2 promoter in granulosa cells
The finding that the gonadotrophin-dependent induction of COX-2 in granulosa cells was dependent on transcriptional events (Wong et al., 1989) provided the rationale for cloning and studying the regulation of the COX-2 promoter. Initial studies revealed that the proximal region located within the first 150–200 base pairs upstream of the transcription start site of the rat and bovine COX-2 promoter was sufficient to confer basal and forskolin/gonadotrophin-inducible activities (Figure 4A) (Sirois and Richards, 1993; Sirois et al., 1993; Liu et al., 1999). A number of consensus cis-acting elements such as C/EBP, ATF/CRE and E-box elements were identified within this region, but electrophoretic mobility shift assays and site-directed mutagenesis studies revealed that the E-box element was playing a predominant role in both species in the regulation of the promoter in granulosa cells (Figure 4B) (Morris and Richards, 1996; Liu et al., 1999). Upstream stimulatory factor (USF)-1 and -2, transcription factors known to bind to the E-box, were detected in rat and bovine granulosa cells. Interestingly, the presence of an amino-terminal truncated form of USF-2 thought to repress gene expression was detected prior to hCG treatment only in bovine granulosa cells, and was proposed as a potential mechanism for the delayed induction of COX-2 in species with a long ovulatory process (Liu et al., 1999). The trans-activating capacity of USF on the COX-2 promoter in granulosa cells, as well as the ability of an amino-truncated USF-2 dominant negative mutant to repress promoter activation, were recently demonstrated experimentally (Figure 4C and D) (Sayasith et al., 2004). Moreover, critical interactions between USF proteins and the E-box element were shown to be conserved as the transcriptional regulation of COX-2 switches from PKA to PKC dependence during cellular luteinization (Wu and Wiltbank, 2002). Other studies revealed that the Il-1β-regulated induction of COX-2 transcripts in whole ovarian dispersates was also dependent on promoter activation (Saito et al., 2001).
COX-2: a marker for ovulation during ovarian stimulation
One undesirable outcome of ovarian stimulation (i.e. induction of multiple ovulation) protocols in cattle is the development of a proportion (up to 20%) of large follicles that fail to ovulate and that ultimately undergo atresia or become cystic (Laurincik et al., 1993; Pruwantara et al., 1994).
To test the hypothesis that COX-2 could be used as a marker of follicular commitment to ovulation during ovarian stimulation, multiple follicular development was stimulated with exogenous FSH, ovulation was induced with hCG, and the pattern of COX-2 expression was characterized (Liu and Sirois, 1998). Results of this study showed that COX-2 expression 24 h after hCG was detected in 76% of follicles >8 mm, but the proportion (24%) of follicles not expressing the enzyme was very similar to the incidence of anovulatory follicles detected by ultrasonography (Laurincik et al., 1993). Interestingly, in contrast to COX-2-positive follicles, COX-2-negative follicles were not luteinized and contained a compact cumulus–oocyte complex, suggesting an apparent overall failure to respond to the gonadotrophin pre-ovulatory signal. Thus, COX-2 expression should provide a valuable marker to unravel the molecular basis behind the development of large anovulatory follicles during ovarian stimulation.
Inhibition of COX-2 action
Considering the central role played by COX enzymes in the production of potent inflammatory mediators such as the prostaglandins, numerous inhibitors (i.e. NSAID) have been developed by the pharmaceutical industry, with several being used to study the role of prostaglandins during ovulation. In recent years, the generation of COX-2- and COX-1-deficient mice has also provided powerful tools to investigate the relative role of each enzyme in various physiological and pathological processes.
Non-selective and selective COX-2 inhibitors
Pharmaceutical drugs able to inhibit follicular COX-2 were actually used >20 years before the discovery of the enzyme. Indeed, the non-selective COX inhibitor indomethacin was administered in the 1970s in numerous species and was shown to block follicular rupture (reviewed in Armstrong, 1981; Murdoch et al., 1993; Priddy and Killick, 1993; Espey and Lipner, 1994). Other non-selective COX inhibitors such as aspirin and naproxen also impaired ovulation, at least in rabbits (Zanagnolo et al., 1996). Likewise, the administration of indomethacin blocked the ovulatory process in rhesus and marmoset monkeys (Wallach et al., 1975b; Maia et al., 1978; Duffy and Stouffer, 2002), and prevented or delayed ovulation in women (Killick and Elstein, 1987; Akil et al., 1996; Athanasiou et al., 1996; Nargund and Wei, 1996). However, in contrast to these studies, ibuprofen had apparently very limited, if any, effect in delaying follicular rupture in 12 women involved in a randomized clinical study (Uhler et al., 2001).
The 1990s brought the development of several selective COX-2 inhibitors that were tested for their effect on the ovulatory process. Celecoxib tended to reduce ovulation in mice (Reese et al., 2001), NS-398 impaired ovulation in vivo and in vitro in rats (Mikuni et al., 1998), and meloxicam blocked follicular rupture in rabbits (Salhab et al., 2001, 2003). A randomized double-blind study in women also showed that rofecoxib delayed follicular rupture without affecting peripheral hormone cyclicity (Pall et al., 2001). Collectively, these studies further underscored the role of COX-2 in ovulation.
Genetic inactivation of COX-2
The most compelling evidence of the importance of COX-2 in the ovulatory process came from genetic studies in which genes encoding COX-1 or COX-2 were inactivated (Dinchuk et al., 1995; Langenbach et al., 1995; Morham et al., 1995). While mice carrying a null mutation for COX-1 were fertile (Langenbach et al., 1995), mice deficient in COX-2 proved to be infertile because of multiple reproductive failures, including a marked anovulatory phenotype (Lim et al., 1997) that could be reversed with exogenous PGE2 (Davis et al., 1999). In general, genetic inactivation of COX-2 resulted in more severe effects on ovulation than pharmacological inhibition of the enzyme (Reese et al., 2001). Interestingly, a recent study showed that the anovulatory phenotype observed in COX-2-deficient mice produced in a C57BL/6J/129 background was not present in COX-2 null mice derived in a CD1 genetic background (Wang et al., 2004). In the latter group, the phenotypic rescue appeared related to the ability of COX-1 to replace a function generally attributed to COX-2 (Wang et al., 2004).
Implications for clinicians managing patients on COX-2 inhibitors
NSAID are among the most widely prescribed drugs worldwide, and their use in women attempting to become pregnant has been associated with reversible female infertility (Akil et al., 1996; Thylan, 1997; Mendonca et al., 2000; Norman, 2001; Deviere, 2002; Stone et al., 2002; Norman and Wu, 2004). It is thought that the use of selective or non-selective COX-2 inhibitors may cause a luteinized unruptured follicle (LUF) syndrome, a condition characterized by the ability of a normally growing pre-ovulatory follicle to luteinize, but not to rupture, in response to the LH surge, resulting in the entrapment of the oocyte and in infertile cycles. Moreover, results from animal studies indicate that inhibition of COX-2 also impairs cumulus expansion, fertilization, decidualization and implantation (Lim et al., 1997; Matsumoto et al., 2001a). Collectively, these findings clearly underscore the risk of taking COX-2 inhibitors while attempting to become pregnant. The potential adverse reproductive consequences of pharmacological inhibition of COX-2 action should therefore be considered given the central role played by the enzyme during ovulation and other reproductive processes, and avoiding NSAID usage during this critical period seems to be a logical recommendation to women consulting for infertility.
COX-2 expression in ovarian cancer
Ovarian cancer is the most common gynecological malignancy in western countries and represents one of the leading causes of death due to cancer in women (Jemal et al., 2004). The majority of primary ovarian tumors is epithelial in origin and derives from the surface epithelium covering the ovary. Although the precise underlying mechanisms responsible for the development of epithelial ovarian cancer remain unclear, various hypotheses have been proposed (Fathalla, 1971; Cramer and Welch, 1983; Ness and Cottreau, 1999; Purdie et al., 2003). Among them, incessant ovulation (Fathalla, 1971; Purdie et al., 2003) and inflammation (Ness and Cottreau, 1999) appear best related to the subject of the present review. A number of factors that shorten the total duration of the women's ovulatory life (i.e. number of ovulations), such as parity, prolonged breast-feeding amenorrhoea and oral contraceptive use, have been associated with the greatest risk reduction for ovarian cancer. It is argued that incessant ovulation causes repeated minor trauma to the epithelial surface of the ovary, and that the inflammatory reaction and mediators (including prostaglandins) associated with the ovulatory process provide recurring oxidative stress that may be mutagenic (Ness and Cottreau, 1999; Purdie et al., 2003). Some evidence suggests that COX-2 could contribute to the early stages of cancer formation, as the enzyme was found to be overexpressed in pre-neoplastic changes of the ovarian surface epithelium (Roland et al., 2003). Borderline and benign tumours have also been reported to express COX-2 (Klimp et al., 2001; Matsumoto et al., 2001b). However, numerous biochemical and epidemiological studies investigating a potential link between COX-2 and ovarian cancer have yielded conflicting results. Several reports documented that COX-2 mRNA and/or protein were detectable in malignant ovarian tumours (Klimp et al., 2001; Matsumoto et al., 2001b; Landen et al., 2003; Li et al., 2004), and that the use of NSAID was associated with a decrease in the risk of ovarian cancer (Cramer et al., 1998; Akhmedkhanov et al., 2001; Moysich et al., 2001). COX-2 expression has been positively correlated with microvessel density of tumours, but negatively correlated with response to chemotherapy and patient survival (Denkert et al., 2002; Ferrandina et al., 2002; Ali-Fehmi et al., 2003). In contrast, other reports were unable to detect COX-2 expression in ovarian tumours (Doré et al., 1999; Gupta et al., 2003; Roland et al., 2003), or to find an association between NSAID use and cancer risk (Rosenberg et al., 2000; Tavani et al., 2000). The precise basis for these conflicting results remain unclear but differences in case selection have been proposed as a possible cause (Gupta et al., 2003), considering that agents used in standard chemotherapy for ovarian cancer can induce COX-2 expression (Subbaramaiah et al., 2000; Cassidy et al., 2002). Other possible reasons for the conflicting results include differences in detection methods, tissue processing and antibody used. Studies in vitro using several ovarian cancer cell lines also showed marked variability in levels of COX-2 expression among cell lines (Denkert et al., 2002; Munkarah et al., 2002; Gupta et al., 2003).
Conclusions
Much progress has been achieved in recent years on our understanding of the control of prostaglandin synthesis in pre-ovulatory follicles. Results from studies presented above have highlighted the central and obligatory role played by the gonodotrophin-dependent induction of COX-2 in ovarian follicles prior to ovulation. Even though COX-2 is generally considered as the rate-limiting enzyme in the prostaglandin biosynthetic pathway, attention will also need to be focused on terminal prostaglandin synthases since, for example, PGES was also shown to be regulated in bovine pre-ovulatory follicles (Filion et al., 2001). The synthesis of biologically active prostaglandins such as PGE2 and PGF2α by follicular cells also raises questions regarding their precise roles during follicular rupture. Given their potential actions as local autocrine, paracrine and perhaps intracrine hormones, the fine molecular mechanisms of prostaglandin action remains a sizeable and exciting challenge.
Ultimately, the clinical implications of COX-2 inhibition to women's fertility should be fully appreciated, considering the obligatory role of the enzyme during the ovulatory process as well as its potential implication in fertilization and implantation. The use of non-selective and selective COX-2 inhibitors should generally be discouraged for women attempting to become pregnant (Norman and Wu, 2004). However, the value of COX-2 inhibition in pathological processes in which the enzyme has been detected such as ovarian cancer and ovarian endometriotic cysts (Ota et al., 2001; Fagotti et al., 2004) will require further investigations.
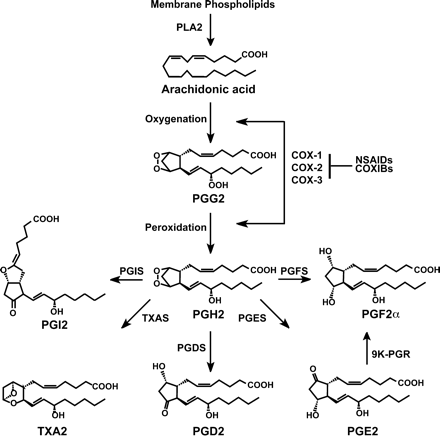
The prostaglandin biosynthetic pathway. PLA2 = phospholipase A2; COX = cyclooxygenase; NSAIDs = non-steroidal anti-inflammatory drugs; COXIBs = COX inhibitors; PGIS = prostacyclin synthase; TXAS = thromboxane synthase; PGDS = PGD2 synthase; PGES = PGE2 synthase; PGFS = PGF2α synthase; 9K-PGR = 9-keto-PGE2 reductase.
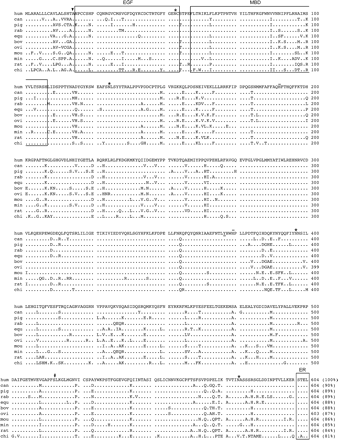
Amino acid sequence of human cyclooxygenase-2 (COX-2) and comparisons with other homologues. The amino acid sequence of COX-2 (hum; GenBank accession number NP_000954) is aligned with the canine (can; AY044905), pig (AY028583), rabbit (rab; AAB71222), equine (equ; AAC07911), bovine (bov; AAC04702), ovine (ovi; P79208), mouse (mou; NP_035328), mink (min; AAC05637), rat (NP_058928), and chicken (chi; P27607) COX-2 homologues. Identical residues are presented as a printed period; the putative signal peptide cleavage site is indicated by an arrowhead; the tyrosine residue (Y371) associated with the cyclooxygenase active site is underlined; histidine residues (H193 and H374) involved in haem binding are overlined; putative N-linked glycosylation sites (N53, N130, N396, N580) are marked with an asterisk; and the aspirin-acetylated serine residue (S516) is indicated by a number sign (#). Boxed regions include an epidermal growth factor (EGF) domain, a membrane-binding domain (MBD), and a short C-terminal domain thought to represent an endoplasmic retention (ER) signal. Numbers on the right refer to the last amino acid residue on that line, and the percentage in parentheses indicates the degree of identity in amino acid residues when comparisons are made with human COX-2.
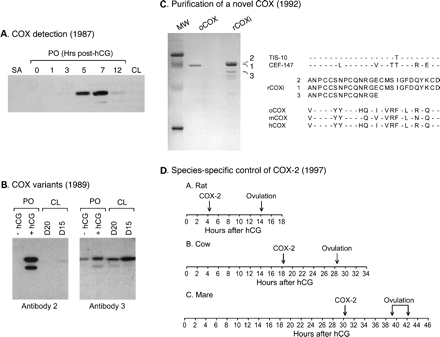
Regulation of cyclooxygenase (COX) expression in pre-ovulatory follicles. (A) First detection of COX expression in rat follicles. Protein extracts were prepared from small antral follicles (SA), from pre-ovulatory follicles (PO) isolated before (0 h) or after (1–12 h) an ovulatory dose of hCG, and from a newly formed corpus luteum (CL; 24 h post-hCG). Results from immunoblot analyses showed the presence of a transient induction of COX after hCG treatment (redrawn and adapted from Hedin et al., 1987). (B) Evidence of distinct COX variants in the ovary. Anti-COX antibodies were produced against purified ovine seminal vesicle COX, and immunoblots were performed on extracts obtained from pre-ovulatory follicles (PO) isolated before (−hCG) or 6 h after ( + hCG) an ovulatory dose of hCG, and from CL obtained on days 15 and 20 of pregnancy. Antibody 2 recognized a COX variant induced in pre-ovulatory follicles isolated after hCG treatment, whereas antibody 3 recognized a COX variant constitutively expressed in all ovarian tissues tested (redrawn and adapted from Wong and Richards, 1991). (C) Purification and characterization of a novel COX isoform. The hCG-induced COX isoform in rat pre-ovulatory follicles (rCOXi) was purified and resolved by one-dimensional SDS–PAGE, transferred to a membrane and stained with Coomassie Blue. Molecular weight markers (MW) and ovine COX (oCOX) were run as standards. Three bands corresponding to immunoreactive rCOXi were used for amino acid microsequencing. Results showed that the purified rat protein was quite different from the ovine, mouse (m) and human (h) COX isoform characterized at the time, but very similar to a novel COX-related cDNA cloned in mice (TIS-10) and chicken (CEF-147) (adapted from Sirois and Richards, 1992). (D) Relationship between time of COX-2 induction, time of ovulation and length of the ovulatory process in rats, cows and mares. Results show that the precise time of COX-2 induction appears related to the species-specific length of the ovulatory process, whereas the interval of time from COX-2 induction to ovulation is highly conserved in these species (adapted from Sirois and Doré, 1997). © 1987, 1991, and 1997, The Endocrine Society, © 1992 The American Society for Biochemistry and Molecular Biology.
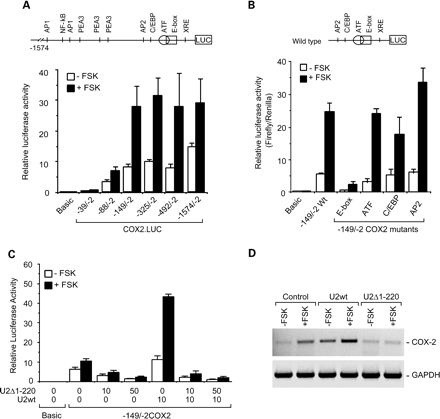
Regulation of the bovine COX-2 promoter in granulosa cells. (A) Deletion analysis of the bovine COX-2 promoter. The bovine COX-2 promoter fragment −1574/−2 (+1 = transcription start site), as well as a series of 5′-deletion mutants, were fused upstream of the firefly luciferase reporter gene (LUC) in the pGL3.Basic vector and transfected into primary cultures of bovine granulosa cells. After transfection, cells were incubated in the absence or presence of forskolin (FSK; 10 mmol/l) for 36 h. Results show that the promoter region located within the first 149 nucleotides upstream of the transcription start site plays a key role in the regulation of the COX-2 promoter in granulosa cells. A schematic representation of the bovine COX-2 promoter with putative consensus cis-elements is shown. (B) Effect of site-directed mutagenesis on bovine COX-2 promoter activity. Plasmid constructs containing the wild type (WT) −149/−2 COX-2 promoter fragment, as well as four site-directed mutants in which point mutations were introduced in the E-box, ATF, C/EBP or AP2 element, were transfected into granulosa cell cultured in the absence or presence of forskolin. Results show that point mutations within the E-box cause a marked decrease in COX-2 promoter activities. (C) Effect of USF-2 on bovine COX-2 promoter activity. Granulosa cells were co-transfected with −149/−2COX2.LUC in the absence or presence of constructs expressing full length wild type bovine USF-2 (U2wt) or an amino-terminal truncated form of USF-2 lacking the first 220 amino acids (U2D1-120). U2D1-120 was designed to contain dimerization and DNA binding domains, but to lack transcription activating domains. Results show that transfections with U2wt alone led to a marked increase in promoter activities, whereas U2D1-120 acted as a dominant negative mutant and blocked endogenous and U2wt-stimulated COX-2 promoter acitivties. (D) Effect of USF-2 on COX-2 transcript expression. Granulosa cells were first transfected with U2wt or U2D1-120, and then cultured for 24 h in the absence or presence of forskolin. RNA samples were prepared and changes in COX-2 and GAPDH (control gene) were analysed by RT–PCR. Results reveal that U2wt overexpression increases, whereas U2D1-120 overexpression represses, COX-2 transcript. (Adapted from Liu et al., 1999; Sayasith et al., 2004.) © 1999 and 2004, The American Society for Biochemistry and Molecular Biology.
Studies from the authors' laboratory were funded by Canadian Institutes of Health Research (CHIR) Grant MT-13190. J.Sirois is supported by a CIHR Investigator Award.
References
Akhmedkhanov A, Toniolo P, Zeleniuch-Jacquotte A, Kato I, Koenig KL and Shore RE (
Akil M, Amos RS and Stewart P (
Ali-Fehmi R, Che M, Khalifeh I, Malone JM, Morris R, Lawrence WD and Munkarah AR (
Ando M, Kol S, Kokia E, Ruutiainen-Altman K, Sirois J, Rohan RM, Payne DW and Adashi EY (
Armstrong DT (
Armstrong DT and Grinwich DL (
Armstrong DT, Grinwich DL, Moon YS and Zamecnik J (
Athanasiou S, Bourne TH, Khalid A, Okokon EV, Crayford TJ, Hagstrom HG, Campbell S and Collins WP (
Boerboom D and Sirois J (
Breyer RM, Bagdassarian CK, Myers SA and Breyer MD (
Cassidy PB, Moos PJ, Kelly RC and Fitzpatrick FA (
Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS and Simmons DL (
Clark MR, Marsh JM and LeMaire WJ (
Clark MR, Marsh JM and LeMaire WJ (
Cramer DW and Welch WR (
Cramer DW, Harlow BL, Titus-Ernstoff L, Bohlke K, Welch WR and Greenberg ER (
Curry TE, Jr, Malik A and Clark MR (
Curry TE, Jr, Bryant C, Haddix AC and Clark MR (
Davis BJ, Lennard DE, Lee CA, Tiano HF, Morham SG, Wetsel WC and Langenbach R (
Denkert C, Kobel M, Pest S, Koch I, Berger S, Schwabe M, Siegert A, Reles A, Klosterhlafen B and Hauptmann S (
Deviere J (
DeWitt DL and Smith WL (
DeWitt DL, el-Harith EA, Kraemer SA, Andrews MJ, Yao EF, Armstrong RL and Smith WL (
Diaz-Infante A, Jr, Wright KH and Wallach EE (
Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, Contel NR, Eng VM, Collins RJ, Czerniak PM et al. (
Dinchuk JE, Liu RQ and Trzaskos JM (
Doré M, Côté LC, Mitchell A and Sirois J (
Downey BR and Ainsworth L (
Duffy DM and Stouffer RL (
Duffy DM and Stouffer RL (
Elvin JA, Clark AT, Wang P, Wolfman NM and Matzuk MM (
Espey LL (
Espey LL and Lipner H (
Fagotti A, Ferrandina G, Fanfani F, Legge F, Lauriola L, Gessi M, Castelli P, Barbieri F, Minelli L and Scambia G (
Fathalla MF (
Ferrandina G, Lauriola L, Zannoni GF, Fagotti A, Fanfani F, Legge F, Maggiano N, Gessi M, Mancuso S, Ranelletti FO et al. (
Filion F, Bouchard N, Goff AK, Lussier JG and Sirois J (
FitzGerald GA (
Funk CD (
Garavito RM and Mulichak AM (
Goff AK (
Gupta RA, Tejada LV, Tong BJ, Das SK, Morrow JD, Dey SK and DuBois RN (
Hedin L and Eriksson A (
Hedin L, Gaddy-Kurten D, Kurten R, DeWitt DL, Smith WL and Richards JS (
Helliwell RJ, Berry EB, O'Carroll SJ and Mitchell MD (
Huslig RL, Malik A and Clark MR (
Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ and Thun MJ (
Joyce IM, Pendola FL, O'Brien M and Eppig JJ (
Killick S and Elstein M (
Kis B, Snipes JA, Isse T, Nagy K and Busija DW (
Klimp AH, Hollema H, Kempinga C, van der Zee AGJ, de Vries AGE and Daemen T (
Kujubu DA, Fletcher BS, Varnum BC, Lim RW and Herschman HR (
Landen CN, Mathur SP, Richardson MS and Creasman WT (
Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD et al. (
Lau IF, Saksena SK and Chang MC (
Laurincik J, Oberfranc M, Hyttel P, Grafenau P, Tomanek M and Pivko J (
Li J, Simmons DL and Tsang BK (
Li S, Miner K, Fannin R, Barrett JC and Davis BJ (
Lim H and Dey SK (
Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM and Dey SK (
Liu J and Sirois J (
Liu J, Carriere PD, Dore M and Sirois J (
Liu J, Antaya M, Boerboom D, Lussier JG, Silversides DW and Sirois J (
Maia H, Jr, Barbosa I and Coutinho EM (
Matsumoto H, Ma W, Smalley W, Trzaskos J, Breyer RM and Dey SK (
Matsumoto Y, Ishiko O, Deguchi M, Nakagawa E and Ogita S (
Mendonca LL, Khamashta MA, Nelson-Piercy C, Hunt BJ and Hughes GR (
Merlie JP, Fagan D, Mudd J and Needleman P (
Mevkh AT, Sud'ina GF, Golub NB and Varfolomeev SD (
Mikuni M, Pall M, Peterson CM, Peterson CA, Hellberg P, Brannstrom M, Richards JS and Hedin L (
Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA et al. (
Morris JK and Richards JS (
Morris JK and Richards JS (
Morris JK and Richards JS (
Moysich KB, Mettlin C, Piver MS, Natarajan N, Menezes RJ and Swede H (
Munkarah AR, Morris R, Baumann P, Deppe G, Malone J, Diamond MP and Saed GM (
Murakami M and Kudo I (
Murdoch WJ, Hansen TR and McPherson LA (
Nakamura T and Sakamoto K (
Nargund G and Wei CC (
Narko K, Ritvos O and Ristimaki A (
Ness RB and Cottreau C (
Norman RJ (
Norman RJ and Wu R (
Olson DM (
Orczyk GP and Behrman HR (
Ota H, Igarashi S, Sasaki M and Tanaka T (
Pall M, Friden BE and Brannstrom M (
Priddy AR and Killick SR (
Pruwantara B, Callesen H and Greve T (
Purdie DM, Bain CJ, Siskind V, Webb PM and Green AC (
Reese J, Zhao X, Ma WG, Brown N, Maziasz TJ and Dey SK (
Richards JS (
Roland IH, Yang WL, Yang DH, Daly MB, Ozols RF, Hamilton TC, Lynch HT, Godwin AK and Xu XX (
Rosen GD, Birkenmeier TM, Raz A and Holtzman MJ (
Rosenberg L, Palmer JR, Rao RS, Coogan PF, Strom BL, Zauber AG, Stolley PD and Shapiro S (
Saito J, Ando M, Sussman D, Negishi H, King G and Adashi EY (
Salhab AS, Gharaibeh MN, Shomaf MS and Amro BI (
Salhab AS, Amro BI and Shomaf MS (
Sayasith K, Bouchard N, Sawadogo M, Lussier JG and Sirois J (
Schwab JM, Schluesener HJ, Meyermann R and Serhan CN (
Schwab JM, Beiter T, Linder JU, Laufer S, Schulz JE, Meyermann R and Schluesener HJ (
Segi E, Haraguchi K, Sugimoto Y, Tsuji M, Tsunekawa H, Tamba S, Tsuboi K, Tanaka S and Ichikawa A (
Shaftel SS, Olschowka JA, Hurley SD, Moore AH and O'Banion MK (
Simmons DL (
Sirois J (
Sirois J and Doré M (
Sirois J and Richards JS (
Sirois J and Richards JS (
Sirois J, Simmons DL and Richards JS (
Sirois J, Levy LO, Simmons DL and Richards JS (
Sirois J, Liu J, Boerboom D and Antaya M (
Smith WL and Marnett LJ (
Smith WL, DeWitt DL and Garavito RM (
Stone S, Khamashta MA and Nelson-Piercy C (
Subbaramaiah K, Hart JC, Norton L and Dannenberg AJ (
Tanabe T and Tohnai N (
Tavani A, Gallus S, La Vecchia C, Conti E, Montella M and Franceschi S (
Tokuyama O, Nakamura Y, Muso A, Honda K, Ishiko O and Ogita S (
Tokuyama O, Nakamura Y, Musoh A, Honda K, Ozaki K and Ishiko O (
Tsafriri A, Koch Y and Lindner HR (
Tsafriri A, Chun SY and Reich R (
Tsai SJ and Wiltbank MC (
Tsai SJ, Wiltbank MC and Bodensteiner KJ (
Uhler ML, Hsu JW, Fisher SG and Zinaman MJ (
Vane JR (
Vane JR (
Wallach EE, Bronson R, Hamada Y, Wright KH and Stevens VC (
Wallach EE, Cruz A, Hunt J, Wright KH and Stevens VC (
Wang H, Ma WG, Tejada L, Zhang H, Morrow JD, Das SK and Dey SK (
Wong WY and Richards JS (
Wong WY and Richards JS (
Wong WY, DeWitt DL, Smith WL and Richards JS (
Wright DH, Abran D, Bhattacharya M, Hou X, Bernier SG, Bouayad A, Fouron JC, Vazquez-Tello A, Beauchamp MH, Clyman RI et al. (
Wu YL and Wiltbank MC (
Wu YL and Wiltbank MC (
Xie WL, Chipman JG, Robertson DL, Erikson RL and Simmons DL (
Zanagnolo V, Dharmarajan AM, Endo K and Wallach EE (
Zor U, Strulovici B, Nimrod A and Lindner HR (
Author notes
1Centre de recherche en reproduction animale and Département de biomédecine vétérinaire and 2Département de pathologie et microbiologie vétérinaire, Faculté de médecine vétérinaire, Université de Montréal, 3200 Sicotte, Saint-Hyacinthe, Québec, Canada J2S 7C6

{kind=link}
{kind=link}
{kind=link}
{kind=link}