





Abstract
Adult T cell leukemia (ATL) is an aggressive neoplastic disease, in which a quarter of the patients develop opportunistic infections due to cellular immunodeficiency. However, the underlying mechanism responsible for the immunosuppression has remained unclear. Recent studies have demonstrated that the leukemia cells from a subset of patients with ATL express Foxp3, a specific marker for CD25+CD4+ regulatory T (Treg) cells, which regulate the immune response by suppressing CD4+ T cell functions. However, whether there is a functional resemblance between ATL cells that have Foxp3 expression and Treg cells is still unknown. In this report, we confirmed the high expression of Foxp3 in leukemia cells from 5 of 12 ATL patients and demonstrated that ATL cells from 3 patients suppressed the proliferation of CD4+ T cells. Similarly, one of six HTLV-I-infected cell lines showed both high Foxp3 expression and suppressive activity. Like Treg cells, the suppression induced by the ATL cells from two patients and the HTLV-infected cell line appeared to be mediated by a cell–cell contact-dependent mechanism. Nevertheless, among the ATL cells that strongly expressed Foxp3, those from two of the five patients showed no apparent suppressive activity. Furthermore, retroviral transfection of Foxp3 did not confer any suppressive function on low Foxp3-expressing HTLV-I-infected cell lines. These results indicate that Foxp3 may be essential but is not sufficient for the Treg-cell-like suppressive activity of ATL cells and HTLV-I-infected cell lines.
Introduction
Adult T cell leukemia (ATL) is a progressive peripheral T lymphocytic malignancy strongly associated with human T cell leukemia virus type 1 (HTLV-I) infection (1, 2). A quarter of ATL patients develop overwhelming opportunistic infections (pneumocystis carinii, cytomegalovirus and fungal infections) as a consequence of defective cellular immunity (2–4). Since patients with ATL do not exhibit bone marrow depression, a well recognized cause of immunodeficiency in other types of leukemia (2, 5), the mechanism(s) may be associated with peripheral immunodeficiency. Nevertheless, the reason for immunosuppression in ATL remains unclear.
Most ATL cells express the helper T cell-associated antigen CD4 and the T cell activation marker CD25 (2, 6). Several previous studies have demonstrated that established HTLV-I-infected T cell lines have characteristics similar to activated CD4 T cells, in terms of their expression of IL-2 receptor subunits and T cell activation markers, such as CD25 and OX40 (2, 6). Thus, leukemia cells in patients with ATL have been thought to be derived from activated CD4 T cells. CD4 and CD25, however, are also characteristic of CD25+CD4+ regulatory T (Treg) cells. Treg cells have been identified recently in both mice and humans as naturally arising professional suppressor T cells that are capable of inhibiting the in vitro proliferation of CD25−CD4+ T cells stimulated via TCRs (7, 8). In addition, a number of in vivo studies in mice have demonstrated that Treg cells can control autoimmune disorders, allograft tolerance and tumor immunity by suppressing T cell-mediated responses (7–9). Despite expressing TCRs and surface IL-2 receptors, Treg cells remain anergic to stimulation via their TCRs. Similarly, most ATL cells are unable to mount a full proliferation response to TCR ligation, even though they too express these receptors. In addition to these similarities between ATL and Treg cells, recent papers have demonstrated that Foxp3, a Treg-cell-specific transcription factor, is expressed in the leukemia cells from a subset of patients with ATL (10–13). Foxp3 expression is indispensable for the development and function of Treg cells (7). From these findings, we hypothesized that the Foxp3-expressing ATL cells may have suppressive activity, and that some of them might have been derived from Treg cells instead of from activated T cells. To address this, we have examined the phenotypic characteristics and immunosuppressive activity of leukemia cells from 12 patients with ATL, and compared them to those of freshly isolated Treg cells and HTLV-I-infected T cell lines. Furthermore, we have investigated the functional association between Foxp3 expression and suppressive function in HTLV-I-infected cell lines retrovirally transfected with the Foxp3 gene.
Methods
Cases
Twelve ATL patients of various ages from Sasebo City General Hospital (Sasebo, Japan) and Tohoku University Hospital (Sendai, Japan) were recruited for this study (Table 1). The diagnosis of ATL was confirmed by the microscopic examination of peripheral blood cells. Furthermore, all of the patients had confirmed positive tests for serum antibodies against ATL antigen. ATL cells (CD25+CD4+ cells) comprised >80% of the PBMCs in most of the patients (Table 1). Among the acute ATL patients, two were demonstrably immunocompromised, even though they, like most of the patients, were receiving appropriate anti-microbial and anti-fungal treatment (Table 1).
ATL patient data
Case ID | Sex | Age | Clinical type | Clinical course | WBC (103 μl−1) | Proportion of ATL cells in PBMCs (%) | Infectious complication |
|---|---|---|---|---|---|---|---|
| 1 | M | 44 | Acute | Onset | 10.9 | 92 | Cutaneous infection |
| 2 | M | 75 | Acute | Relapse | 10.4 | 88 | Oral candida |
| 3 | M | 62 | Acute | Onset | 145.5 | 90 | No |
| 4 | M | 70 | Acute | Relapse | 8.4 | 97 | No |
| 5 | M | 41 | Acute | Onset | 102.8 | 90 | No |
| 6 | M | 58 | Chronic | Onset | 177 | 96 | No |
| 7 | F | 68 | Acute | Onset | 58 | 83 | No |
| 8 | M | 69 | Chronic | Onset | 19.5 | 82 | No |
| 9 | M | 69 | Chronic | Remission | 60.3 | 94 | No |
| 10 | F | 55 | Acute | Onset | 39 | 90 | No |
| 11 | M | 68 | Acute | Onset | 9.9 | 47 | No |
| 12 | M | 58 | Acute | Onset | 13.4 | 80 | No |
Case ID | Sex | Age | Clinical type | Clinical course | WBC (103 μl−1) | Proportion of ATL cells in PBMCs (%) | Infectious complication |
|---|---|---|---|---|---|---|---|
| 1 | M | 44 | Acute | Onset | 10.9 | 92 | Cutaneous infection |
| 2 | M | 75 | Acute | Relapse | 10.4 | 88 | Oral candida |
| 3 | M | 62 | Acute | Onset | 145.5 | 90 | No |
| 4 | M | 70 | Acute | Relapse | 8.4 | 97 | No |
| 5 | M | 41 | Acute | Onset | 102.8 | 90 | No |
| 6 | M | 58 | Chronic | Onset | 177 | 96 | No |
| 7 | F | 68 | Acute | Onset | 58 | 83 | No |
| 8 | M | 69 | Chronic | Onset | 19.5 | 82 | No |
| 9 | M | 69 | Chronic | Remission | 60.3 | 94 | No |
| 10 | F | 55 | Acute | Onset | 39 | 90 | No |
| 11 | M | 68 | Acute | Onset | 9.9 | 47 | No |
| 12 | M | 58 | Acute | Onset | 13.4 | 80 | No |
ATL patient data
Case ID | Sex | Age | Clinical type | Clinical course | WBC (103 μl−1) | Proportion of ATL cells in PBMCs (%) | Infectious complication |
|---|---|---|---|---|---|---|---|
| 1 | M | 44 | Acute | Onset | 10.9 | 92 | Cutaneous infection |
| 2 | M | 75 | Acute | Relapse | 10.4 | 88 | Oral candida |
| 3 | M | 62 | Acute | Onset | 145.5 | 90 | No |
| 4 | M | 70 | Acute | Relapse | 8.4 | 97 | No |
| 5 | M | 41 | Acute | Onset | 102.8 | 90 | No |
| 6 | M | 58 | Chronic | Onset | 177 | 96 | No |
| 7 | F | 68 | Acute | Onset | 58 | 83 | No |
| 8 | M | 69 | Chronic | Onset | 19.5 | 82 | No |
| 9 | M | 69 | Chronic | Remission | 60.3 | 94 | No |
| 10 | F | 55 | Acute | Onset | 39 | 90 | No |
| 11 | M | 68 | Acute | Onset | 9.9 | 47 | No |
| 12 | M | 58 | Acute | Onset | 13.4 | 80 | No |
Case ID | Sex | Age | Clinical type | Clinical course | WBC (103 μl−1) | Proportion of ATL cells in PBMCs (%) | Infectious complication |
|---|---|---|---|---|---|---|---|
| 1 | M | 44 | Acute | Onset | 10.9 | 92 | Cutaneous infection |
| 2 | M | 75 | Acute | Relapse | 10.4 | 88 | Oral candida |
| 3 | M | 62 | Acute | Onset | 145.5 | 90 | No |
| 4 | M | 70 | Acute | Relapse | 8.4 | 97 | No |
| 5 | M | 41 | Acute | Onset | 102.8 | 90 | No |
| 6 | M | 58 | Chronic | Onset | 177 | 96 | No |
| 7 | F | 68 | Acute | Onset | 58 | 83 | No |
| 8 | M | 69 | Chronic | Onset | 19.5 | 82 | No |
| 9 | M | 69 | Chronic | Remission | 60.3 | 94 | No |
| 10 | F | 55 | Acute | Onset | 39 | 90 | No |
| 11 | M | 68 | Acute | Onset | 9.9 | 47 | No |
| 12 | M | 58 | Acute | Onset | 13.4 | 80 | No |
Cell lines
The ILT-Mat, TCL-Mor, TCL-Kan and MT-2 cell lines are in vitro-established HTLV-I-infected CD4+ T cell lines described previously (14, 15). TL-Om1 and EDγ-16 were derived from leukemia cells isolated from patients with ATL (14, 16, 17). JPX9, which is an HTLV-I tax transfectant cell line derived from Jurkat, a human acute lymphocytic leukemia T cell line, was previously described (18). In brief, JPX9 cells were established by stably introducing a metallothionein promoter-directed p40tax expression vector, pMAXRHneo-I, into Jurkat cells. Thus, the p40tax product could be induced in response to CdC12 in these cells. Tax expression in JPX9 cells was confirmed by reverse transcription (RT)–PCR as described previously (19). These cells were maintained in RPMI 1640 medium supplemented with 10% FCS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. The ILT-Mat culture medium included 500 nM recombinant human IL-2 (Ajinomoto, Kanagawa).
Retroviral transduction with the Foxp3 gene
To establish lines from cells transfected with the Foxp3 gene, human full-length Foxp3 cDNA was cloned from primary Treg cells and inserted into a GFP-expressing retrovirus vector pMXs-IG, yielding pMXs-IG-Foxp3. Packaging cells were transfected with the pMXs-IG-Foxp3 plasmid by Ca3(PO4)2 precipitation. The retrovirus-containing supernatant was collected after a 48-h culture at 37°C. EDγ-16 and ILT-Mat were suspended in the retrovirus-containing supernatant, spun for 60 min at 1200 × g and cultured in the complete medium. After 2 weeks of culture, the GFP-positive cells were isolated by FACSAria (BD, Bioscience). The cloning primers for the full-length Foxp3 cDNA were forward primer 5′-CCGCTCGAGCAAGGACCCGATGCCCAACC and reverse primer 5′-CCGCTCGAGCTCTGCCTCCCACCAGTTTG.
In vitro T cell responses
CD4+ T cells were isolated from PBMCs by using a CD4+ T cell Isolation Kit (Miltenyi Biotec, Gladbach, Germany). CD25+CD4+ Treg and conventional CD25−CD4+ T cells were labeled with anti-CD25-biotin (IMMUNOTECH, Marseille Cedex, France) and further separated by an AutoMACS (Miltenyi Biotec). Purified CD25−CD4+ T cells (5 × 104) were stimulated with 10 μg ml−1 soluble anti-CD3 mAb for 5 days in the presence of PBMC-derived dendritic cells (DCs; 5 × 103). To prepare DCs, monocytes were isolated from PBMCs by plastic adherence and cultured in X-VIVO-15 (BioWhittaker, Walkersille, MD) supplemented with 30 ng ml−1 IL-4 and granulocyte macrophage colony-stimulating factor (PeproTech, London, UK) for 7 days. To prepare ATL cells, whole PBMCs from all the patients except for patient 11 were used as ATL cells because leukemia cells comprised 80–96% of PBMC in 11 of the 12 patients (see Table 1). In the case of patient 11, in whom leukemic cells accounted for 43% of PBMC, CD25+CD4+ cells were purified as described above. Purified CD25+CD4+ T (Treg) or ATL cells were added to the culture at the indicated dose. After 5 days of culture, the CD25−CD4+ T cells were assayed for 3Hthymidine ([3H]TdR) uptake. Alternatively, CD25−CD4+ T cells were labeled with 5,6-carboxy-fluorescein succinimidyl ester (CFSE) and assayed for CFSE intensity. In some experiments, neutralizing antibody against IL-10 (BD Biosciences), cytotoxic T lymphocyte antigen 4 (CTLA-4) (IMMUNOTECH) or TGFβ (R&D System) (10 μg ml−1 each) was added to the culture.
Trans-well experiments
CD25−CD4+ T cells (2 × 105) were cultured with DCs (1 × 104) in the presence of anti-CD3 mAb (10 μg ml−1) in 24-well plates. ATL or Treg cells, and DCs (1 × 104) were either added directly to the wells or placed in trans-well chambers. After a 5-day culture, the amount of proliferation of the CD25−CD4+ T cells was determined by [3H]TdR uptake in 96-well plates. In other proliferation studies, purified CD25−CD4+ T cells were labeled with CFSE (Molecular Probes, Eugene, OR) by incubation with 2.5 μM CFSE in protein-free PBS for 10 min at 37°C and then with a 10-fold volume of RPMI 1640 medium containing 10% FCS for 1 min. Cells were then washed twice with chilled PBS. The cell division of CFSE-labeled cells was estimated by their decreased fluorescence intensity, using a flow cytometer.
Establishment of HTLV-I-infected T cell clones derived from CD25−CD4+ T cells
Purified CD25−CD4+ T cells (purity > 99%) from healthy donors were co-cultured with 20 000-rad irradiated MT-2 cells in RPMI 1640 medium supplemented with 10% FCS. Two months later, live cells were collected and cultured at limiting dilution in the presence of IL-2, to establish HTLV-I-infected cell clones. Two clones (4-2 and 4-20) were obtained in two independent infections. These cells were confirmed to be positive for HTLV-I infection by immunostaining with the GIN14 mAb, which recognizes HTLV-I core proteins (6).
Real-time PCR
Total RNA was isolated by Trizol Reagent (Invitrogen, Carlsbad, CA), and converted to cDNA by the SuperScript III First-Strand Synthesis System for RT-PCR kit (Invitrogen) using an oligo(dT)15 primer. To detect Foxp3, the TaqMan probe and primer was used. Real-time PCR was performed using an ABI 7700 Sequence Detection System (Applied Biosystem, Foster, CA). The expression of each gene was normalized to the copies of GAPDH mRNA from the same sample.
Detection of tax gene
Total RNA was isolated from ATL cells and JPX9 cells as described above, and treated with DNase I (Sigma, US) before RT. cDNA was generated from the extracted total RNA as described above. Primers for tax (19) are as follows: forward primer 5′-CCCACTTCCCAGGGTTTGGACAGA, reverse primer 5′-CTGTAGAGCTGAGCCGATAACGCG. Primers for GAPDH are as follows: forward primer 5′-CCACATCGCTCAGACACCAT, reverse primer 5′-GCCATCACGCCACAGTTTCC.
Results
Phenotypic analysis of ATL cells
To confirm recent observations, we first examined the expression of Foxp3 in leukemia cells isolated from patients with ATL, by real-time PCR. The transcript level of Foxp3 in the ATL cells of 5 of 12 patients (patients 1, 6, 7, 8, and 9) was comparable to the level in freshly isolated Treg cells (Table 2 and Fig. 1A). Similar results were obtained in three recent reports (10–12). In addition, the average Foxp3 expression in ATL cells was significantly higher than that in activated conventional CD4+ T cells, in which low Foxp3 expression was observed (Fig. 1A). Furthermore, no Foxp3 expression in the leukemia cells from a patient with acute T lymphocytic leukemia (T-ALL) could be detected. Thus, from a phenotypic standpoint, we concluded that the ATL cells from more than one-third of the patients we examined resembled Treg cells rather than activated T cells.
![ATL cells are phenotypically and functionally similar to Treg cells. (A) Relative expression of the Foxp3 transcript in ATL cells. Total RNA was isolated from ATL (n = 12), Treg (n = 6), freshly isolated CD25−CD4+ T (n = 4) and activated CD25−CD4+ T (n = 6) cells by Trizol Reagent (Invitrogen, Carlsbad, CA), and converted to cDNA. Activated T cells were prepared by culturing of CD25−CD4+ T cells for 2 days in the presence of coated anti-CD3 mAb (5 μg ml−1) and soluble anti-CD28 mAb (2 μg ml−1). The relative expression of the Foxp3 transcript was estimated by real-time PCR. As an endogenous control, a set of primers and a probe specific for human GAPDH mRNA (Applied Biosystems) was used. In each amplification, the cDNAs were quantified using relative standard curves. All results were normalized with respect to the internal control. The significance of the data was evaluated by Student's t-test (* represents p < 0.05). (B) Suppressive activity of ATL cells. CD25−CD4+ T cells (5 × 104) were stimulated with anti-CD3 mAb in the presence of DCs. ATL cells or Treg cells and DCs were added at the indicated ratio directly to the culture or were placed in trans-well chambers. [3H]TdR incorporation of cells cultured in the lower well during the last 4 h of a 5-day culture was measured as an indicator of cell proliferation and is expressed as the mean (±SD) of triplicate cultures. This figure shows four representative ATL samples, including suppressive ATL cells from patients 1, 2 and 9, and non-suppressive ATL cells from patient 7. (C) Suppressive activity of ATL cells. Co-culture and trans-well experiments using CFSE-labeled CD25−CD4+ T cells were performed as described above. The CFSE intensity of CD25−CD4+ T cells after a 5-day culture was measured with a flow cytometer. The frequency (%) of cells failing to undergo cell division is indicated in the figure. Representative results of the suppressive ATL cells from patients 1, 2 and 9, and non-suppressive ATL cells from patient 7 are shown. (D) The neutralization of regulatory molecules did not affect the ATL-cell-mediated suppression. CD25−CD4+ T cells (5 × 104) were stimulated with anti-CD3 mAb in the presence of Treg cells (5 × 104) or ATL cells from patient 9 (5 × 104 or 2.5 × 105). Neutralizing antibodies (10 μg ml−1) for CTLA-4, TGFβ or anti-IL-10 were added to the cultures. [3H]TdR incorporation during the last 4 h of a 5-day culture was measured as an indicator of cell proliferation and is expressed as the mean (±SD) of triplicate cultures. Similarly, addition of these neutralizing antibodies did not affect the suppression mediated by ATL cells from patients 1 and 2 (data not shown).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/intimm/18/2/10.1093/intimm/dxh366/2/m_intimmdxh366f01_ht.gif?Expires=1716345823&Signature=VTqLVbX-kDMV0wazFy2eVn8FiZjf75BEuwhIkRlY7c6ROGD282VNma9P1fXpRhuOjUuldAIsrV9RjJOyOzyFIHCCXeHLNAVcdkrkbH9xe02lLlnPvK94YciF2sF0Gq8bJen8QibpRsfBi3siG7kXzavn7~J3uWxo4MFvcJwC3nogWEocbnQattgRRf5skPSY~Vk~Qr5ww-ug~kX2D1rHQs~zUwFXSWCmTu60VJZWyFeGz-26tU4x6AqA4EP95Tsvhu1-6jrWurEsm4bcUKCb34mGoJ2jJtLbmErL-MP7jd9HHqhuTG38Aywqlj1OId6EoeNTWaBLejsgfI-dQRCWtw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
ATL cells are phenotypically and functionally similar to Treg cells. (A) Relative expression of the Foxp3 transcript in ATL cells. Total RNA was isolated from ATL (n = 12), Treg (n = 6), freshly isolated CD25−CD4+ T (n = 4) and activated CD25−CD4+ T (n = 6) cells by Trizol Reagent (Invitrogen, Carlsbad, CA), and converted to cDNA. Activated T cells were prepared by culturing of CD25−CD4+ T cells for 2 days in the presence of coated anti-CD3 mAb (5 μg ml−1) and soluble anti-CD28 mAb (2 μg ml−1). The relative expression of the Foxp3 transcript was estimated by real-time PCR. As an endogenous control, a set of primers and a probe specific for human GAPDH mRNA (Applied Biosystems) was used. In each amplification, the cDNAs were quantified using relative standard curves. All results were normalized with respect to the internal control. The significance of the data was evaluated by Student's t-test (* represents p < 0.05). (B) Suppressive activity of ATL cells. CD25−CD4+ T cells (5 × 104) were stimulated with anti-CD3 mAb in the presence of DCs. ATL cells or Treg cells and DCs were added at the indicated ratio directly to the culture or were placed in trans-well chambers. [3H]TdR incorporation of cells cultured in the lower well during the last 4 h of a 5-day culture was measured as an indicator of cell proliferation and is expressed as the mean (±SD) of triplicate cultures. This figure shows four representative ATL samples, including suppressive ATL cells from patients 1, 2 and 9, and non-suppressive ATL cells from patient 7. (C) Suppressive activity of ATL cells. Co-culture and trans-well experiments using CFSE-labeled CD25−CD4+ T cells were performed as described above. The CFSE intensity of CD25−CD4+ T cells after a 5-day culture was measured with a flow cytometer. The frequency (%) of cells failing to undergo cell division is indicated in the figure. Representative results of the suppressive ATL cells from patients 1, 2 and 9, and non-suppressive ATL cells from patient 7 are shown. (D) The neutralization of regulatory molecules did not affect the ATL-cell-mediated suppression. CD25−CD4+ T cells (5 × 104) were stimulated with anti-CD3 mAb in the presence of Treg cells (5 × 104) or ATL cells from patient 9 (5 × 104 or 2.5 × 105). Neutralizing antibodies (10 μg ml−1) for CTLA-4, TGFβ or anti-IL-10 were added to the cultures. [3H]TdR incorporation during the last 4 h of a 5-day culture was measured as an indicator of cell proliferation and is expressed as the mean (±SD) of triplicate cultures. Similarly, addition of these neutralizing antibodies did not affect the suppression mediated by ATL cells from patients 1 and 2 (data not shown).
Phenotypic and functional analysis of ATL cells
Case ID | Relative Foxp3 expression | Suppressive activity | Expression of Tax |
|---|---|---|---|
| 1 | 12 | + | + |
| 2 | 1.1 | + | + |
| 3 | 2.3 | − | − |
| 4 | 0.32 | − | + |
| 5 | 2.7 | − | − |
| 6 | 6.7 | − | − |
| 7 | 34 | − | − |
| 8 | 9.8 | ND | − |
| 9 | 11.1 | + | + |
| 10 | 0.6 | − | − |
| 11 | 0.4 | ND | − |
| 12 | 2.3 | − | − |
| T-ALL | 0.16 | − | − |
Case ID | Relative Foxp3 expression | Suppressive activity | Expression of Tax |
|---|---|---|---|
| 1 | 12 | + | + |
| 2 | 1.1 | + | + |
| 3 | 2.3 | − | − |
| 4 | 0.32 | − | + |
| 5 | 2.7 | − | − |
| 6 | 6.7 | − | − |
| 7 | 34 | − | − |
| 8 | 9.8 | ND | − |
| 9 | 11.1 | + | + |
| 10 | 0.6 | − | − |
| 11 | 0.4 | ND | − |
| 12 | 2.3 | − | − |
| T-ALL | 0.16 | − | − |
Phenotypic and functional analysis of ATL cells
Case ID | Relative Foxp3 expression | Suppressive activity | Expression of Tax |
|---|---|---|---|
| 1 | 12 | + | + |
| 2 | 1.1 | + | + |
| 3 | 2.3 | − | − |
| 4 | 0.32 | − | + |
| 5 | 2.7 | − | − |
| 6 | 6.7 | − | − |
| 7 | 34 | − | − |
| 8 | 9.8 | ND | − |
| 9 | 11.1 | + | + |
| 10 | 0.6 | − | − |
| 11 | 0.4 | ND | − |
| 12 | 2.3 | − | − |
| T-ALL | 0.16 | − | − |
Case ID | Relative Foxp3 expression | Suppressive activity | Expression of Tax |
|---|---|---|---|
| 1 | 12 | + | + |
| 2 | 1.1 | + | + |
| 3 | 2.3 | − | − |
| 4 | 0.32 | − | + |
| 5 | 2.7 | − | − |
| 6 | 6.7 | − | − |
| 7 | 34 | − | − |
| 8 | 9.8 | ND | − |
| 9 | 11.1 | + | + |
| 10 | 0.6 | − | − |
| 11 | 0.4 | ND | − |
| 12 | 2.3 | − | − |
| T-ALL | 0.16 | − | − |
Suppressive activity of ATL cells
We next examined the suppressive activity of ATL cells on the proliferation and cell division of CD25−CD4+ (conventional CD4) T cells by determining the levels of DNA synthesis and CFSE intensity, respectively. When cultured at an equal ratio, ATL cells from patients 1, 2 and 9 induced moderate inhibition of the CD3-stimulated CD25−CD4+ T cell proliferative responses that was less than the inhibition induced by Treg cells (Fig. 1B). In addition, the frequency of cells failing to undergo cell division was higher when ATL cells from patients 1 and 9 were co-cultured (16 and 15%, respectively) than when CD25−CD4+ T cells alone were added (9%) (Fig. 1C). Furthermore, ATL-cell-induced suppression was markedly enhanced when the ratio of the ATL cells from patients 1 and 9 to CD25−CD4+ T cells was increased 5-fold (Fig. 1B and C). Interestingly, patient 7's ATL cells, which had the highest Foxp3 expression, did not affect the proliferation or cell division of conventional CD4 T cells, even when a higher ratio of ATL cells was added to the co-culture (Fig. 1B and C).
Mechanisms responsible for the ATL-cell-mediated suppression
It has been reported that, following in vitro culture, ATL cells secrete the regulatory cytokines IL-10 and TGFβ (19, 20). On the other hand, Treg cells can mediate suppressive activity in a cytokine-independent manner (7). We therefore examined whether a panel of inhibitory cytokines or factors could mediate the ATL-cell-induced suppression. The addition of neutralizing antibodies for CTLA-4, TGFβ or IL-10 to a co-culture of ATL cells from patient 9 with responder T cells did not affect the inhibitory activity of the ATL cells (Fig. 1D). These results indicate that a cytokine-independent mechanism may be responsible for the suppression. Based on the above results, we reasoned that ATL-cell-induced suppression may mimic the cell–cell contact-dependent mechanism by which Treg cells mediate suppression. To test this possibility, we next performed a series of trans-well experiments. Separating ATL cells from the CD25−CD4+ responder T cells abolished the suppressive activity of the ATL cells from patients 1 and 9 (Fig. 1B and C). Note, however, that the suppression caused by the ATL cells from patient 2 was not fully abrogated by separation from the responder T cells (Fig. 1B and C).
Foxp3 expression and the suppressive activity of HTLV-I-infected cell lines
We also measured the Foxp3 expression in some ATL cell lines (EDγ-16 and TL-Om1) and in vitro-established HTLV-I-infected cell lines (MT-2, ILT-Mat, TCL-Mor, TCL-Kan). Except for MT-2, none of these cell lines expressed Foxp3 mRNA (Fig. 2A). We then examined the suppressive activity of these cell lines by co-culturing them with CFSE-labeled CD25−CD4+ T cells. The cell division profiles of CD25−CD4+ T cells co-cultured with MT-2 were similar to those obtained with Treg cells (Fig. 2B). In contrast, the TL-Om1 (Fig. 2B and C) cells and other cell lines did not inhibit cell division and cell proliferation (data not shown). Furthermore, to investigate the mechanism for the suppressive activity of MT-2, we performed a trans-well experiment. As we had seen with the Treg cells, the separation of the MT-2 from the CD25−CD4+ T cells abolished the MT-2-cell-induced suppression of the division of conventional CD4 T cells and restored the CD4 T cell number (Fig. 2B and C). In addition, as with ATL-induced suppression, none of the neutralizing antibodies for CTLA-4, TGFβ or IL-10 changed the inhibitory activity of the MT-2 cells (data not shown).
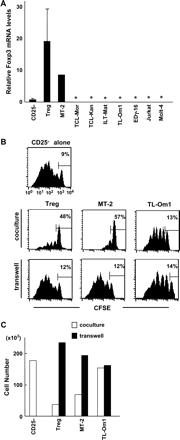
Foxp3 expression and suppressive activity of HTLV-I-infected cell lines. (A) Foxp3 expression in ATL, HTLV-I-infected, and non-infected T cell lines. Foxp3 expression in T cell lines was determined by real-time PCR as described in Fig. 1A. The mRNA expression of Foxp3 was normalized to the copies of GAPDH mRNA from the same sample. Asterisks represent undetectable levels of the Foxp3 transcript. (B) Suppressive activity of HTLV-I-infected cell lines. CFSE-labeled CD25−CD4+ T cells (5 × 104) were stimulated with soluble anti-CD3 mAb in the presence of irradiated DCs. Treg cells, MT-2 cells or TL-Om1 cells (2.5 × 104 each) were added directly to the cultures or placed into trans-well chambers. After 5 days of culture, the CFSE intensity was measured by FACS as an indicator of cell division. The frequency (%) of cells failing to undergo cell division is indicated in the figure. (C) Trans-well separation abrogates the MT-2-cell-associated suppression of CD4 T cell proliferation. The same experiments were performed as described in (B). The number of live CFSE-labeled CD4+ T cells was counted 5 days after CD3 stimulation. Similar results were obtained in three independent experiments.
Forced expression of Foxp3 failed to produce suppressive activity in HTLV-I-infected cell lines
Since the Treg-like ATL cells (from patients 1 and 9) and MT-2 cell lines, all of which expressed high Foxp3 expression, showed regulatory activity, we next addressed the role of Foxp3 in the suppressive function of ATL cells. An ATL cell line (EDγ-16) and an HTLV-I-infected T cell line (ILT-Mat) were transfected with the Foxp3 gene using a retroviral vector (Fig. 3A). Although deliberate Foxp3 expression converts primary conventional CD4 T cells to Treg-like suppressor cells in vitro (7), Foxp3 expression in these ATL and HTLV-I-infected cell lines failed to induce any apparent suppressive function (Fig. 3B). These results suggest that Foxp3 expression is not sufficient to elicit the Treg cell function in ATL and HTLV-I-infected cells. This idea is supported by the negligible suppressive activity of the ATL cells from patient 7, even though they expressed the highest Foxp3 level among the ATL cells used in this study (Table 2, Fig. 1B and C).
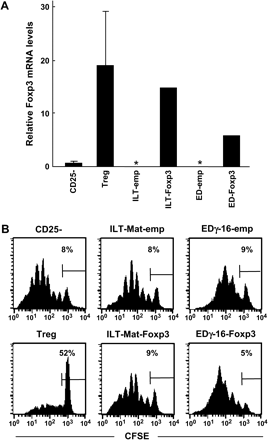
Forced expression of Foxp3 cannot convert HTLV-I-infected cells to Treg-like suppressive cells. (A) Foxp3 expression in HTLV-I-infected cell lines transfected with the Foxp3 gene. Foxp3 expression in Foxp3-transfected ILT-Mat and EDγ-16 cells was determined in comparison with that in empty-vector-transfected cells by real-time PCR, as described in Fig. 1A. The mRNA expression of Foxp3 was normalized to the copies of GAPDH mRNA from the same sample. Asterisks represent undetectable levels of the Foxp3 transcript. (B) Suppressive activity of Foxp3-transfected cell lines. Co-culture of CFSE-labeled CD25−CD4+ T cells (5 × 104) with the Foxp3-transfected cell lines (2.5 × 104) was performed as described in Fig. 2(B). After 5 days of culture, the CFSE intensity was measured by FACS, as an indicator of cell division. The frequency of cells failing to undergo cell division is indicated in the figure.
T cell activation could not induce Foxp3 expression
Several reports suggest that activated T cells that are stimulated under certain conditions acquire Foxp3 expression (21, 22). Others, however, have been unable to reproduce the Foxp3 induction in these activated T cells (23). We therefore examined the Foxp3 induction in primary activated CD4 T cells, which we stimulated in a variety of ways, according to previous reports, and failed to detect any obvious Foxp3 induction in these activated T cells (Fig. 4A), in agreement with Yagi et al. (23).
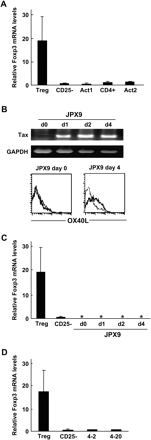
Foxp3 expression was not induced during T cell activation or HTLV-I infection. (A) Foxp3 expression was not be induced in activated T cells. Several recent reports demonstrated that certain procedures for T cell activation can induce Foxp3 expression. Therefore, according to the methods described in these papers, CD25−CD4+ T cells were activated with coated anti-CD3 and soluble anti-CD28 for 1 day and then cultured in the absence of stimulants for three more days (Act1) (21). Alternatively, CD4+ T cells were stimulated with allogeneic APCs in the presence of IL-2 and TGFβ for 6 days (Act2) (23). Foxp3 expression in Treg cells and activated CD4+ T cells before (CD25− or CD4+) and after stimulation (Act 1–2) was measured by real-time PCR as described in Fig. 1A. (B) Tax induction in Jurkat cells. JPX9 cells were treated with 30 μM CdCl2. Tax expression and induction of OX40L, a target for tax, were confirmed at the indicated days after treatment. Thick and thin lines in the lower panels represent OX40L staining and control staining, respectively. (C) HTLV-I tax induction did not affect the Foxp3 expression in Jurkat cells. JPX9 cells were collected at the indicated days after stimulation, and their Foxp3 expression was measured by real-time PCR, as described above. Asterisks represent undetectable levels of the Foxp3 transcript. (D) HTLV-I infection of CD25−CD4+ T cells does not change their Foxp3 expression. The Foxp3 expression in freshly isolated CD25−CD4+ T cells and two independent HTLV-infected cell clones (4-2 and 4-20) that were established from purified CD25−CD4+ T cells by co-culture with MT-2 cells was estimated by real-time PCR, as described above.
HTLV-I Tax expression or HTLV infection failed to induce Foxp3 expression in T cells
Since the tax gene in HTLV-I has been considered an essential transactivator for the tumorigenesis of HTLV-I-infected T cells through its induction of several cellular genes (2), we also examined whether Tax could induce Foxp3 expression. Although tax gene induction in a human T cell line, Jurkat, induced the surface expression of OX40L (Fig. 4B), which is a good target for Tax (24, 25), Tax could not induce Foxp3 expression in Jurkat cell lines (Fig. 4C). Finally, to examine whether HTLV-I infection could induce Foxp3 expression in conventional CD4 T cells, purified CD25−CD4+ T cells were infected with HTLV-I. Figure 4(D) shows that HTLV-I infection did not change the Foxp3 expression in two independent CD25−CD4+ T cell clones. These results suggest that HTLV-I infection may not be associated with high Foxp3 expression in ATL cells.
Discussion
In the present study we detected high Foxp3 expression, which is a good marker for CD25+CD4+ Treg cells, in 5 of 12 ATL cell isolates (Table 2). In addition, two of the five Foxp3-expressing ATL cell isolates possessed Treg-like characteristics, not only in having high Foxp3 expression but also in having a suppressive function on CD25−CD4+ T cells that is mediated by cell–cell contact (Fig. 1B and C). Although four recent papers have reported Foxp3 expression in ATL cells (10–13), here we show the first evidence that ATL cells have a Treg-like regulatory function. Furthermore, MT-2 cells, which express Foxp3 at high levels, also exhibited Treg-like suppressive activity. These results indicate that some ATL cells have an immunosuppressive activity resembling that of Treg cells. Considering that cellular immunodeficiency is observed in a quarter of patients with ATL (2–4), we hypothesize that immunosuppressive activity by ATL cells may contribute to the immunodeficiency. The immunodeficiency-associated infection seen in two patients (patients 1 and 2) (Table 1), whose cells were suppressive, may support this idea, although the suppressive mechanism of the ATL cells from patient 2 might be different from that of Treg cells. Although it may seem at first glance that our experimental system is artificial because it used four times more ATL cells than Treg cells to show the suppressive activity, many patients have a large number of ATL cells, which could be sufficient to provide the suppressive activity in vivo. As shown in Table 1, leukemia cells account for ∼90% of the PBMCs in most patients with ATL, in contrast to the much lower population (1–2% of PBMCs) of primary Treg cells in healthy donors. Thus, when the ATL cells have Treg-like suppressive effects, they may contribute to the immunosuppression in patients with ATL, because of their high population in PBMCs.
In both humans and mice, the retroviral gene transfer of Foxp3 converts conventional CD4 T cells (CD25−CD4+ T cells) to Treg-like suppressor cells (23, 26). Thus, Foxp3 expression has been thought to be essential and sufficient for Treg cell function (7). In our study, two ATL cells (from patients 1 and 9) and one HTLV-I-infected cell line (MT-2), all of which showed high Foxp3 expression, had Treg-like suppressive activity that is mediated by a cell–cell contact mechanism (Fig. 1B and C). In contrast, ATL cells (from patient 2) with a lower level of Foxp3 expression were able to suppress T cell responses even in the presence of a transmembrane (Fig. 1B and C). While a close association between Foxp3 expression and Treg-like suppressive activity can be inferred from this result, it is clear nevertheless that ATL cells derived from patient 2 may harbor an alterative mechanism of suppression. To directly address the functional association between Foxp3 expression and suppressive activity of ATL cells, deliberate suppression of Foxp3 expression in ATL cells such as the use of siRNAs may be required. However, this may be challenging as freshly isolated ATL cells do not proliferate in vitro and are therefore resistant to any gene-transfer treatment. In addition, to our knowledge, no study has demonstrated that deliberate knock-down of Foxp3 expression in Treg cells abrogates their suppressive activity. A recent report by Sereti et al. shows that overexpression of two Foxp3 isoforms on CD25−CD4+ T cells, could induce the generation of phenotypic Treg cells but with poor suppressive activity (27). Thus, functional roles of Foxp3 expression for Treg-like suppression by not only ATL cells but also by Treg cell remain completely unresolved.
Some of the ATL cells (from patients 6 and 7), in spite of their high Foxp3 expression, did not show any inhibitory activity. In addition, the forced expression of Foxp3 in non-Foxp3-expressing T cell lines could not confer the suppressive activity (Fig. 3B). These results indicate that Foxp3 is not sufficient for the suppressive activity of ATL cells, ATL cell lines and HTLV-I-infected cell lines. Other factor(s) in addition to Foxp3 may be essential for the suppressive function. During a long neoplastic process, such factor(s) might be lost by the ATL cells, rendering them non-suppressive in spite of their high Foxp3 expression.
With regard to the origin of ATL cells, we speculate that there might be at least two distinct subsets of CD25+ ATL cells in terms of Foxp3 expression. The ones with Treg-like features may be derived from primary Foxp3-expressing cells, because HTLV-I infection, Tax induction and other mitogenic stimulations of T cells could not induce Foxp3 expression in our system. Furthermore, Tax expression does not correlate with Foxp3 expression in ATL cells (Table 2). In contrast, the leukemia cells from some patients with ATL (patients 4, 10 and 11) showed a low level of Foxp3 and negligible regulatory activity, which resemble the characteristics of activated T cells. In addition, the TL-Om1 and EDγ-16 cell lines, which were derived from ATL cells, demonstrated neither high Foxp3 expression nor the suppressive function. Thus, another subset of ATL cells may be derived from activated T cells, as previously thought. An interesting question is whether the origins of ATL cells can be classified into two different T cell types, activated T and Treg cells. The analysis of in vitro-established HTLV-I-infected cell lines can help us explore the origin of ATL cells. Most of the HTLV-I-infected cell lines were established by using whole peripheral T cells, including both Treg and non-Treg T cells, as the infection targets, which is closer to the physiological condition than the use of an isolated cell population. Among the six HTLV-I-infected cell lines we checked, MT-2 cells expressed Foxp3 and harbored the Treg-cell-like function, suggesting that MT-2 cells may have originated from Treg cells. Our preliminary results demonstrated that HTLV-I could infect purified CD25+CD4+ T cells and that the HTLV-I-infected Treg cells still showed high Foxp3 expression (data not shown). In contrast, HTLV-I infection failed to induce Foxp3 expression in purified CD25−CD4+ T cells (Fig. 4D). These results support the idea that ATL and HTLV-I-infected cell lines that sustain high Foxp3 expression may be derived from Treg cells. Since Treg cells are anergic to stimulation with antigen, HTLV-I, which is a typical retrovirus, may selectively infect proliferating conventional CD4 T cells in vitro. This is the likely reason so few HTLV-I-infected Treg cell lines were obtained. To investigate properly the leukemogenesis of Treg cells as the origin of ATL cells, appropriate methods for the HTLV-I infection of Treg cells may be essential, although it may be difficult to render Treg cells susceptible to HTLV-I retrovirus by inducing cell division. Nevertheless, further studies on the cellular origins of ATL cells will improve our understanding of how ATL and its associated immunosuppression develop in human patients.
Transmitting editor: T. Saito
This work was supported in part by a grant-in-aid for scientific research on priority areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and a grant-in-aid for scientific research on priority areas from the Japan Society for the Promotion of Science.
References
Hinuma, Y., Nagata, K., Hanaoka, M. et al.
Uchiyama, T.
Shimoyama, M.
Yasunaga, J., Sakai, T., Nosaka, K. et al.
Sugamura, K., Fujii, M., Kannagi, M., Sakitani, M., Takeuchi, M. and Hinuma, Y.
Sakaguchi, S.
Shevach, E. M.
Ndhlovu, L. C., Takeda, I., Sugamura, K. and Ishii, N.
Karube, K., Ohshima, K., Tsuchiya, T. et al.
Ishida, T., Iida, S., Akatsuka, Y. et al.
Matsubara, Y., Hori, T., Morita, R., Sakaguchi, S. and Uchiyama, T.
Kohno, T., Yamada, Y., Akamatsu, N. et al.
Sugamura, K., Nakai, S., Fujii, M. and Hinuma, Y.
Miyoshi, I., Kubonishi, I., Yoshimoto, S. and Shiraishi, Y.
Maeda, M., Shimizu, A., Ikuta, K. et al.
Ishii, N., Asao, H., Kimura, Y. et al.
Nagata, K., Ohtani, K., Nakamura, M. and Sugamura, K.
Mori, N., Gill, P. S., Mougdil, T., Murakami, S., Eto, S. and Prager, D.
Niitsu, Y., Urushizaki, Y., Koshida, Y. et al.
Walker, M. R., Kasprowicz, D. J., Gersuk, V. H. et al.
Zheng, S. G., Wang, J. H., Gray, J. D., Soucier, H. and Horwitz, D. A.
Yagi, H., Nomura, T., Nakamura, K. et al.
Miura, S., Ohtani, K., Numata, N. et al.
Higashimura, N., Takasawa, N., Tanaka, Y., Nakamura, M. and Sugamura, K.
Hori, S., Nomura, T. and Sakaguchi, S.
Author notes
3Department of Dermatology, 4Department of Rheumatology and Hematology and 5Institute for Animal Experimentation, Tohoku University Graduate School of Medicine, Sendai 980-8575, Japan

{kind=link}
{kind=link}
{kind=link}
{kind=link}