





Abstract
Reliability of genotyping is an issue for studies using non-invasive sources of DNA. We emphasize the importance of refining DNA extraction methods to maximize reliability and efficiency of genotyping for such DNA sources. We present a simple and general method to quantitatively compare genotyping reliability of various DNA extraction techniques and sample materials used. For bighorn sheep (Ovis canadensis) fecal samples we compare different fecal pellet materials, different amounts of fecal pellet material, and the effects of eliminating two DNA extraction steps for four microsatellite loci and four samples heterozygous at each locus. We evaluated 192 PCR outcomes for each treatment using indices of PCR success and peak height (signal strength) developed from analysis output of sequencer chromatograms. Outermost pellet material produced PCR results almost equivalent to DNA extracted from blood. Where any inner pellet material was used for DNA extraction, PCR results were poorer and inconsistent among samples. PCR success was not sensitive to amount of pellet material used until it was decreased to 15 mg from 60 mg. Our PCR index provides considerably more information relative to potential genotyping errors than simply comparing genotypes derived from paired fecal and blood or tissue samples. Our DNA extraction method probably has wide applicability to herbivores that produce pelleted feces where samples dry rapidly after deposition.
Feces as a source of DNA is attractive because of the ease of sampling and the potential for unprecedented sample sizes. While recent studies using nuclear DNA from feces have established the viability of this approach (Ernest et al 2000; Kohn et al. 1999; Taberlet et al. 1996, 1997; Wasser et al. 1999), some also have demonstrated potential pitfalls, involving significant percentages of PCR reactions that fail or produce incorrect results due to allelic dropout (the loss of one allele for heterozygous loci) or false alleles (Huber et al. 2002, 2003; Taberlet et al. 1996, 1999; Taberlet and Luikart 1999), often necessitating large numbers of replicate PCRs for reliable genotyping. In contrast, the use of feces as a source of mitochondrial DNA appears relatively uncomplicated due to the high ratio of mitochondria to nuclei (Kovach et al. 2003). Methodological studies concerning application of fecal DNA to nuclear markers have had two basic focuses: how to avoid the inherent problems with better DNA extraction methods (Fernanado et al. 2003, Flagstad et al. 1999) and how to maximize the probability of obtaining correct genotypes when faced with significant rates of allelic dropout and false alleles (Creel et al. 2003, Miller et al. 2002, Taberlet et al. 1996). Here we emphasize the importance of the former approach and provide an example and indices useful for evaluating different extraction procedures relative to reliability of genotypes obtained.
Our example involves bighorn sheep (Ovis canadensis), an herbivore for which blood and tissue samples are mostly difficult and costly to obtain. Herbivores potentially pose a fecal DNA challenge because of inhibitory effects of plant secondary compounds on PCR reactions, and PCR success may depend on maximizing the concentration of intact DNA from the target animal, while minimizing secondary compounds derived from plants in the diet (Fernando 2003). Flagstad et al. (1999) described a simple and successful method using magnetic beads to extract DNA from intestinal cells washed from the outside surface of feces of mid-sized herbivores. They compared their method with alternative fecal pellet material and other extraction methods, none of which were as successful as their method. An alternative extraction method they tested utilized guanadinium thiocyanate and silica as incorporated in commercial extraction kits. We have used such kits to extract DNA from bighorn sheep feces since 1998 and have refined our methods to where reliability is very high and comparable to the methods proposed by Flagstad et al. (1999), as well as to DNA extracted from blood.
Here we test our approach by evaluating the effects of varying which fecal material is used and different DNA extraction procedures on PCR success. The DNA extraction steps we test are ones we have added to a commercial procedure that has appeared to improve PCR success. The tests we present offer a method for researchers to evaluate the success of extraction methods using feces or other noninvasive approaches.
One standard test is to verify that DNA derived from feces yields the same genotypes as DNA derived from blood or tissue from the same animal (Fernando et al. 2003, Flagstad et al. 1999). However, where this comparison involves fecal samples manually removed from the colon of captured animals, results may not represent field-collected samples due to the disruption of the fecal pellet surface and the colon wall during sample collection. While we provide such a comparison, a more important question concerns the proportion of PCRs from field-collected samples that produce correct genotypes and the resultant number of replicate PCRs needed to produce reliable results (Taberlet et al. 1996). We consider the other tests we present as more useful for evaluating DNA extraction methods, and an alternative to tests involving quantitative PCR (Morin et al. 2001). It is our contention that efforts made to optimize DNA extraction methods will yield a high payoff by reducing needed replications of DNA extractions and PCRs and minimizing data analytical procedures to correct for genotype uncertainties.
Methods
DNA Extraction Procedure
For blood samples we have extracted DNA from the buffy coat using procedures in the Qiagen blood and tissue kit (TM). For fecal samples we have used Qiagen stool minikits (TM) with some modifications. It appears to be widely recognized that the outermost fecal material may yield the least degraded DNA and the lowest concentration of PCR inhibitors (Fernando et al. 2003, Flagstad et al. 1999). We also have focused on that fecal material for the same reason. Flagstad et al. (1999) used 100 mg of homogenized fecal material in one of their tests that produced poor results. We have encountered problems, including persistent pigment, in DNA extractions using that amount of material. Our method begins instead with 60 mg carefully scraped with a razor blade from the outer mucosal layer of bighorn sheep fecal pellets in 2ml screwtop microcentrifuge tubes. To this we add 1 mm zirconia/silica beads (Biospec Products) and 1.6 ml of the initial extraction buffer (ASL). We then use a bead-beater8 (Biospec Products) to homogenize the material at medium speed for 45s followed by full speed (3300 rpm; Miller et al. 1999) for 15s. High speed bead-beating was limited to 15s to avoid degrading DNA (Miller et al. 1999). Bead-beating is followed by heating for 10 min at 56°C. The Qiagen kit procedure is then followed with two alterations: (1) after addition of ethanol the samples are left for 1h to maximize DNA precipitation; and (2) the elution buffer (AE) is heated to 70°C, allowed to incubate in the spin columns for 3 min, and the eluate is centrifuged through the spin columns a second time to maximize DNA collection.
DNA concentration was not quantified. We avoided contamination by (1) using aerosol resistant pipettor tips for critical procedures in DNA extractions and PCR preparations, (2) setting up PCRs in an appropriate clean hood, and (3) preparing fecal samples in a separate location from PCR setups with glove changes between samples.
Comparisons With Blood
We used the following 11 variable microsatellite markers and annealing temperatures to compare results of our fecal extraction method with DNA extracted from blood for 7 wild bighorn sheep from the Sierra Nevada of California using paired samples collected from the same captured sheep: OarHH47 (Henry et al. 1993) multiplexed with MAF209 (Buchanan and Crawford 1992a; 51°C); OarCP20 (Ede et al. 1995; 55°C); MAF33 (Buchanan and Crawford 1992b; 55°C); MAF48 (Buchanan et al. 1991; 55°C); OarHH62 (Ede et al. 1994) multiplexed with OarFCB11 (Buchanan and Crawford 1993; 56°C); MAF36 (Swarbrick et al. 1991) multiplexed with OarFCB304 (Buchanan and Crawford 1993; 56°C); and MAF65 (Buchanan et al. 1992) multiplexed with OarAE16 (Pentry et al. 1993; 61°C). The range of allele sizes was 80–143 bp. Two PCRs were run for each blood and each fecal sample, and two additional PCRs of each were run to resolve any discrepancies. PCRs were set up as 20μL reactions, but received an extra 1.2μL of water to counteract dry down. Reaction conditions were: 0.52μL DNA extract, 1× PCR buffer (Applied Biosystems), 2.25mM MgCl2, 160μM dNTPs, 8 μg bovine serum albumin (New England BioLabs), 80nM each primer, and 0.7 units of Amplitaq Gold DNA polymerase (Applied Biosystems). To minimize pipetting error, DNA was added as 5.2 μL of DNA diluted 1:10. PCRs were carried out in 96 well polycarbonate plates with silicone sealing mats (Costar) carefully sealed in each well followed by sealing the entire mat to the plate with Scotch Magic Tape (TM). Cycling was carried out on an Eppendorf Mastercycler gradient thermocycler (Brinkman Instruments) with 96°C heated lid under conditions of 7.5 min at 93°C followed by 40 cycles of 95°C for 30s, 51–61°C depending on locus (see above) for 30s, and 72°C for 30s, with a final 2 min at 60°C. Forward primers contained flourescent dye labels and PCR products were electrophoresed on an ABI PRISM 377 DNA sequencer (Applied Biosystems) using Singel Long Ranger burst packs for the gel matrix and tamra 350 size standards (Applied Biosystems). Varying amounts of PCR products by locus were combined based on previous experience to run multiple loci per lane and dried completely. Each well then received 1.5 μL formamide, 0.35 μL tamra 350 size standards (Applied Biosystems), and 0.30 μL dextran dye (Applied Biosystems), was heated to 90°C for 2 min, immediately cooled to <0°C, and 1.5μl was loaded in each lane of the sequencer. Chromatograms were analyzed using the program GeneScan 3.0 (Perkin-Elmer Applied Biosystems). Our criterion for scoring alleles was the existence of characteristic microsatellite stutter bands.
Fecal DNA Extraction Experiments
We conducted four extraction experiments, each with three treatments (Table 1) in which we varied (1) what part of the fecal pellet was used, (2) how much fecal material was used, and (3) procedures used in the extraction process. One experiment used the full extraction process and compared three different fecal pellet materials: scraped outer mucosal layer; inner material that included none of the outer mucosal layer; and whole pellet that included outer and inner material taken as a slice of the pellet perpendicular to its long axis. The second experiment varied the amount of pellet material used (60mg, 30 mg, and 15 mg) for the full process applied to the outer pellet material. The third and fourth experiments both compared the full extraction process with the elimination of 1 and 2 steps of the extraction process (1h ethanol precipitation; 1h ethanol precipitation and bead-beating), one for scraped outer pellet material, and the other for whole pellet material. Because the two treatments involving the full extraction process applied to outer and whole pellet material were used in multiple experiments, the total number of different treatments was nine (Table 1; Figures 1 and 2).
Summary of the four experiments conducted
Experiment | Constant Features | Treatments | ||||
|---|---|---|---|---|---|---|
| 1 | full process, 60mg | outer pellet | whole pellet | inner pellet | ||
| 2 | outer pellet, full process | 60mg | 30mg | 15mg | ||
| 3 | outer pellet, 60mg, | full process | no extended DNA precipitation | no extended DNA precipitation or beadbeating | ||
| 4 | whole pellet, 60mg, | full process | no extended DNA precipitation | no extended DNA precipitation or beadbeating | ||
Experiment | Constant Features | Treatments | ||||
|---|---|---|---|---|---|---|
| 1 | full process, 60mg | outer pellet | whole pellet | inner pellet | ||
| 2 | outer pellet, full process | 60mg | 30mg | 15mg | ||
| 3 | outer pellet, 60mg, | full process | no extended DNA precipitation | no extended DNA precipitation or beadbeating | ||
| 4 | whole pellet, 60mg, | full process | no extended DNA precipitation | no extended DNA precipitation or beadbeating | ||
Summary of the four experiments conducted
Experiment | Constant Features | Treatments | ||||
|---|---|---|---|---|---|---|
| 1 | full process, 60mg | outer pellet | whole pellet | inner pellet | ||
| 2 | outer pellet, full process | 60mg | 30mg | 15mg | ||
| 3 | outer pellet, 60mg, | full process | no extended DNA precipitation | no extended DNA precipitation or beadbeating | ||
| 4 | whole pellet, 60mg, | full process | no extended DNA precipitation | no extended DNA precipitation or beadbeating | ||
Experiment | Constant Features | Treatments | ||||
|---|---|---|---|---|---|---|
| 1 | full process, 60mg | outer pellet | whole pellet | inner pellet | ||
| 2 | outer pellet, full process | 60mg | 30mg | 15mg | ||
| 3 | outer pellet, 60mg, | full process | no extended DNA precipitation | no extended DNA precipitation or beadbeating | ||
| 4 | whole pellet, 60mg, | full process | no extended DNA precipitation | no extended DNA precipitation or beadbeating | ||
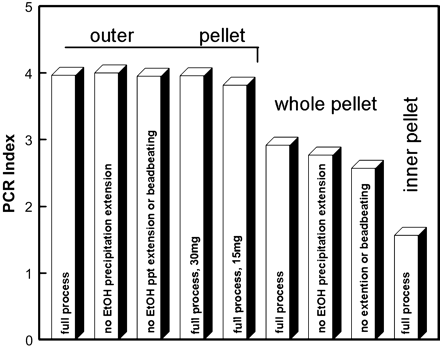
PCR index for all treatments with samples combined. Each value is the mean of 192 PCR outcomes in each treatment. DNA extractions used 60mg of pellet material except where noted. See text for additional details.
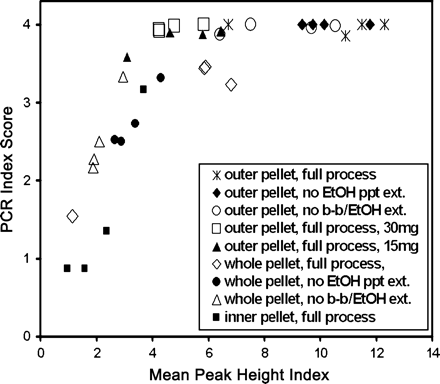
The relationship between PCR index and mean peak height index. For the PCR index the four points in each treatment each represent 12 replicates of four loci (48 total reactions). For the peak height index each point is the mean value of the 12 replicates for each sample. DNA extractions used 60mg of pellet material except where noted. EtOH ppt ext = extended ethanol precipitation; b-b = bead-beating.
The number of treatments that could be run was limited by the amount of outer and whole pellet material available. The outcome of experiment 1 was the basis for using only outer pellet material in experiment 2 and not doing further experiments with inner pellet material. Experiments 3 and 4 allowed the standard Qiagen extraction procedure to be the control that could be compared with our full extraction process in a way that might allow the effects of the two additional procedures to be measured by difference.
Four loci (MAF36, OarFCB304, MAF48, and OarHH62) were evaluated in all extraction tests. Because they covered the entire size range of the loci we used, all could be run in a single lane on the sequencer; additionally, the first two could be multiplexed. Since 1998 we have used the principles of Taberlet et al. (1996) to develop reliable genotypes for 221 fecal samples from bighorn sheep in the central and southern Sierra Nevada and 20 from two neighboring desert mountain ranges to the east. We searched those databases to find four samples that were heterozygous for all four loci to allow consistent comparisons of rates of allelic dropout among samples. Because of lower genetic diversity in the Sierra Nevada only one sample (B76-8) came from that range, collected at 3600m in an alpine habitat. The other three samples (INY 1–6, 1–13, and 1–15) came from a desert slope at 1300m in the Inyo Mountains 40 km to the east. The two habitats are very different, including the forage species available to the sheep. All four samples came from known habitat of bighorn sheep, but none was collected fresh from sheep seen; thus, all were dry when collected. On the basis of loss of surface sheen, the sample from the Sierra Nevada may have been less than a week old, while those from the Inyo Mountains appeared to be 1–2 months old when collected. None of the samples appeared to have been rained on. The samples were stored away from sunlight in paper bags in a dry desert environment until processed four months later for the Inyo Mountains samples and 19 months later for the Sierra Nevada sample.
PCR ingredients and cycling conditions were as previously described. PCR products were treated identically for each treatment as described above and electrophoresed on an ABI PRISM 377 sequencer. For each of the nine treatments, 12 replicate PCRs were run for each sample for all four loci, yielding 48 test results per sample and 192 per treatment. A prior power analysis indicated that this total sample size (192) would allow resolution of about a 6.7% difference in rates of reaction outcomes compared with 16.7% for a sample size of 48. Because of a curvilinear relationship in which resolution gains decline with increasing sample size increments, larger sample sizes were not warranted.
Analyses
We classified results into one of five categories for each locus of each replicate: (1) good (heterozygote with the first peak virtually as high or higher than the second peak and allele sizes no more than 1bp different from correct sizes); (2) reversed peaks (first peak enough lower than the second peak that it would be interpreted as a stutter band if the two alleles differed by only 2 bp; (3) allelic dropout (one allele missing); (4) false allele (one or more alleles ≥2 bp different from the correct size); and (5) failed reactions (no evident amplification).
These five categories form an ordinal series relative to PCR outcome and were given the following ordinal codings: 0 = failed reaction; 1 = false allele(s); 2 = allelic dropout; 3 = reversed peaks; and 4 = good reactions. Each PCR result was coded accordingly and the mean value was calculated for the 48 PCR results for each sample in each treatment, and for the 192 outcomes for entire treatments, as indices of PCR success. By our definition of PCR outcome categories, reversed peaks will be misclassified as allelic dropout where alleles differ by only 2 bp, resulting in an underestimated PCR index value. Only one of our samples (INY1-13) had such close alleles, but it had two sets of alleles that were 2 bp apart. Examination of our data for that sample indicated that the number of PCR results potentially misclassified as allelic dropout varied among treatments from 0 to 5. This translates to a maximum index underestimate of 0.1, which is negligible.
We used peak heights from chromatogram analysis output as a second measure to compare treatments. For consistency, we used only the 123 bp allele of MAF48 because it was the smallest allele for that locus in each of the four samples. To eliminate potential variance from pipetting error when loading gel lanes, we used the ratio of the peak height for that allele to the peak height of the 100 bp size standard as our signal strength index, given that the ratio of PCR products to size standards in the loading mixture was invariant. Failed reactions were assigned a value of zero for this index.
We tested significance of differences in the proportions of good versus other PCR results using a G test (Sokal and Rohlf 1981). We also used ANOVA with the number of good reactions as the dependent variable and the four samples as the source of within variance, with Bonferroni post hoc comparisons. We used a two-way ANOVA (treatments and the four samples) with Bonferroni post hoc comparisons to test peak height differences.
Results
Comparisons with Blood
Paired blood and fecal samples yielded identical genotypes for both replicates of all samples with the exception of two multiplexed loci (AE16 and MAF65) from one individual that gave consistent, but different genotypes for both replicates. Two additional replicate PCRs for both blood and fecal DNA all matched the results from the fecal DNA in the first runs, indicating that the DNA from blood produced false alleles in the first runs. One of those was from a well that showed notable dry-down. Both of these loci previously have shown tendencies occasionally to amplify false alleles that are alleles present in the populations studied.
Fecal DNA Extraction Experiments
Outer pellet material yielded consistently good results. Only when the outer pellet material was decreased to 15 mg did results begin to show a notable drop in PCR success (Figure 1). For the three treatments that used 60mg of outer pellet material (two eliminated extra extraction steps), 8 of 12 samples yielded perfect PCR index scores of four and only 15 of 576 total amplifications were classified as less than good. Of those 15, 13 were reversed peaks and the remaining two were from one multiplexed reaction that was probably due to improper reaction conditions rather than amount of template DNA. Decreasing the amount of outer pellet material yielded a small, but measurable decrease in PCR success, especially at 15 mg, (Figure 1; P = .022 for 60 mg versus 15 mg, P = .061 for 60 mg versus 30 mg; ANOVAs of good amplifications).
Peak heights exhibited a somewhat different pattern of variation for treatments involving outer pellet material (Figure 2); values for 30 mg and 15 mg both were notably lower compared with 60mg (P < .001), but not different from each other (P = 1.000).
There were obvious differences in PCR index values among the different pellet materials used when all treatments were compared (Figure 1; P < .001 for all comparisons; G tests for full extraction process); PCR amplification success varied inversely with the proportion of inner pellet material used. The use of inner pellet material had a similar effect on the peak height index (Figure 2; P < .001 for all comparisons for full extraction process).
Different extraction procedures for outer pellet material yielded no significant differences for PCR outcomes (P = .177; ANOVA of number of good results). The peak height index similarly showed no effects from extending ethanol precipitation (P = 1.000), but exhibited a notable decline when bead-beating also was eliminated (Figure 2; P = .013 relative to full process and 0.021 relative to no extension of ethanol precipitation). When samples were combined for each treatment involving whole pellet material, the pattern suggested improvements in PCR results from the additional extraction steps (Figure 1), and the G-test for the three treatments was significant (P = .006). However, where any inner pellet material was included in DNA extractions, the four individual samples exhibited widely varying patterns for different treatments, including one sample (INY 1–15) for which the additional extraction steps substantially lowered the PCR success; however, the highest PCR success for that sample included bead-beating (Figure 3), indicating an interaction effect between bead-beating and extended alcohol precipitation. Because of the wide variation among samples, the ANOVA of number of good outcomes comparing extraction procedures for whole pellet material was not significant (P = .474). In contrast, peak heights produced significant differences for comparisons with the full extraction process for whole pellet material (Figure 2; P = .012 for full process versus no extension of ethanol precipitation and P < .001 for full process versus no extension of ethanol precipitation or bead-beating); but the two altered extraction procedures were not different from each other (P = .151).
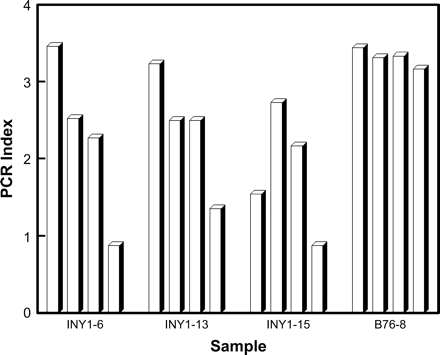
PCR index for treatments involving whole and inner pellet material by sample. Treatments for each sample from left to right are: whole pellet, full process; whole pellet, no extension of ethanol precipitation; whole pellet, no bead-beating or extended ethanol precipitation; and inner pellet, full process.
PCR results also showed considerable variation among samples where only inner pellet material was used (Figure 3). Consequently, while the ANOVA of good PCR outcomes comparing the three different pellet materials was significant (P = .008), the only significant posthoc difference was outer pellet versus whole pellet (P = .008).
The low PCR index value for full extraction process of whole pellet material for sample INY 1–15 (Figure 3) was identified in ANOVA of good results as a statistical outlier. Consequently, two additional replicate extractions from whole pellet material were carried out for this sample along with the same PCRs. Both replicate DNA extractions produced PCR index values close to the first (1.54, 1.56, and 1.62). This statistical outlier apparently represented important information rather than noise. Additionally, the closeness of the PCR index values among those three replicates suggests that this index is a sensitive and consistent measure of PCR success.
The extended alcohol precipitation incorporated in our full DNA extraction process was not only inconsistent in its effect on PCR outcome, it also increased the variance among samples in the peak height index for outer and whole pellet material (Figure 2). For outer pellet material, this increased variance was due to a notable decline in the peak height index for a different sample (B76-8) than the one affected for whole pellet material. Overall, the peak height index exhibited an asymptotic relationship with the PCR index (Figure 2).
Discussion
Our results, and those of Flagstad et al. (1999) and Fernando et al. (2003), show that DNA extracted from feces of some herbivores can yield reliable results with few replicates. There are multiple factors that might affect PCR results from fecal DNA. Our experiments focused primarily on the influences of using different parts of the fecal pellet, different amounts of fecal material, and two extraction steps. When only the very outer pellet material was used, PCR amplifications were consistently excellent. In contrast, when any inner pellet material was included PCR success declined, accompanied by increased variation among samples. Differences in secondary compounds associated with different diets may account for that variation. While our limited sampling did not represent the full range of variation in habitats utilized by bighorn sheep, it did include two habitats that are floristically very different. There was also some variation among samples in the time they were exposed to the environment before collection that co-varied with habitat differences. The notable result was an apparent lack of a habitat/exposure effect when only outer pellet material was used.
Rates of allelic dropout and false alleles dictate the number of replicate PCRs that will be needed for each sample to develop reliable genotypes (Taberlet et al 1996). Where we used 30–60 mg of outer pellet material and included bead-beating in the DNA extraction, 99.8% of PCRs produced genotypes, and 99.7% of those were correct genotypes. This exceeds the 99% reliability that Taberlet et al. (1996) sought and would require no replicate PCRs to achieve their standard. Nevertheless, we included a second PCR for each sample to assure correct genotyping and ran two additional replicates where results were not consistent. We treated DNA derived from blood samples the same way due to occasional evidence of allelic dropout and false alleles, as seen here and noted by Fernando et al. (2003). With our current extraction methods, unweathered fecal samples approach the reliability of blood and tissue as sources of DNA.
Our methods will probably yield similarly reliable outcomes for other ungulate species that produce pelleted feces that dries quickly after deposition. Bighorn sheep typically live in arid habitats where fecal pellets dry rapidly after deposition. The outermost layer is the first to dry, and therefore should experience the least microbial degradation of DNA. Consequently, it probably is important to dry fresh samples as quickly as possible. Fecal pellets of herbivore species that live in wetter environments may undergo more DNA degradation if they are not collected fresh and dried immediately. However, Frantzen et al. (1998) investigated different storage procedures for baboon feces under conditions more humid than our study and concluded that simple dry storage under their prevailing humidity was as good as other methods investigated.
Our earlier extraction procedure included a small amount of inner pellet material and we added bead-beating and extended ethanol precipitation to our extraction procedure because they appeared to improve results. In the current experiments, these procedures had little effect on PCR success for outer pellet material, while each produced small overall gains in the PCR index for whole pellet material (Figure 1). However, extended alcohol precipitation caused a notable decrease in the PCR success for whole pellet material from one sample (Figure 3) leading to increased variance among samples. That procedure similarly increased variances in the peak height index for outer and whole pellet material (Figure 2). Because of this inconsistency in the effects of extended alcohol precipitation, it cannot be recommended in general; however this procedure may improve results in some cases. In contrast, where bead-beating has an effect, it appears only to be a positive effect on signal strength and PCR success.
Not all herbivores produce feces as distinct pellets, and the droppings of some may be similar to whole pellet material, regardless of how they are treated. Investigators working with species that do not provide ideal outer pellet material may need to try different procedures for more problematic samples, including extended alcohol precipitation.
Our best results used 60 mg of scraped outer pellet material; however, while 30 mg caused a proportional (54%) decline in signal strength, PCR success was little affected. This suggests that the outer pellet layer contained a significant amount of high quality template DNA that may allow some flexibility in the amount used. Careful scraping of the outside of pellets is a time-consuming and tedious process. The flexibility in the amount of material needed may allow more efficient methods of sampling the outer pellet layer, such as that proposed by Flagstad et al. (1999).
For studies using noninvasive sampling techniques, optimization of DNA extraction procedures will minimize genotyping errors and maximize efficiency of laboratory effort. In situations where tested procedures may not apply, we recommend using a process similar to the one demonstrated here to compare different extraction procedures, including (1) selection of samples known to be heterozygous at all test loci to facilitate comparisons of rates of allelic dropout; (2) sufficient replication of test PCRs to have adequate resolution of potential differences in reaction outcome rates; and (3) use of an index of PCR success.
Adequate quantitative optimization of DNA extraction methods may alleviate the need to compare paired fecal and blood/tissue samples. Minimally, the results of optimization tests will serve as a basis for calculating the minimum number of replicate PCRs needed for each sample to arrive at the desired level of genotyping accuracy (Taberlet et al. 1996). Our experience suggests that for reliable genotyping at least one replicate PCR should be run even for DNA derived from blood.
Corresponding Editor: Ryder Oliver
We thank Patagonia, the Genetic Resources Conservation Program at the University of California at Davis, the San Diego and national chapters of Safari Club International, and the Biological Resources Division of the U.S. Geological Survey for initial funding of this research, and O. Ryder for making the genetics lab available at the Center for the Reproduction of Endangered Species at the San Diego Zoo. The final phase of this research was funded by the California Department of Fish and Game as part of the Sierra Nevada Bighorn Sheep Recovery Program and was greatly facilitated by the donation of DNA sequencers by the R. W. Johnson Pharmaceutical Research Institute. This paper was improved by helpful comments from three anonymous reviewers.
References
Buchanan FC and Crawford AM,
Buchanan FC and Crawford AM,
Buchanan FC and Crawford AM,
Buchanan FC, Swarbrick PA, and Crawford AM,
Buchanan FC, Swarbrick PA, and Crawford AM,
Creel S, Spong G, Sands JL, Rotella J, Zeigle J, Joe L, Murphy KM, and Smith D,
Ede AJ, Pierson CA, and Crawford AM,
Ede AJ, Peirson CA, Henry H, and Crawford AM,
Ernest HB, Penedo MCT, May BP, Syvanen M, and Boyce WM,
Fernando PT, Vidya NC, Pajapakse C, Dangolla A, and Melnick DJ,
Flagstad O, Roed K, Stacy JE, and Jakobsen KS,
Frantzen MAJ, Silk JB, Ferguson JWH, Wayne RK, and Kohn MH,
Henry HM, Pentry JM, Pierson CA, and Crawford AM,
Huber S, Bruns U, and Arnold W,
Huber S, Bruns U, and Arnold W,
Kohn MH, York EC, Kamradt DA, Haught G, Sauvajot RM, and Wayne RK,
Kovach AI, Litvaitis MK, and Litvaitis JA,
Miller CR, Joyce P, and Waits LP,
Miller DN, Bryant JE, Madsen EL, and Ghiorse WC,
Morin PA, Chambers KE, Boesch C, and Vigilant L,
Pentry JM, Henry HM, Ede AJ, and Crawford AM,
Swarbrick PA, Buchanan FC, and Crawford AM,
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, Waits LP, and Bouvet J,
Taberlet P, Camarra JJ, Griffin S, Uhres E, Hanotte O, Waits LP, Dubois-Paganon C, Burke T, and Bouvet J,
Taberlet P and Luikart G,
Taberlet P, Waits LP, and Luikart G,

{kind=link}
{kind=link}
{kind=link}