





Abstract
The remarkable responsiveness of dog morphology to selection is a testament to the mutability of mammals. The genetic sources of this morphological variation are largely unknown, but some portion is due to tandem repeat length variation in genes involved in development. Previous analysis of tandem repeats in coding regions of developmental genes revealed fewer interruptions in repeat sequences in dogs than in the orthologous repeats in humans, as well as higher levels of polymorphism, but the fragmentary nature of the available dog genome sequence thwarted attempts to distinguish between locus-specific and genome-wide origins of this disparity. Using whole-genome analyses of the human and recently completed dog genomes, we show that dogs possess a genome-wide increase in the basal germ-line slippage mutation rate. Building on the approach that gave rise to the initial observation in dogs, we sequenced 55 coding repeat regions in 42 species representing 10 major carnivore clades and found that a genome-wide elevated slippage mutation rate is a derived character shared by diverse wild canids, distinguishing them from other Carnivora. A similarly heightened slippage profile was also detected in rodents, another taxon exhibiting high diversity and rapid evolvability. The correlation of enhanced slippage rates with major evolutionary radiations suggests that the possession of a “slippery” genome may bestow on some taxa greater potential for rapid evolutionary change.
The speed, magnitude, and diversity of the responses of dog morphology to selection are awe inspiring. The explosive radiation of dog morphologies under domestication reveals the evolutionary potential embedded in the dog genome and may serve as a model of the mammalian radiation of the past 100 million years. Due to their very recent emergence, dog breeds lack the fog of the myriad neutral genetic variations that obscure the geneticist's view of functional differences between natural species, and so dogs provide us with a rare opportunity to determine the mutational origins of phenotypic change in mammals. The mutational origins of this genetic variation include point mutation, transposable element insertion, and repeat slippage mutation, but the relative contributions of these and other mutational processes are unknown (Clark et al. 2006; Fondon and Garner 2004; Mosher et al. 2007; Sutter et al. 2007; Wang and Kirkness 2005). We have found that some of the morphological variation among breeds is attributable to tandem repeat length variation in genes involved in development. A comparison of orthologous repeats in the coding regions of developmental genes showed that the dog repeat was more pure, that is, it had fewer interruptions to the canonical repeat sequence than humans for 31 of 36 repeats examined; the remaining 5 were equal, and 3 of these 5 had perfect purity in both species (Fondon and Garner 2004). Such a lopsided interspecies difference in repeat purity seemed unlikely to be the result of locus-by-locus selection, but the fragmentary nature of the dog genome sequence available at the time precluded reliable investigation of genome-wide processes. The completion of a high-quality dog genome sequence now enables a comprehensive analysis of this question. Does the increase in repeat purity detected in a sample of dog developmental genes reflect the effects of selection at those loci or is it a consequence of genome-wide elevation of microsatellite repeat slippage mutation rates in dogs?
Microsatellites are stretches of tandemly repeated sequences of short sequence motifs of 6 or fewer nucleotides. Microsatellites frequently exhibit polymorphism for a number of repeat motifs, and they possess a characteristic life cycle: while single-nucleotide base substitutions gradually degrade the repetitive character of a repeat, these impurities are periodically removed during repeat length mutation events that occur primarily via a “copy-and-paste” DNA strand slippage mechanism (Gragg et al. 2002). These 2 processes (point mutation and slippage) work in opposition to each other in an unstable dynamic: the acquisition of point mutations suppresses their removal by reducing slippage rates, whereas purifying slippage events tend to increase the likelihood of further slippage (Harr et al. 2000; Kruglyak et al. 1998; Schlotterer 2000). If either extreme at a locus is maladaptive, selection can operate to remove these alleles on a locus-by-locus basis. Alternatively, basal microsatellite slippage mutation rates can increase genome wide as the result of changes in the DNA damage repair apparatus (de Wind et al. 1995; Grady et al. 2001; Sia et al. 2001). Direct measurements of germ-line slippage mutation rates in mammals lack the precision to detect modest rate differences that may have large effects over evolutionary time scales. However, evidence of even subtle differences in mutation spectra will accrue in the genome, and the relative quantities of pure and impure microsatellites in genomes are a reflection of historical basal germ-line repeat slippage mutation rates (Kruglyak et al. 2000; Ellegren 2004; Schlotterer et al. 2006). To distinguish between locus-specific and genome-wide sources of elevated repeat purity in dogs, we compared the repetitive content of the dog genome with that of humans and other mammals, examining the relative quantities of pure and impure microsatellites in particular, and we identified the phylogenetic origins and extent of this trait by comparative sequencing of a large panel of diverse carnivores. Our results suggest that episodic fluctuations in the basal meiotic slippage mutation rate may contribute to differences in the inherent evolvability of some taxa.
Methods
Comparison of Genome-Wide Repeat Content and Purity
Two independent methods for repeat detection in completed dog and human genomes were employed (build numbers 1 and 35, respectively). In the first, all nonoverlapping occurrences of 21 consecutive bases conforming to uninterrupted microsatellites, single interruptions, or double interruptions (spaced 2–6 bases apart) were enumerated for entire genomes. The second approach utilized a more sophisticated repeat detection algorithm, Tandem Repeats Finder (TRF) (Benson 1999), to identify microsatellites 24–45 nucleotides long, with up to 3 interruptions in any arrangement (TRF score setting: 2, 5, 5, 20). The minimum lengths analyzed were set by technical constraints: due to the presence of very large numbers of such sequences in mammalian genomes, exhaustive analysis of repeats of shorter lengths was computationally impractical. Because there are distinct effects of mutations in various DNA replication and repair genes on different types of microsatellites (Sia et al. 2001), we performed comparisons of microsatellite classes with distinct repeat unit lengths and sequences separately (e.g., dinucleotide repeats were divided into 4 groups: ACn, AGn, ATn, and CGn—the other dinucleotides being related to one of these by cyclic permutation, complementation, or both). Subsequent analyses of chimpanzee and mouse genomes were performed using the same methods. There is a disproportionately large number of polyadenine (poly-A) and A-rich repeats in mammals due to the frequent incorporation of poly-A tails of retroposed sequences such as SINEs and pseudogenes. Humans possess over 300 000 copies of the AluL element, which commonly has a long, variable poly-A tail (Price et al. 2004). This and other classes of repeats embedded within or propagated primarily by mobile DNA elements rather than replication slippage were detected, in either genome, on the basis of their frequently occurring in proximity to and on a characteristic strand with recognized mobile elements and were excluded from further analysis. Omitted repeat classes included An, ACn, AGn, and A-rich repeats of longer periods (AANn, AAANn, AAAANn, and AAAAANn). Manual inspection revealed that these A-rich repeats, which represented the vast majority of all repeats of unit length longer than 6, nearly always comprised degenerated poly-A tails of retroposed sequences. As our intent was to investigate the properties of slippage mutation and not retrotransposition or poly-A synthesis and there were not significant numbers of non–A-rich repeats of unit lengths greater than 6, longer unit repeats were not considered further.
Comparative Sequencing of Carnivore Coding Repeats
We sequenced 55 repeat-containing coding regions from 42 species of mammals, representing most major Carnivore families and subfamilies, and measured repeat purity at these orthologous trinucleotide repeats for all species. There is a well-known statistical artifact of ascertainment bias in comparative studies of microsatellites: repeats chosen for analysis on the basis of their length or purity in one species (the focal species) will tend to be longer or more pure in this species than in any nonfocal species to which they are compared (Amos et al. 2003). Repeat loci were selected for analysis on the basis of their predicted homopolymer amino acid sequence in primates. This will result in an ascertainment bias toward longer repeat length in primates, and no ascertainment bias among any members of a taxon with a common ancestor separating them from primates, as is the case for the Carnivora (Vowles and Amos 2006). Any observed increases in purity of a carnivore over a primate will be a conservative estimate due to ascertainment bias in the opposite direction. Amplification primers were designed complementary to conserved regions flanking the chosen repeats, and attempts were made to maximize the quantity of flanking nonrepetitive sequence in amplicons to facilitate accurate alignment, detection of contaminants, evaluation of idiosyncrasies in local mutation spectra, and to capture any nearby repeats that were not part of the original selection criteria. Chimpanzee homopolymers of at least 7 uninterrupted alanines, prolines, or glycines, in addition to a small number of histidine and glutamine repeats were chosen without regard to the composition of the nucleotide repeats encoding them. Homopolymers of these amino acids are the most common types and reflect the distribution of repeat types in the original dog study that accurately represented the genome-wide differences between dogs and humans. Purity was computed as the per nucleotide number of perfect matches to the canonical repeat unit divided by the total length of the repeat (repeat boundaries defined by amino acid sequence), averaged over all loci examined. To avoid biasing results toward better-represented clades, the canonical repeat unit was determined for each species independently, for example, if a given 9 alanine repeat had 5 gcg codons and 4 gcc codons in species A, it would be counted as a gcg9 with 4 interruptions (purity = 0.852), whereas if the orthologous polyalanine in species B had 4 gcg and 5 gcc codons, it would be scored as a gcc9 with 4 interruptions (also purity = 0.852). Note that the theoretical minimum purity for repeats of amino acids with 4 possible codons ranges from 0.75 to 0.8, depending on repeat length, and is higher for amino acids with fewer synonymous codons. Theoretical minima are rarely observed in natural amino acid repeats, and typical purities are much higher.
This panel of repeats was sequenced for 1 or 2 individuals of the following species: domestic dog (Canis lupus familiaris), gray wolf (Canis lupus), coyote (Canis latrans), red fox (Vulpes vulpes), swift fox (Vulpes velox), Arctic fox (Alopex lagopus), gray fox (Urocyon cinereoargenteus), island gray fox (Urocyon littoralis), spectacled bear (Tremarctos ornatus), polar bear (Ursus maritimus), brown bear (Ursus arctos), black bear (Ursus americanus), walrus (Odobenus rosmarus), California sea lion (Zalophus californianus), hog-nosed skunk (Conepatus mesoleucus), striped skunk (Mephitis mephitis), river otter (Lontra spp.), sea otter (Enhydra lutris), American badger (Taxidea taxus), wolverine (Gulo gulo), fisher (Martes pennanti), American marten (Martes americana), raccoon (Procyon lotor), ringtail (Bassariscus astutus), domestic cat (Felis silvestris), jaguarundi (Herpailurus yaguarondi), margay (Leopardus wiedii), Canadian lynx (Lynx canadensis), bobcat (Lynx rufus), caracal (Caracal caracal), serval (Leptailurus serval), puma (Puma concolor), leopard (Panthera pardus), cheetah (Acinonyx jubatus), spotted hyena (Crocuta crocuta), aardwolf (Proteles cristatus), meerkat (Suricata suricatta), dwarf mongoose (Helogale parvula), and ring-tailed mongoose (Galidia elegans). Failure rates for polymerase chain reaction amplifications after at least 2 attempts ranged from 0% to 25% across species, with an average of 49 repeats represented for each species. Sequences for primate orthologues (human, chimpanzee, and rhesus) were obtained from National Center for Biotechnology Information.
In addition to the focal repeat, any other repeats of at least 5 amino acids (of any type) appearing in the amplicon exhibiting any length variation among carnivores was also scored. The requirement that repeats exceed 4 residues and display some length variation among species was intended to filter out loci at which slippage either is not tolerated or occurs at such a low frequency (in all taxa) as to be incapable of accruing any signal to inform the analysis (Harr et al. 2000). Three repeats were excluded on this basis.
Statistical Analyses
All data were analyzed using pairwise comparison methods in which data for each repeat (or repeat class for whole-genome data) in one species was compared with its counterpart in another species. Several factors must be taken into account for evaluation of the results of whole-genome enumeration of pure and impure repeats, including the potential for differing mutational spectra for various repeat types, nonslippage sources of repeat propagation, the completeness of the genome sequence, and differences in genome size. A relationship between genome size and repetitive content is well known, but increasing repeat content (due to changes in mutation spectra) is a major driver of increases in genome size among metazoans, and so controlling for genome size would effectively throw the baby out with the bathwater when our intent is to infer differences in mutation rates and spectra from repeat content (Dieringer and Schlotterer 2003). However, the divergence of dogs and humans is sufficiently recent that relative genome sizes have changed little, and analyses performed both with and without normalizing by genome size (by dividing raw counts by total sequence length) yielded similar results. Repeats types commonly propagated by mobile element insertions in either genome were eliminated from all analyses for both genomes. Because the analysis entailed whole-genome enumeration of all occurrences of repeats, rather than a sampling scheme, sampling error is not a concern (hence no error bars in Figure 1). A paired-sample t-test was used to formally evaluate significance, but the lopsided nature of the results rendered statistical testing superfluous.
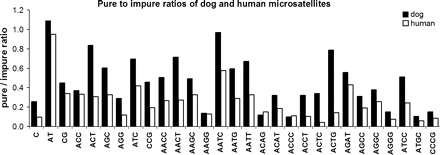
Dogs have elevated purity of mono-, di-, tri-, and tetranucleotide repeats when compared with humans (P < 0.0001, paired-sample t-test). Black bars: dogs; white bars: humans. Repeats of 24–45 bases with 0–3 interruptions in the dog and human genomes were identified using TRF. Microsatellite classes propagated by mobile DNA elements in either species and classes for which either species had an insignificant number (<5) of pure occurrences were eliminated.
Unlike whole-genome comparisons, inferences of slippage rate differences from repeat purity comparisons for orthologous coding repeats among carnivores are subject to sampling error, and estimating this error is problematic due to the limited sample size and significant departures of the distributions pairwise differences from normality. Under these conditions t-tests are unreliable, and the nonparametric Wilcoxon matched-pair signed-rank test (a.k.a. Wilcoxon paired-sample test), which uses bootstrapped significance values to determine the probability of similarly skewed rank orders, was employed as a more rigorous means of evaluating the significance to interspecies repeat purity differences. Moreover, as genomes cannot be assumed to be at equilibrium and the purity differences have been accumulated over evolutionary time scales, it is not valid to infer precise current meiotic slippage rates from these data; even relative rate inferences should be viewed as qualitative, rather than quantitative measures unless supported by other indicators such as interspecific differences in polymorphism.
Results
Genome-Wide Purity Differences between Humans and Dogs
The ratio of numbers of perfectly pure repeats to impure repeats for all mono-, di-, tri-, and tetranucleotide repeats that are not associated with mobile elements are presented in Figure 1. The dog has a higher pure–impure ratio for all these mono-, di-, and trinucleotide repeat types and 18 out of 20 tetranucleotide types. The difference is highly significant (P < 0.0001, paired-sample t-test) and is sufficient to account for the finding in our earlier studies of a limited set of coding repeats in developmental genes in which 31 of 36 repeats were more pure in dogs (Fondon and Garner 2004). This result was robust to the particulars of counting methodology; using the simpler repeat detection technique (21 base window with up to 2 mismatches), or a broader range of TRF window sizes (up to 75 nucleotides) did not substantially affect the results (P < 0.0001, not shown). The trend is broad and continues into larger repeat unit sizes, with dogs' advantage in purity ratios and overall numbers of repeats (both pure and impure) fading as the repeat unit length increases (Table 1). To provide perspective for the differences between dogs and humans and help assess their significance, identical analyses were performed for the chimpanzee, which is known to have a marginally lower microsatellite mutation rate. Humans had a slight edge over chimpanzees in repeat number (not statistically significant), but the human–chimp differences in repeat numbers and purity were more than an order of magnitude smaller than dogs' increase over humans (Table 1). Because differences in genomic repeat quantity and purity reflect basal germ-line rates of slippage mutation (Harr et al. 2000; Kruglyak et al. 2000; Schlotterer et al. 2006), we conclude that the basal slippage mutation rate for microsatellites is significantly higher for dogs than humans, and the difference in repeat purity previously observed in a sample of coding sequences is explained by a genome-wide elevation in germ-line slippage events and not attributable to locus-specific selection, natural or otherwise.
Differences in the number and ratios of pure and impure repeats among mammalian genomes
| No. of pure repeatsa | Pure/impure normalizeda,b | |||||||
| Unit length | Dog | Human | Chimp | Mouse | Dog | Human | Chimp | Mouse |
| 1 | 355 | 19 | 18 | 153 | 2.67 | 1.00 | 1.08 | 1.44 |
| 2 | 5442 | 5460 | 4782 | 7790 | 1.15 | 1.00 | 0.98 | 1.43 |
| 3 | 1974 | 975 | 733 | 3239 | 1.91 | 1.00 | 0.95 | 1.84 |
| 4 | 6017 | 2515 | 2249 | 10551 | 1.93 | 1.00 | 0.98 | 2.50 |
| 5 | 969 | 564 | 522 | 2124 | 1.38 | 1.00 | 1.16 | 2.02 |
| 6 | 1667 | 736 | 554 | 2194 | 1.35 | 1.00 | 1.03 | 1.54 |
| No. of pure repeatsa | Pure/impure normalizeda,b | |||||||
| Unit length | Dog | Human | Chimp | Mouse | Dog | Human | Chimp | Mouse |
| 1 | 355 | 19 | 18 | 153 | 2.67 | 1.00 | 1.08 | 1.44 |
| 2 | 5442 | 5460 | 4782 | 7790 | 1.15 | 1.00 | 0.98 | 1.43 |
| 3 | 1974 | 975 | 733 | 3239 | 1.91 | 1.00 | 0.95 | 1.84 |
| 4 | 6017 | 2515 | 2249 | 10551 | 1.93 | 1.00 | 0.98 | 2.50 |
| 5 | 969 | 564 | 522 | 2124 | 1.38 | 1.00 | 1.16 | 2.02 |
| 6 | 1667 | 736 | 554 | 2194 | 1.35 | 1.00 | 1.03 | 1.54 |
Repeat types propagated primarily by nonslippage mechanisms (e.g., transposon association) have been excluded (An, ACn, AGn, AANn, AAANn, etc.).
Normalized to human by dividing purity for each species by human purity (i.e., [∑puredog/∑impuredog]/[∑purehuman/∑impurehuman]). Repeat types with fewer than 3 occurrences with perfect purity in any one species are excluded from average purity calculations in all species to eliminate spurious distortions of ratios resulting from using small values in ratio calculations.
Differences in the number and ratios of pure and impure repeats among mammalian genomes
| No. of pure repeatsa | Pure/impure normalizeda,b | |||||||
| Unit length | Dog | Human | Chimp | Mouse | Dog | Human | Chimp | Mouse |
| 1 | 355 | 19 | 18 | 153 | 2.67 | 1.00 | 1.08 | 1.44 |
| 2 | 5442 | 5460 | 4782 | 7790 | 1.15 | 1.00 | 0.98 | 1.43 |
| 3 | 1974 | 975 | 733 | 3239 | 1.91 | 1.00 | 0.95 | 1.84 |
| 4 | 6017 | 2515 | 2249 | 10551 | 1.93 | 1.00 | 0.98 | 2.50 |
| 5 | 969 | 564 | 522 | 2124 | 1.38 | 1.00 | 1.16 | 2.02 |
| 6 | 1667 | 736 | 554 | 2194 | 1.35 | 1.00 | 1.03 | 1.54 |
| No. of pure repeatsa | Pure/impure normalizeda,b | |||||||
| Unit length | Dog | Human | Chimp | Mouse | Dog | Human | Chimp | Mouse |
| 1 | 355 | 19 | 18 | 153 | 2.67 | 1.00 | 1.08 | 1.44 |
| 2 | 5442 | 5460 | 4782 | 7790 | 1.15 | 1.00 | 0.98 | 1.43 |
| 3 | 1974 | 975 | 733 | 3239 | 1.91 | 1.00 | 0.95 | 1.84 |
| 4 | 6017 | 2515 | 2249 | 10551 | 1.93 | 1.00 | 0.98 | 2.50 |
| 5 | 969 | 564 | 522 | 2124 | 1.38 | 1.00 | 1.16 | 2.02 |
| 6 | 1667 | 736 | 554 | 2194 | 1.35 | 1.00 | 1.03 | 1.54 |
Repeat types propagated primarily by nonslippage mechanisms (e.g., transposon association) have been excluded (An, ACn, AGn, AANn, AAANn, etc.).
Normalized to human by dividing purity for each species by human purity (i.e., [∑puredog/∑impuredog]/[∑purehuman/∑impurehuman]). Repeat types with fewer than 3 occurrences with perfect purity in any one species are excluded from average purity calculations in all species to eliminate spurious distortions of ratios resulting from using small values in ratio calculations.
Evolutionary Origins of Elevated Slippage Mutation Rates
When did this property of the dog genome arise? One possibility is that it is a consequence of human selection for those animals that best responded to breeding efforts. Because length variation in tandem repeats within genes contributes to phenotypic variation and coding repeats are concentrated in genes important for development, any mutation among early dogs which increased repeat slippage rates might have been highly adaptive under these conditions of strong directional selection. Alternatively, this trait may have predated domestication as a natural feature of the wolf genome and might have contributed to an inherent domesticability of wolves.
If dogs' elevated repeat purity arose during domestication, then wild canids will lack this property; if this trait preceded domestication, then it should be exhibited by wolves and perhaps other closely related taxa. To distinguish between these possibilities, we sequenced 55 trinucleotide repeat-containing coding regions from 42 species of mammals, representing most families and subfamilies of Carnivora, and measured repeat purity at these orthologous repeats for all species. Ascertainment bias was controlled by selecting repeat loci on the basis of their predicted amino acid sequence in primates, producing ascertainment bias toward higher length and purity in primates, and no bias among any members of the Carnivora (Vowles and Amos 2006, see Methods). The results are summarized in Figure 2.
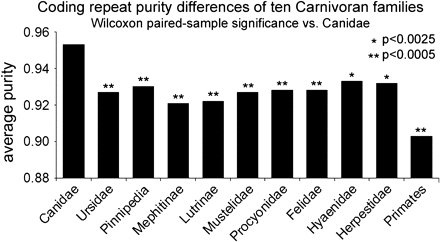
Elevated slippage mutation predates the canid radiation and dog domestication. Average repeat purity was determined by comparative sequencing of 55 orthologous trinucleotide repeat-coding regions for 42 mammals and assaying the number of interruptions to the canonical repeat sequence for each species. Canid repeats were significantly more pure than all noncanids (nonparametric Wilcoxon paired-sample rank test, see Methods for details). Note that the theoretical minimum purity for repeats of amino acids with 4 possible codons ranges from 0.75 to 0.8, and with 2 codons, this theoretical minimum is 0.83.
The repeat sequences in wolves are nearly identical to their dog orthologues in overall purity and the location and identity of interruptions. Indeed, all wild canids examined (gray wolves, coyotes, red, Arctic, swift, gray, and island gray foxes) have levels of purity similar to dogs; however, the positions and identities of the impurities vary among evolutionarily more distant canids. All other carnivores have significantly lower purities (Figure 2).
A phylogenetic reconstruction of the patterns of impurity losses and gains shows a general trend of accelerated loss of ancestral impurities in the canid lineage; therefore, the differences are not due to an increase in the rate of new point mutations in noncanids (Harr et al. 2000). In addition, whereas the overall quantities of impurity losses are common to all canids, several of the individual purification events within canids are clade specific (Figure 3). Thus, the purification of repeats in the canid lineage was not the result of a brief burst of slippage in deep history but has unfolded over several million years.

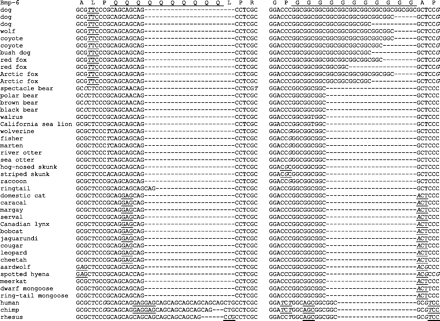
Purification of canid repeats has unfolded over millions of years. Individual ancestral impurities (GCA codons, asterisks) have been lost multiple times in different canid lineages. The dog-like canids (except the bush dog) have mostly retained the ancestral impurities; domestic dogs are polymorphic for the loss of the second impurity. The red fox clade (including red, Arctic, and swift foxes) shows more extensive impurity loss, with a minimum of three independent loss events. The gray fox clade has retained all three ancestral impurities. Ancestral impurities were inferred from their presence in multiple clades of both the Caniformes and Feliformes sub-orders (they are conserved in some members of all major taxa except the pinnipeds). The length of the uninterrupted portion of the ancestral sequence was defined by consensus, but varies among taxa and the length depicted here may not reflect a genuine ancestral state. Despite sequencing only one or two individuals for each of these species, several length polymorphisms were discovered.
Despite the small numbers of individuals sequenced for each species (n = 1 or 2), several polymorphisms for repeat length and the loss of ancestral impurities were observed in foxes, coyotes, wolves, and dogs (but were less common in noncanid taxa). The small numbers of individuals sequenced per species and differences in population structure and history preclude drawing any firm conclusions from differences in length polymorphism rates among taxa; however, the presence of polymorphisms for the loss of ancestral impurities at several loci for multiple canids indicates that the purification of repeats that has occurred over the course of canid evolution is still ongoing in present-day populations. Differences in repeat lengths observed between canid species were often smaller than the within-species variation.
Not all classes of repeats exhibited the same level of canid-specific purification or polymorphism. Although most repeat classes were not present in sufficient numbers to be analyzed independently, one exception is the ccgn repeat. In its various cyclic permutations on either DNA strand, the ccgn repeat may encode for polyalanine, polyglycine, polyproline, or polyarginine, and each of these amino acid repeats may also be encoded by other codon repeats. Although amino acid repeats were selected for analysis without regard to how they were encoded, almost all repeats of alanine, proline, or glycine were found to be encoded by ccgn repeats. Only 1 of 27 polyalanines was not comprised primarily of runs of gcc or gcg, and this lone exception was highly degenerate (mean purity ∼0.8, near the theoretical minimum) and was the only polyalanine longer than 6 repeats to be completely invariant among carnivores. None of the 14 polyglycines or 3 polyprolines was encoded by anything other than ccgn. Considering only ccgn repeats marginally increases the average purity difference and its statistical significance between canids and all other families; an unfortunate consequence of selecting repeats blind with respect to their DNA sequence is that non-ccgn repeats were not represented in sufficient numbers to permit meaningful comparisons of their purity levels.
One potential explanation for the prominence of ccgn among amino acid repeats and their enhanced purity in canids is that these repeats are inherently more slippage prone than other trinucleotide repeats. A physical basis for a slippage process specific to these triplet repeats has been described in which slipped-strand structures, intermediates of the slippage mutation pathway, of ggcn repeats are stabilized by formation of a quadruplex DNA structure (Sinden et al. 2002). Although the dog genome-wide purity ratio for this repeat class is not exceptional, falling near the middle of the range for dogs, the overall quantities of these and nontriplet microsatellites with potential for forming quadruplex structures (i.e., repeats with runs of 3 or more guanines) are highly elevated. Another possibility is that changes in CpG methylation may be involved, as loss of CpG methylation is known to destabilize repeats, but the effects appear to be in trans and may have little to do with CpG methylation of the repeats themselves (Gorbunova et al. 2004). Pure CpG-containing repeats are highly enriched in dogs. Dogs have ∼7.5-fold more pure CpG-containing hexamers than humans do, but only ∼1.7-fold more pure hexamers that do not contain CpGs (507 and 67 with CpGs, 1160 and 669 without CpGs). However, because a similar enrichment is also observed for GC-rich repeats that lack CpGs, cis-effects of DNA methylation of the repeats themselves probably cannot be a direct cause of the overabundance of ccgn-encoded amino acid repeats or can they fully account for enhanced purity or polymorphism of ccgn repeats in canids. Humans are known to have marginally higher lengths and mutation rates for cagn repeats than other primates, but this is thought to be driven by distinct mutational processes (Vowles and Amos 2006). Whereas dogs have higher pure to impure ratios of cagn repeats than humans (Figure 1), humans have more such repeats than dogs (1.4-fold more pure, 1.3-fold more impure), a pattern not observed for any other triplet. Alignments of flanking ccgn and cagn repeats from the propeptide domain of bmp-6 (Figure 4) illustrates the characteristic differences in variation between these 2 classes of repeats in primates and canids, indicative of distinct slippage profiles in these taxa.
Clade-specific expansion of ccgn and cagn repeats in canids and primates. Repeats comprised ccg (or cgg on the opposite strand), showing little or no length variation among noncanid lineages, often exhibit extensive variation among canids. A similar phenomenon is observed for cagn repeats in primates. The bone morphogenetic protein-6 gene reflects both these phenomena simultaneously, with nearby polyglutamine and polyglycine repeats in the propeptide region displaying clade-specific expansions.
Discussion
Simple sequence repeats are generated from initially nonrepetitive or only weakly repetitive DNA, primarily by polymerase slippage mutations during DNA synthesis. Once established, microsatellites experience frequent slippage mutation, at rates that are a function of the repeat unit sequence, length, and purity. When point mutations occur within an otherwise perfect repeat, they suppress slippage mutation rates by disrupting local self-similarity necessary for the misalignment of the slipped-strand precursor to length mutation. Conversely, the “copy-and-paste” nature of the slippage mutation process has the effect of removing these impurities and restoring the repetitive character of the repeat. Because the creation, expansion, and purification of repeats are all directly dependent on the basal slippage mutation rate, relative slippage rates can be inferred from comparisons of genomic repetitive content and purity (Harr and Schlotterer 2000; Kruglyak et al. 2000; Schlotterer et al. 2006; Vowles and Amos 2006). Through comparisons of the entire genomic complement of simple sequences in the human and recently completed dog genomes, we show that the increased purity initially observed for a few dozen dog coding repeats is reflective of a genome-wide increase in slippage rates and not necessarily the result of locus-specific selection as initially indicated by analysis of the fragmentary standard poodle genome sequence (Fondon and Garner 2004).
The radical diversification of dog morphology under domestication has been accompanied by extraordinary diversification of coding repeats in genes controlling morphology. Dog Hox genes show tremendous breed-to-breed variation in coding repeat lengths, with length ranges well outside those observed for natural populations of wolves or coyotes. It is possible that the genome-wide increase in dog slippage mutation rates is a by-product of the intense and ever-changing directional selection dogs have experienced under domestication. Under such conditions, a mutation that resulted in an increase in the production of new genetic variation might have been of considerable adaptive value and have been indirectly favored. Alternatively, possession of this trait by wolves might have made them more domesticable or more responsive to breeders' efforts to modify them. By extending our repeat purity analysis to wild canids and noncanid carnivores, we find that elevated slippage rates were already present in dogs' wild predecessors, having arisen in the canid lineage prior to the divergence of the extant Canidae.
Although all the extant canids are descended from only the most recent of 3 major canid evolutionary radiations, they exhibit a wide range of morphological variation. The repeated radiations and diversity of fossil and extant canid forms—from bat-eared foxes to stilt-legged maned wolves to diminutive bush dogs and such unusual canids as the raccoon dog—is in sharp contrast to the natural history of cats, where the relative uniformity of the extant and fossil species has prevented reliable phylogenetic classification on the basis of morphology, and the contemporary understanding of cat phylogeny is based on molecular characters (Johnson et al. 2006). The rise of slippage mutation rates in the canid lineage may have contributed to canids' apparent evolutionary malleability. If so, similar rises might have accompanied other major mammalian evolutionary radiations, such as those of bats or rodents. Conducting the repeat content and purity analysis on the mouse and rat genomes produced results very similar to dogs (Table 1), and the similarities in the patterns of changes apparent in each class of microsatellite that have arisen independently in the rodent and canid lineages imply related mechanistic origins (Table 1). Sequencing a panel of 10 laboratory mouse strains for coding repeats in developmental genes revealed high levels of allele length variation similar to that observed among breeds of dogs (20 of 31 genes polymorphic among these 10 strains, unpublished results, Fondon, John), providing independent support that mice also experience frequent slippage mutations in developmental genes which may also contribute to phenotypic differences among races of mice.
Interestingly, the Hyaenidae (hyenas and aardwolves) showed the highest purity levels of the noncanid carnivores despite being more closely related to cats. It is intriguing that these species share dog-like morphologies and lifestyles as well, and underscores the role that extragenomic factors, such as generalist (e.g., scavenger/opportunist/predators like canids and hyenas) versus specialist (dedicated predators such as cats) lifestyles play in prepositioning animals to exploit novel niches. Such extragenomic and genomic components of evolvability might be expected to interact, favoring the emergence of elevated mutation regimes in taxa that frequently invade and adapt to new niches or inhabit fluctuating environments.
Slipped-strand mutation intermediates of ccgn and cagn repeats adopt distinct structures, and it seems likely that distinct mutational mechanisms may underlie lineage-specific changes in repeat mutation profiles in canids and primates. The enrichment of ccg- and cag-derived repeats in genes of different functional classes may offer means by which the generation of allelic variation might be enhanced for one aspect of phenotype relative to another. Such an enrichment might be expected to arise as an indirect consequence of extended periods of directional selection on a particular aspect of phenotype, such as morphology or brain function, without invoking the sort of forward-looking or anticipatory measures disallowed by evolutionary theory.
Conclusions
The increased slippage rate of dogs is a derived character of the canid lineage predating domestication, having appeared abruptly between the canid divergence and modern canid radiation, and it has been preserved in modern species. Our findings suggest that one or more molecular “defects” in the DNA replication or repair apparatus arose before the major evolutionary radiation of extant canids, leading to increased slippage rates and repeat length variation. Such molecular events are not without precedent, as Drosophila and yeast mutants with elevated slippage rates have been described, and a class of human tumors is characterized by elevated microsatellite slippage rates (Flores and Engels 1999; Sia et al. 2001). Previous work has shown a role for repeat length variation in morphological and behavioral variation in mammals (Goodman et al. 1997; Fondon and Garner 2004; Hammock and Young 2005); high slippage rates may therefore have been of adaptive value in generating phenotypic variation on which selection—both natural and artificial—could act (Kashi and King 2006). This would suggest that other canids might be as amenable to domestication as dogs, and it is of note that the domestication of the silver fox (V. vulpes) was accomplished in fewer than 30 generations and was accompanied by surprising increases in morphological and coat color variation (Trut 1997).
Unlike point mutations, length mutations in microsatellites in genes frequently result in incremental effects on gene function and phenotype. In principle, increases in phenotypic variation of similar magnitude could also be generated by a genome-wide increase in point mutation rates, but the genetic load of this more haphazard process might be too high for populations possessing the high anatomical and physiological complexity and low reproductive rates of vertebrates. Not all mammalian genes are equally likely to harbor slippage-prone tandem repeats in their coding sequences. Mammalian coding repeats are highly concentrated in a few classes of regulatory genes, particularly those involved in development, to the exclusion of proteins where they are unlikely to provide adaptive value, such as core metabolism enzymes (Lavoie et al. 2003). A mechanism for specifically accelerating repeat mutation, whether regulated in response to stresses or simply stochastic (e.g., mutations in DNA mismatch repair), might be of significant adaptive value in fluctuating evolutionary landscapes (Ruden et al. 2005). Whether this is in fact an oft-used trick for accelerating the rate at which mutations of potential adaptive utility occur will become apparent as more genomes are sequenced.
We thank D. Clifton, W. Murphy, R. C. Fleischer, B. Jacobson, and the University of Alaska Museum for tissue samples. This work was supported by the Sara and Frank McKnight Fellowship in Biochemistry (J.W.F.), the P. O'B. Montgomery Chair in Biochemistry (H.R.G.), by the Robert A. Welch foundation (R.R.), and the Mallinckrodt Foundation Scholar Award (R.R.). R.R. is an investigator of the Howard Hughes Medical Institute.
References
Author notes
Corresponding Editor: Elaine Ostrander
This paper was delivered at the 3rd International Conference on the Advances in Canine and Feline Genomics, School of Veterinary Medicine, University of California, Davis, CA, August 3–5, 2006.

{kind=link}
{kind=link}
{kind=link}
{kind=link}