




Abstract
Background : We previously developed an estrogen receptor (ER)–positive breast cancer cell line (MCF-7:5C) that is resistant to long-term estrogen deprivation and undergoes rapid and complete apoptosis in the presence of physiologic concentrations of 17β-estradiol. Here, we investigated the role of the mitochondrial apoptotic pathway in this process. Methods : Apoptosis in MCF-7:5C cells treated with estradiol, fulvestrant, or vehicle (control) was investigated by annexin V–propidium iodide double staining and 4′,6-diamidino-2-phenylindole (DAPI) staining. Apoptosis was also analyzed in MCF-7:5C cells transiently transfected with small interfering RNAs (siRNAs) to apoptotic pathway components. Expression of apoptotic pathway intermediates was measured by western blot analysis. Mitochondrial transmembrane potential (ψ m ) was determined by rhodamine-123 retention assay. Mitochondrial pathway activity was determined by cytochrome c release and cleavage of poly(ADP-ribose) polymerase (PARP) protein. Tumorigenesis was studied in ovariectomized athymic mice that were injected with MCF-7:5C cells. Differences between the treatment groups and control group were determined by two-sample t test or one-factor analysis of variance. All statistical tests were two-sided. Results: MCF-7:5C cells treated with estradiol underwent apoptosis and showed increased expression of proapoptotic proteins, decreased ψ m , enhanced cytochrome c release, and PARP cleavage compared with cells treated with fulvestrant or vehicle. Blockade of Bax, Bim, and p53 mRNA expression by siRNA reduced estradiol-induced apoptosis relative to control by 76% [95% confidence interval (CI) = 73% to 79%, P <.001], 85% [95% CI = 90% to 80%, P <.001], and 40% [95% CI = 45% to 35%, P <.001], respectively, whereas blockade of FasL by siRNA had no effect. Estradiol caused complete regression of MCF-7:5C tumors in vivo. Conclusion: The mitochondrial pathway of apoptosis plays a critical role in estradiol-induced apoptosis in long-term estrogen-deprived breast cancer cells. Physiologic concentrations of estradiol could potentially be used to induce apoptosis and tumor regression in tumors that have developed resistance to aromatase inhibitors.
It is generally believed that the balance between proliferation and apoptosis influences the response of tumors to treatments such as chemotherapy ( 1 ) , radiotherapy ( 2 ) , and hormonal therapy ( 3 ) . It has been suggested that, when these treatments fail, dysregulation of apoptosis may be the cause. Apoptosis is a form of programmed cell death that is executed by a family of proteases called caspases, which can be activated either by cell-surface death receptors (i.e., the extrinsic pathway) or by perturbation of the mitochondrial membrane (i.e., the intrinsic pathway) ( 4 ) . Components of the extrinsic pathway include the death receptors FasR/FasL, DR4/DR5, and tumor necrosis factor (TNFR), whereas the intrinsic pathway centers on the mitochondria, which contain key apoptogenic factors such as cytochrome c and apoptosis-inducing factor (AIF) ( 4 ) . In the intrinsic pathway, the integrity of mitochondrial membranes is controlled primarily by a balance between the antagonistic actions of the proapoptotic and antiapoptotic members of the Bcl-2 family. Bcl-2 family proteins comprise three principal subfamilies: 1) antiapoptotic members, including Bcl-2/Bcl-x L , which possess the Bcl-2 homology (BH) domains BH1, BH2, BH3, and BH4; 2) proapoptotic members, such as Bax, Bak, and Bok, which have the BH1, BH2, and BH3 domains; and 3) BH3-only proteins, such as Bid, Bim, Bad, Bik, and Puma, which generally possess only the BH3 domain ( 5 ) . The Bcl-2 family of proteins regulates apoptosis by altering mitochondrial membrane permeabilization and controlling the release of cytochrome c .
Many estrogen receptor (ERα)–positive human breast cancer cells require estrogen for their proliferation and undergo apoptotic cell death when deprived of estrogen ( 6 ) . As such, the current strategy for treating ERα-positive breast cancer is to block the action of estrogen on tumor cells. There are three general approaches: 1) inhibiting estrogen binding to the ERα using an antiestrogen such as tamoxifen ( 7 ) , 2) blocking estrogen synthesis using an aromatase inhibitor such as exemestane ( 8 ) , or 3) reducing ERα protein levels using a pure antiestrogen such as fulvestrant (ICI 182780 or Faslodex) ( 9 ) . In addition to stimulating growth of ER-positive breast cancer cells, estradiol also promotes cell survival, and it has been suggested that the ability of estradiol to act as a survival factor for breast cancer may occur, in large part, through its prevention of apoptosis via the activation of antiapoptotic proteins such as Bcl-2 and Bcl-x L . Indeed, estradiol has been shown to inhibit apoptosis in several cell types, including neurons ( 10 ) , endothelial cells ( 11 ) , epithelial cells of the female reproductive tract ( 12 ) , and hormone-dependent human breast cancer MCF-7 cells ( 13 , 14 ) .
Recent evidence also suggests that estradiol is capable of inducing programmed cell death (i.e., apoptosis) in certain cell types, including bone-derived cells ( 15 ) , immune system cells, and breast cancer cells that have developed resistance following extensive antihormonal therapy ( 16 – 20 ) . In these models of drug resistance, our laboratory ( 16 – 18 ) and those of other investigators ( 19 ) have found that physiologic concentrations of estradiol induce cell death and rapid tumor regression rather than stimulate growth. These data are particularly interesting because high doses of synthetic estrogens such as diethystilbestrol (DES) or ethinyl estradiol have been used effectively to treat breast cancer patients for the last 60 years ( 21 ) . However, the idea that low doses of estradiol can induce cell death under appropriate circumstances is novel. Furthermore, the mechanism by which estrogen promotes apoptosis is not understood. Previous studies ( 21 – 23 ) have demonstrated a link between estradiol-induced apoptosis and activation of the FasR/FasL death-signaling pathway. Song et al. ( 19 ) reported that estradiol treatment statistically significantly increased the expression of FasL in a long-term estrogen-deprived (LTED) breast cancer cell model that was sensitive to estradiol-induced apoptosis, and Osipo et al. ( 24 ) and Liu et al. ( 25 ) reported an increase in FasR expression in tamoxifen-resistant tumors and raloxifene-resistant cells, respectively, during estradiol-induced apoptosis and tumor regression. More recently, however, we ( 22 ) have found no evidence to suggest a link between estradiol-induced apoptosis and activation of the FasR/FasL signaling pathway in MCF-7:5C, an estrogen-deprived breast cancer cell line that is resistant to estrogen withdrawal.
Therefore, in the present study, we determined the role of the mitochondrial (intrinsic) pathway in estradiol-induced apoptosis in MCF-7:5C cells. Members of the Bcl-2 family along with the tumor suppressor protein p53, which are important regulators of mitochondrion-mediated apoptosis, were examined in estradiol-treated MCF-7:5C cells. The effect of fulvestrant on the growth of MCF-7:5C cells was also examined, and its efficacy as a growth inhibitor was compared to that of estradiol. Fulvestrant was selected because it is an estrogen receptor antagonist with no agonist effects and is currently licensed in the United States for the treatment of postmenopausal women with hormone-sensitive advanced breast cancer following progression on prior antiestrogen therapy ( 23 ) .
M ATERIALS AND M ETHODS
Cells and Tissue Culture Conditions
The ER-positive human breast cancer MCF-7 cells were obtained from Dr. Dean Edwards (University of Texas, San Antonio), and were maintained in full serum medium (RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 m M glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 1× nonessential amino acids [all from Invitrogen, Carlsbad, CA], and bovine insulin at 6 ng/mL [Sigma-Aldrich, St. Louis, MO]). Estrogen-independent MCF-7:5C ( 26 ) cells were cloned from wild type MCF-7 cells and were also maintained in estrogen-free RPMI 1640 medium supplemented with 10% dextran-coated charcoal-stripped fetal bovine serum (SFS) and other supplements as in the full serum medium. MCF-7:5C cells stably overexpressing Bcl-x L or the vector control pBabe were kindly provided by Dr. Navdeep Chandel (Northwestern University, Chicago, IL) and were maintained in phenol red–free RPMI 1640 medium supplemented with 10% SFS and 1-μg/mL puromycin (Sigma-Aldrich).
Cell Proliferation Assays
Wild-type MCF-7 cells were switched from phenol red RPMI 1640 medium supplemented with 10% fetal bovine serum to phenol red–free RPMI 1640 medium for 4 days prior to beginning the proliferation assay. Because MCF-7:5C cells were routinely maintained in estrogen-free RPMI 1640 medium, no medium switch was required for these cells. On the day of the experiment (day 0), MCF-7 and MCF-7:5C cells were cultured in estrogen-free RPMI containing 10% SFS at a density of 2 × 10 4 cells per well in 24-well plates. After 24 hours (day 1), the medium was replaced with fresh RPMI 1640 medium, and various concentrations of estradiol (10 −14 to 10 −8M ), fulvestrant (10 −14 to 10 −6M ), or <0.1% ethanol vehicle (control) were added. The compound-containing medium was replaced with fresh compound-containing medium on days 3 and 5, and the experiment was stopped on day 7. The DNA content of the cells, a measure of proliferation, was determined as previously described ( 27 ) using a Fluorescent DNA Quantitation kit (Bio-Rad Laboratories, Hercules, CA). In brief, cells were harvested after the appropriate treatment, sonicated in 0.1× phosphate-buffered saline (PBS) (Invitrogen) for 10 seconds, and incubated with a Hoechst dye mixture (BioRad Laboratories) for 1 hour. Total DNA was measured using a VersaFluor fluorometer (Bio-Rad Laboratories). For each analysis, six replicate wells were used and at least three independent experiments were performed.
Annexin V Analysis of Apoptosis
The annexin V–FITC-labeled Apoptosis Detection Kit I (Pharmingen, San Diego, CA) was used to detect and quantify apoptosis by flow cytometry according to the manufacturer's instructions. In brief, MCF-7:5C cells (1 × 10 6 cells/mL) were seeded in 100-mm dishes and cultured overnight in estrogen-free RPMI 1640 medium containing 10% SFS. The next day, cells were treated with <0.1% ethanol vehicle (control), estradiol (1 n M ), or fulvestrant (1 μ M ) for 72 hours and then harvested in cold PBS (Invitrogen) and collected by centrifugation for 10 min at 500 × g . Cells were then resuspended at a density of 1 × 10 6 cells/mL in 1× binding buffer (HEPES buffer, 10 m M , pH 7.4, 150 m M NaCl, 5 m M KCl, 1 m M MgCl 2 , and 1.8 m M CaCl 2 ) and stained simultaneously with FITC-labeled annexin V (25 ng/mL; green fluorescence) and propidium iodide (PI) (50 ng/mL). PI was provided as a 50-μg/mL stock (Pharmingen) and was used as a cell viability marker. Cells were analyzed using a fluorescence-activated cell sorter (FACS) flow cytometer (Becton Dickinson, San Jose, CA), and the data were analyzed with CellQuest software.
4′,6-Diamidino-2-Phenylindole Staining
MCF-7:5C cells were grown (overnight) in RPMI 1640 medium containing 10% SFS and then treated with ethanol vehicle (i.e., control), 1 n M estradiol alone, or 1 μ M fulvestrant for 96 hours. The cells were then washed in PBS, fixed with 4% paraformaldehyde for 20 minutes at room temperature, and washed again in PBS. Cells were then treated with 1 μg/mL of 4′,6-diamidino-2-phenylindole (DAPI) (Sigma Chemical Co.) for 30 minutes, washed again with PBS for 5 minutes, and treated with 50 μL of VectaShield (Vector Laboratories, Burlingame, CA). Stained nuclei were visualized and photographed using a Zeiss fluorescence microscope (Provis AX70; Olympus Optical Co., Tokyo, Japan). Apoptotic cells were morphologically defined by cytoplasmic and nuclear shrinkage and by chromatin condensation or fragmentation.
Electron Microscopy Analysis
For electron microscopic observation, MCF-7:5C cells were grown (overnight) in RPMI 1640 medium containing 10% SFS and then treated with estradiol (1 n M ), fulvestrant (1 μ M ), or ethanol vehicle (<0.1%) for 96 hours. Floating cells were then harvested together with adherent cells and centrifuged at 200 × g for 5 min. The cell pellets were fixed overnight in a 0.2 M sodium cacodylate buffer solution (pH 7.4) containing 2% glutaraldehyde at 4 °C. Samples were then post-fixed in cacodylate-buffered 1% osmium tetroxide, dehydrated, and embedded in Epon 812 (Structure Probe, Inc., West Chester, PA) for ultra-thin sectioning. The ultra-thin sections were stained with uranyl acetate and lead citrate and viewed with a JEOL JEM-1220 120-kV transmission electron microscope (Peabody, MA) in the Northwestern University Cell Imaging Facility.
Western Blot Analysis
After treatment with estradiol or fulvestrant, cells were washed twice with PBS and lysed in ice-cold lysis buffer (50 m M Tris–HCL, pH 7.5, 150 m M NaCl, 0.5% Nonidet P40, 1 m M phenylmethylsulfonyl fluoride [PMSF], 1 m M NaF, 1 m M sodium orthovanadate, 1 m M dithiothreitol [DTT], 10 μ M β-glycerophosphate, and 2-μg/μL complete protease inhibitor cocktail), as previously described ( 28 ) . Lysates were centrifuged at 12 000 × g for 30 min at 4 °C, and protein concentration was measured with the Bio-Rad Protein Assay kit (Bio-Rad Laboratories). Thirty micrograms of protein was mixed with sodium dodecyl sulfate (SDS) sample buffer, denatured by boiling, and separated on 15%, 12%, or 7.5% SDS-polyacrylamide gel gels. Proteins were then electroblotted to nitrocellulose membranes and blocked for 1 hour at room temperature in TBS-T (50 m M Tris–HCL, pH 7.5, 150 m M NaCl, 0.1% Tween-20) buffer containing 5% nonfat milk. Membranes were then incubated overnight at 4 °C with the respective primary antibodies. Antibodies against Bax (sc-493), Bak (sc-7873), Bcl-2 (sc-509), Bcl-x L (sc-8392), Bim (sc-11425), ERα (sc-544), MDM2 (sc-5304), Noxa (sc-22764), p53 (sc-126), and Puma (sc-19187) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against caspase 7 (product 556541), caspase 8 (product 556466), caspase 9 (product 556510), and poly(ADP-ribose) polymerase (PARP) (product 556494) were purchased from BD Pharmingen (San Jose, CA). The antibody against β-actin (product AC-15) was obtained from Sigma-Aldrich (St. Louis, MO). Anti-mouse or anti-rabbit secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology) was used to visualize the stained bands with an enhanced chemiluminescence (ECL) visualization kit (Amersham, Arlington Heights, IL).
PARP Cleavage
The intact PARP molecule (116 kDa) is activated by breaks in DNA and is cleaved by caspases to yield a p85 fragment that is a characteristic marker of apoptosis ( 29 ) . For detection of PARP cleavage, MCF-7:5C cells were treated with either ethanol vehicle (0.1%; control) or 1 n M estradiol for 24, 48, 72, and 96 hours, and lysates were analyzed by western blotting as described above. Membranes were probed with an anti-PARP monoclonal antibody from BD Pharmingen, which recognizes both the 116-kDa intact form of PARP and the 85-kDa cleavage fragment.
Real-Time Reverse Transcription–Polymerase Chain Reaction
Total RNA was reverse transcribed with TaqMan reverse transcription reagents (Applied Biosystems, Hayward, CA) and random hexamers as the primers. Primers and probes for human transforming growth factor alpha (TGF-α) and trefoil factor 1 (pS2) were designed with Primer Express TM1.5 software (Applied Biosystems) set at default parameters to select the optimal primer and probe sets for this system. Both TGF-α and pS2 are considered estrogen-responsive genes; hence, ERα activity was assessed by measuring the estrogen-responsive expression of TGF-α and pS2 mRNA in MCF-7:5C cells following estradiol or fulvestrant treatment. Eukaryotic 18S ribosomal RNA (Applied Biosystems, product 4319413E) was used as an internal control, and each total cDNA sample was normalized to the content of 18S mRNA. The sequences for all primers and probes are as follow: TGF-α forward primer, 5′-GCCTGTAACACACATGCAGTGA-3′; TGF-α reverse primer, 5′-TTTCCAAAGGACTGA CTTGGAAG-3′; TGF- α probe, 5′-FAM-AGGCCTCACATATACGCCTCCCTAGAAGTG-QSY7–3′; pS2 forward primer, 5′-GAGGCCCAGACAGAGACGTG-3′; pS2 reverse primer, 5′-CCCTGCAGAAGTGTCTAAAATTCA-3′; pS2 probe, 5′-FAM-CTGCTGTTTCGACGAC ACCGTTCG-QSY7–3′; p53 forward primer, 5′-CCCCAGCCAAAGAGAAACC-3′; and p53 reverse primer, 5′-TCCAAGGCCTCATTCAGCTCT-3′. All probes were labeled with 6-carboxyfluorescein (FAM) as the reporter and with QSY7 (a nonfluorescent diarylrhodamine derivative; MegaBases, Chicago, IL) as the quencher.
The reverse transcription–polymerase chain reaction (RT-PCR) portion of the reaction was performed with the TaqMan Universal PCR Master Mix reagent kit (Applied Biosystems, Branchburg, NJ). A 25-μL reaction mixture, containing 50 ng of total cDNA, 100 n M probe, and 200 n M primers, was prepared using 2× TaqMan Universal PCR master mix (Applied Biosystems). RT-PCR was performed with the ABI-Prism 7700 sequence detection system (Applied Biosystems). The PCR was run in a sealed 96-well optical plate, and the thermal cycler conditions were as follow: initial denaturation at 50 °C for 2 minutes and 95 °C for 10 minutes, followed by 40 cycles of a denaturation step at 95 °C for 15 seconds, and a primer annealing/extension step at 60 °C for 1 minute. RT-PCR results were quantitated by normalizing the cycle threshold (CT) values (which are proportional to mRNA copy number) of TGF-α, pS2, or p53 mRNA to the CT values of the endogenous control 185. The relative quantitation number was then calculated by subtracting the average CT for TGF-α, pS2, or p53 from the corresponding average CT for 185.
Small Interfering RNA Transfection
For transient transfections, MCF-7:5C cells were seeded at a density of 50–70% in 12-well plates in estrogen-free Opti minimal essential medium (Opti-MEM) containing 10% SFS. The following day, cells were transfected with 100 n M small interfering RNAs (siRNAs) for p53 (Cell Signaling, product number 6230), Bax (Dharmacon, product number M-003308–00), Bim (Dharmacon, product number M-004383–00, Noxa (Dharmacon, product number M-005275–01), Puma (Dharmacon, product number M-004380–00), or FasL (Dharmacon Research, Lafayette, CA) using Lipofectamine 2000 transfection reagent (Invitrogen, product number 116668–019), according to the manufacturer's recommended protocol. Scramble siRNA was purchased from Dharmacon and was used as a control (Silencer negative control siRNA, product number 4611). The cells were harvested 48 hours posttransfection and analyzed by western blot (as described above) and/or by RT-PCR (as described above). Transfected cells were also treated with estradiol for an additional 72 hours, and apoptotic cells were measured using annexin V staining (as described above).
Mitochondrial/Transmembrane Potential (Δψ m ) and Cytochrome c Release
Mitochondrial membrane potential was measured by flow cytometry using the cationic lipophilic green fluorochrome rhodamine-123 (Rh123) (Molecular Probes). Disruption of ΔΨ m is associated with a lack of Rh123 retention and a decrease in fluorescence. Briefly, MCF-7:5C cells were washed twice with PBS and incubated with 1-μg/mL Rh123 at 37 °C for 30 min. Cells were then washed twice with PBS, and Rh123 intensity was determined by flow cytometry. Cells with reduced fluorescence (less Rh123) were counted as having lost some of their mitochondrial membrane potential.
For detection of cytochrome c , MCF-7:5C cells (1 × 10 7 ) were treated with either ethanol vehicle (<0.1%) or 1 n M estradiol for 48 hours and then harvested by centrifugation at 800 × g at 4 °C for 15 min. After washing three times with ice-cold PBS, cell pellets were resuspended in HEPES-buffer (20 m M HEPES, 10 m M KCl, 1.5 m M MgCl 2 , 1 m M EDTA, 1 m M EGTA, 1 m M DTT, 0.1 m M PMSF, pH 7.5) containing 250 m M sucrose, homogenized with a homogenizer, and centrifuged at 800 × g at 4 °C for 15 min. The supernatants were centrifuged at 10 000 × g for 15 min at 4 °C, and the mitochondrial pellets were dissolved in SDS sample buffer (25 μL), subjected to 15% SDS- polyacrylamide gel electrophoresis, and analyzed by immunoblotting with monoclonal antibodies against cytochrome c (BD Biosciences; product 556433) and cytochrome oxidase subunit IV (COXIV) (Molecular Probes; product A21437). Aliquots (25 μL) of the supernatant (i.e., cytosolic fraction) were also analyzed for cytochrome c expression by western blotting. An anti-mouse secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology) was used to visualize the stained bands with an ECL visualization kit (Amersham). COXIV, which is a mitochondrion-specific protein, was used as a control to demonstrate that mitochondrial protein fractionation was successfully achieved.
Caspase 8 Activity
A caspase 8 fluorometric assay kit (Chemicon International, Inc., Temecula, CA), which contains the caspase 8–specific substrate Acetyl-Ile-Glu-Thr-Asp-p-Nitroaniline (Ac-IETD-pNA), was used to evaluate caspase 8 enzyme activity in MCF-7:5C cells following 1 n M estradiol treatment (96 and 120 hours), according to the manufacturer's instructions. The broad-spectrum caspase inhibitor, benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone (z-VAD-fmk), and the caspase 8–specific inhibitor, z-Lle-Glu(OMe)-Thr-Asp(OMe)-fluoromethyl ketone (z-IETD-FMK), were purchased from Calbiochem (La Jolla, CA).
Neutralization of the Fas Receptor and Fas Ligand
Cells (1 × 10 5 ) were seeded in 12-well plates in RPMI 1640 medium containing 10% SFS and after 24 hours were treated with an anti-FasR neutralizing antibody (ZB4) at a final concentration of 250 ng/mL for 12 hours, which we have previously determined to be the optimal treatment in terms of Fas neutralization of MCF-7:5C ( 22 ) . Cells were then treated with 1 n M estradiol for an additional 48 and 72 hours, and apoptosis was determined by annexin V staining. ZB4 is very effective at antagonizing FasR and has previously been shown to block drug-induced apoptosis in breast and cervical cancer cells ( 30 ) . Similar experiments were performed with the FasL neutralizing antibodies NOK1 and NOK2 at a final concentration of 500 ng/mL.
Growth of MCF-7:5C Tumors In Vivo
Approximately 1 × 10 7 MCF-7:5C cells suspended in saline solution (product 20012027; Invitrogen) were bilaterally inoculated into mammary fat pads of 30 ovariectomized BALB/c nu/nu mice (Harlan Sprague Dawley, Madison, WI), as described previously ( 31 ) . Tumors were measured weekly with vernier calipers, and the cross-sectional tumor area was calculated by multiplying the length ( l ) by the width ( w ) by π and dividing the product by four (i.e., lw π/4). When the untreated MCF-7:5C tumors reached a mean cross-sectional area of 0.23 cm 2 (at approximately 4 weeks), groups of 10 mice were randomly assigned to the following groups: 1) 0.3-cm estradiol capsule, 2) fulvestrant (5 mg/0.1 mL of peanut oil injected subcutaneously twice a week), and 3) vehicle control. For the estradiol treatment, 0.3-cm silastic estradiol capsules (Baxter HealthCare, Mundelein, IL), were implanted subcutaneously in the mice and replaced after 8 weeks of treatment. These capsules produced a mean serum estradiol level of 83.8 pg/mL ( 32 ) , which is similar to postmenopausal serum levels of estradiol. For the fulvestrant treatment, fulvestrant (AstraZeneca, Cheshire, UK) was dissolved in 100% ethanol and diluted in peanut oil and injected twice a week. For the vehicle control group, the mice did not receive any treatment. All animal handling procedures were in accordance with the approved guidelines of the Animal Care and Use Committee of Northwestern University. The experiment was followed for 10 weeks (data shown for only 8 weeks), and at the end of the study, all animals were killed by CO 2 asphyxiation. For detection of apoptosis in vivo, MCF-7:5C tumors were removed from control and estradiol-treated mice on day 5, fixed in formalin, and embedded in paraffin blocks. No tumors were collected at the end of the study (week 10) because estradiol treatment caused complete tumor regression.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End-Labeling Assay
The terminal deoxynucleotidyl transferase–mediated dUTP nick end–labeling (TUNEL) assay followed by tetramethylrhodamine deoxyuridine 5′-triphosphate (TMR) staining was used to detect apoptosis in vivo. An in situ cell death assay with the TMR red detection kit (Roche Diagnostic, Indianapolis, IN) was performed on paraffin-embedded MCF-7:5C tumor sections (described above), according to the manufacturer's instructions. The TUNEL assay was performed in triplicate and repeated three times with six independent tumors from each treatment group. The percentage of apoptosis was calculated by dividing the number of TMR-positive cells or TUNEL-positive cells by the total number of epithelial cells (identified by DAPI staining) and multiplying the result by 100.
Statistical Analysis
All data are expressed as the mean (with 95% confidence interval [CI]) of at least three determinations, unless otherwise stated. The differences between the treatment groups and the control group (for all analyses except tumor growth) were determined by two-sample t test or one-factor analysis of variance (ANOVA). Generalized Estimating Equation methods ( 33 ) were used to compare tumor growth rates among the three treatment groups using data collected in weeks 5–8. First, left and right tumor areas were averaged for each mouse at each time point. This measure was the dependent variable for subsequent analyses. Second, treatment time was log transformed to approximately linearize the growth curves. For each treatment group, slopes were estimated from the tumor growth data. Cage was included in the analysis as a clustering factor. Finally, pairwise between-group comparisons were made using robust standard errors. The type 1 error for the pairwise tests was adjusted for multiple comparisons using Bonferroni's method (i.e., α = .05/3, or .0167). All statistical tests were two-sided.
R ESULTS
Growth of LTED Breast Cancer Cells In Vitro
Wild-type MCF-7 human breast cancer cells ( 34 ) are a permanent cell line that contains estrogen receptors. These cells have retained estrogen responsiveness for a sustained period of continuous cell culture and show estrogen-dependent stimulation of cell proliferation by natural estrogens or inhibition by antiestrogens ( 35 , 36 ) . Therefore, we compared the growth characteristics of wild-type MCF-7 cells with those of LTED MCF-7:5C cells in the presence of estradiol or the pure antiestrogen fulvestrant. Figure 1, A shows that estradiol treatment increased the growth of wild-type MCF-7 cells in a concentration-dependent manner, with maximum stimulation at 10 −10M . In contrast, fulvestrant treatment reduced the growth of MCF-7 cells, with maximum inhibition at 10 −6M . Figure 1, B shows that estradiol at 10 −9M statistically significantly reduced the growth of MCF-7:5C cells, as measured by DNA content, from 11.5 μg [95% CI = 11 to 12 μg] in the absence of estradiol to 1.7 μg [95% CI = 1.4 to 2 μg, P <.001] ( Fig. 1, B ), whereas fulvestrant (10 −6M ) reduced the growth of these cells from 11.3 μg [95% CI = 11.1 to 11.5 μg] to 4.4 μg [95% CI = 4.2 to 4.6 μg]. The difference in the inhibitory effects of fulvestrant at 10 −6M and estradiol at 10 −9M was statistically significant ( P = .002). Overall, these results show that there are dramatic differences between MCF-7 cells and MCF-7:5C cells in their response to estradiol—specifically, that LTED altered MCF-7:5C cells in such a way that estrogen induces cell death rather than cell growth.
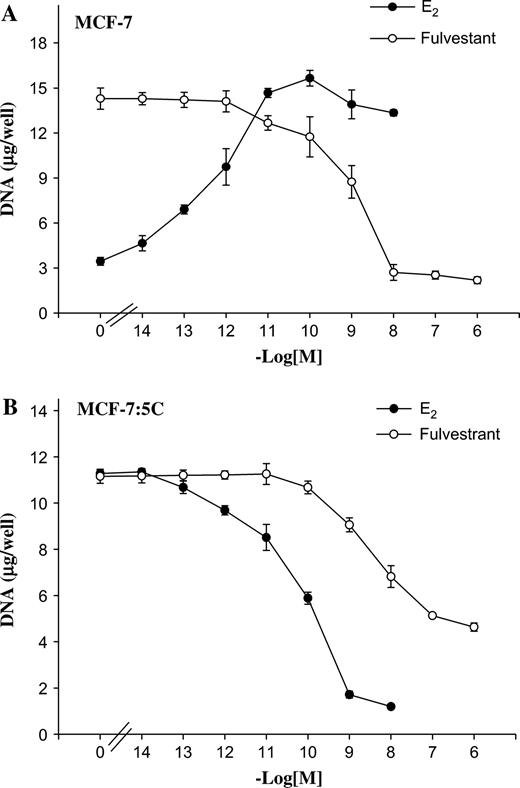
Effects of estradiol (E 2 ) and fulvestrant (Ful) on the growth of MCF-7 cells. A ) Wild-type MCF-7 cells. B ) Long-term E 2 -deprived MCF-7:5C cells. For growth assays, approximately 2 × 10 4 cells were seeded in 24-well plates and then treated with various concentrations of E 2 (10 −14 to 10 −8M ) or fulvestrant (10 −14 to 10 −6M ) for 7 days. Cells were harvested on day 7, and total DNA (micrograms/well) was quantitated. The experiment was repeated three times. The data represent the mean of three independent experiments with 95% confidence intervals.
To assess ERα-mediated transcriptional activation in MCF-7:5C cells following estradiol or fulvestrant treatment (24 hours), the endogenous estrogen-responsive genes TGF-α and pS2 were measured by real-time PCR. Expression of TGF-α mRNA is normally induced by estradiol in MDA-MB-231 human breast cancer cells stably transfected with the wild-type ERα ( 37 ) . The pS2 gene is often used as a prognostic marker in breast cancer cells and has been widely used in studies of ER action, and it is suggested that estrogen regulates the expression of pS2 through an imperfect ERE in the pS2 promoter ( 38 ) . Our results showed that estradiol treatment increased TGF-α and pS2 mRNA expression in MCF-7:5C cells that was blocked completely by the pure antiestrogen fulvestrant (data not shown). We also found that MCF-7:5C cells expressed higher levels of ERα protein than did wild-type MCF-7 cells, as assayed by western blotting (data not shown). These results demonstrate that resistance to LTED did not alter the ability of the ER to regulate the expression of estrogen-responsive genes.
Effect of Estradiol and Fulvestrant on Apoptosis in MCF-7:5C Cells
Because a decrease in cell proliferation may result from the induction of apoptosis, we investigated whether the estradiol- or fulvestrant-induced growth inhibition of MCF-7:5C cells was due to an increase in apoptosis. MCF-7:5C cells were treated with ethanol vehicle (i.e., control), estradiol (1 n M ), or fulvestrant (1 μ M ) for 72 hours, and annexin V–FITC and PI fluorescence was determined by flow cytometry ( Fig. 2, A ). In the control and fulvestrant-treated groups, only 2.8% (95% CI = 2.3% to 3.3%) and 3.8% (95% CI = 3.3% to 4.3%), respectively, of cells stained positive for annexin V, whereas, in the estradiol-treated group, 77.6% [95% CI = 71% to 84.2%] of cells stained positive for annexin V. In additional experiments (not shown), cells were treated with estradiol plus fulvestrant or estradiol plus the universal caspase inhibitor z-VAD-FMK. In both cases estradiol-induced apoptosis in MCF-7:5C cells was blocked, suggesting the involvement of the ERα and caspases in this process.
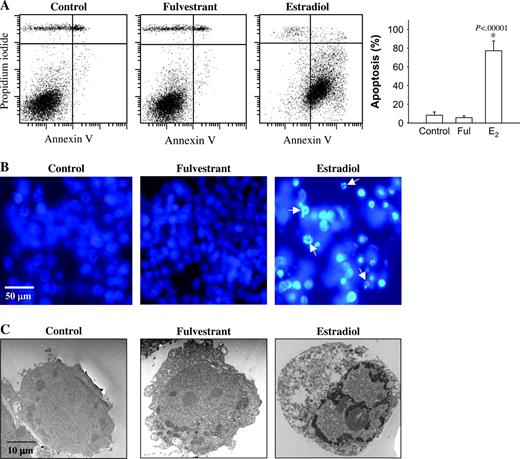
Effects of estradiol and fulvestrant on apoptosis in MCF-7:5C cells. A ) Annexin V staining for apoptosis. MCF-7:5C cells were seeded in 100-mm plates at a density of 1 × 10 6 per plate. After 24 hours, the cells were treated with ethanol vehicle ( control ), 1 μ M fulvestrant (Ful), or 1 n M estradiol (E2) for 72 hours, and then cells were stained with FITC–annexin V and propidium iodide (PI) and analyzed by flow cytometry. Representative cytograms are shown for cells treated with ethanol vehicle (control), estradiol (1 n M ), or fulvestrant (1 μ M ). Viable cells are FITC − and PI − , early apoptotic cells are FITC + and PI − , and late apoptotic cells are FITC + and PI + . Right panel = Quantitation of apoptosis (percentage of all cells that were apoptotic). * P <.001 for estradiol-treated cells compared with control. The data represent the means of three independent experiments, and the error bars show upper 95% confidence intervals. B ) Fluorescent microscopic analysis of apoptotic cells stained with 4′,6-diamidino-2-phenylindole (DAPI). Round and/or shrunken nuclei of DAPI-stained cells ( white arrows ) are hallmarks of apoptosis. Experiments were repeated three times with similar results. Representative slides are shown. Scale bars = 50 mm. C ) Electron microscopic analysis of MCF-7:5C cells treated with ethanol, 1 μ M fulvestrant, or 1 n M estradiol for 96 hours. Experiments were repeated three times with similar results. Representative slides are shown. Scale bars = 10 μm.
To confirm the apoptosis-inducing effect of estradiol on MCF-7:5C cells, we stained cells with DAPI. In contrast to untreated control cells and fulvestrant-treated cells, which had very little condensed or fragmented chromatin, the majority of estradiol-treated cells displayed apoptotic features, including condensed nuclei, membrane blebbing, and nuclear fragmentation ( Fig. 2, B ).
Electron microscopy was used to characterize the structural changes associated with estradiol-induced apoptosis in MCF-7:5C cells. Cells treated with vehicle or fulvestrant showed no signs of apoptosis ( Fig. 2, C ), displaying a uniform rounded shape and no condensation of chromatin. There were also very few vesicles in the cytoplasm and little debris in the extracellular space of these cells. In contrast, cells treated with estradiol displayed classic apoptotic nuclei, with completely condensed chromatin at the inner side of the nuclear envelope. Estradiol-treated cells also showed membrane blebbing and formed apoptotic bodies, which are morphologic hallmarks of apoptosis. Together, these findings suggest that estradiol induces apoptotic death in MCF-7:5C cells; by contrast, fulvestrant inhibits growth through a nonapoptotic pathway.
FasR/FasL Signaling Pathway in Estradiol-Induced Apoptosis in MCF-7:5C Cells
Song et al. ( 19 ) reported that estradiol-induced apoptosis in an LTED breast cancer cell line is mediated by the FasR/FasL signaling pathway. We performed several analyses to investigate whether this result was also the case in MCF-7:5C cells. First, western blot analysis was performed to measure FasR and FasL protein expression in wild-type MCF-7 cells and MCF-7:5C cells. As Fig. 3, A shows, MCF-7:5C cells expressed a higher basal level of FasR protein than did wild-type MCF-7 cells. Moreover, estradiol further increased FasR protein expression, but not FasL protein expression, to a level that was markedly higher than that of control. However, estradiol induction of FasR did not occur until after 5 days of treatment (data not shown), after apoptosis had commenced.
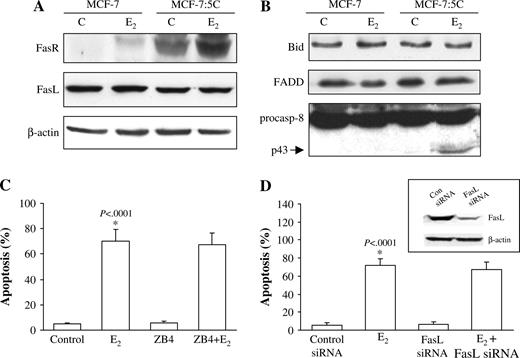
Effect of estradiol on FasR/FasL signaling pathway in MCF-7 and MCF-7:5C cells. A ) Western blot analysis for FasR and FasL protein expression in parental MCF-7 cells and MCF-7:5C cells following 96 hours of treatment with either ethanol (vehicle) (C) or 1 n M estradiol (E 2 ). FasR = 45 kDa; FasL = 38 kDa; NS = non-specific band. B ) Western blot analysis of Bid, FADD, and caspase 8 protein expression in parental MCF-7 cells and MCF-7:5C cells following 96 hours of treatment with either 1 n M E 2 or ethanol. p43 is the cleaved (i.e., activated) form of caspase 8 (casp-8). C ) Effect of Fas neutralizing antibody ZB4 on E 2 -induced apoptosis in MCF-7:5C cells. Cells were preincubated with the ZB4 antibody for 12 hours and then treated with 1 n M E 2 for 96 hours. Percentage of cells that were apoptotic was determined by annexin V staining. Results shown are the means of three independent experiments with 95% confidence intervals. D ) Reduction of FasL expression in MCF-7:5C cells by small interfering RNA (siRNA). Cells were transfected with a scrambled control siRNA or FasL-specific siRNA. After 48 hours, the cells were analyzed by western blotting for FasL protein expression ( insert ) or were treated with estradiol (or ethanol) for an additional 72 hours, after which apoptosis was assessed by annexin V staining.
We next investigated whether Fas-associated death domain (FADD), caspase 8, and Bid, which are important components of the FasR/FasL death signaling pathway, are activated by estradiol. In MCF-7:5C cells, estradiol treatment increased the cleavage of caspase 8 (cleaved fragment, p43) but did not alter FADD or Bid protein levels or Bid cleavage (i.e. activation) ( Fig. 3, B ). Estradiol-induced cleavage of caspase 8, however, occurred after 5 days of estradiol treatment and was not associated with apoptosis induction. In addition, inhibition of caspase 8 activity using the caspase 8 inhibitory peptide z-IETD-fmk failed to block estradiol-induced apoptosis or cytochrome c release (data not shown), which suggests that activation of caspase 8 is not a critical event in estradiol-induced apoptosis. The ability of the universal caspase inhibitor z-VAD-FMK to completely block estradiol-induced apoptosis in MCF-7:5C cells indicates that other caspases are involved in this process.
Because the Fas protein was induced by estradiol in MCF-7:5C cells, we investigated whether this induction was required for estradiol-induced apoptosis. Treatment of cells with neutralizing antibody against FasR (ZB4) did not prevent estradiol-induced apoptosis of MCF-7:5C cells ( Fig. 3, C ). Finally, we reduced FasL expression in MCF-7:5C cells using siRNA. As shown in Fig. 3, D (insert), transfection of MCF-7:5C cells with FasL-specific siRNA led to a marked reduction in FasL protein levels, whereas transfection with control siRNA did not have this effect, indicating that the inhibition was sequence specific. Inhibition of FasL expression, however, did not block estradiol-induced apoptosis in MCF-7:5C cells ( Fig. 3, D ). Moreover, FasL neutralizing antibodies (NOK1/NOK2) also did not prevent estradiol-induced apoptosis (data not shown). Overall, these findings indicate that estradiol induction of apoptosis in MCF-7:5C cells did not require activation of FasR or FasL and suggest that the extrinsic death receptor pathway is not a critical mediator of estradiol-induced apoptosis.
Role of the Mitochondrial Pathway in Estradiol-Induced Apoptosis in MCF-7:5C Cells
To examine the role of the mitochondrial (i.e., intrinsic) pathway in estradiol-induced apoptosis in MCF-7:5C cells, we used Western blot analysis to measure the levels of the proapoptotic Bax, Bak, and Bim and antiapoptotic Bcl-2 and Bcl-x L proteins, all components of the intrinsic pathway, after treatment with either estradiol or fulvestrant. Figure 4, A shows that there was a marked increase in the levels of Bax, Bak, and Bim proteins in estradiol-treated MCF-7:5C cells compared with vehicle-treated (control) cells. Estradiol treatment, however, did not increase the protein level of Bax, Bak, or Bim in wild-type MCF-7 cells ( Fig. 4, A ), which is consistent with its antiapoptotic activity in these cells.
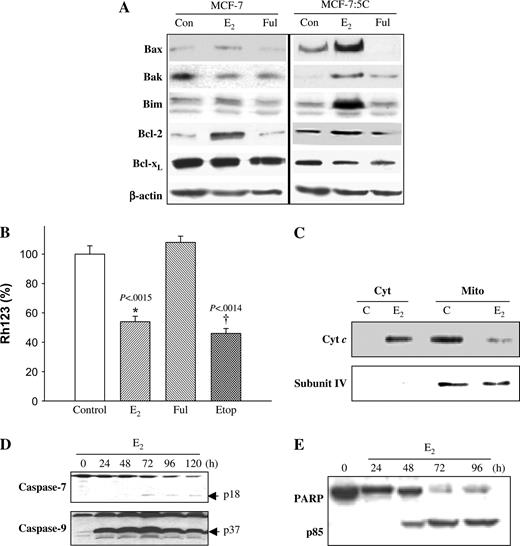
Effect of estradiol on Bcl-2 family protein expression and mitochondrial function in MCF-7:5C cells. A ) Western blot analysis for Bax, Bak, Bim, Bcl-2, and Bcl-x L protein expression in parental MCF-7 cells and MCF-7:5C cells following 48 hours of treatment with ethanol (Con), 1 n M estradiol (E 2 ), or 1 μ M fulvestrant (Ful). Equal loading was confirmed by reprobing with an antibody against β-actin. B ) Loss of mitochondrial potential (i.e., mitochondrial integrity) in MCF-7:5C cells was determined by rhodamine-123 (Rh123) retention assay. The percentage of cells retaining Rh123 in each treatment group was compared with untreated control. * P <.001 is the E 2 -treated group compared with the control group. C ) Cytosolic and mitochondrial fractions were generated as described in “Materials and Methods.” Equivalent protein loading was verified using anti-cytochrome c oxidase IV (subunit 4) antibody. D ) Activation of caspase 7 (casp-7) and caspase 9 (casp-9) was assessed by Western blot using specific antibodies. The upper band of caspase 7 represents the full-length protein, and the lower band (p18) represents the cleaved activated product. E ) PARP cleavage was determined by western blotting using a rabbit polyclonal PARP antibody. MCF-7:5C cells were treated with 1 n M E 2 for 24, 48, 72, and 96 hours. Full-length PARP is approximately 116 kDa; cleaved (active) PARP is 85 kDa.
A key step in the mitochondrion-dependent apoptotic pathway is the disruption of the mitochondrial membrane, leading to loss of mitochondrial transmembrane potential (Δψ m ) ( 39 ) . Therefore, we examined the effect of estradiol on Δψ m by Rh123 retention. Figure 4, B shows that estradiol treatment statistically significantly reduced Rh123 fluorescence in MCF-7:5C cells by 45% [95% CI = 40% to 50%, P = .002] compared with control, suggesting that the mitochondrial pathway is involved in estradiol-induced apoptosis. By contrast, fulvestrant did not have a statistically significant effect on Δψ m ( Fig. 4, B ); however, when fulvestrant was combined with estradiol, estradiol-induced loss of Δψ m was blocked completely (data not shown). This finding suggests that estradiol-induced loss of Δψ m requires the estrogen receptor.
During mitochondrion-mediated apoptosis, cytochrome c is released from the mitochondria into the cytosol, where it promotes caspase activation ( 40 ) . Therefore, we examined the effects of estradiol on cytochrome c release in MCF-7:5C cells using western blot analysis. Figure 4, C shows that in vehicle-treated (control) cells, cytochrome c was detected primarily in the mitochondria and was undetectable in the cytosol; however, estradiol treatment (48 hours) led to a statistically significant increase in cytochrome c release from the mitochondria to the cytosol that coincided with cleavage (i.e., activation) of caspase 9 (active product p37) ( Fig. 4, D ) and cleavage of PARP (active product p85) ( Fig. 4, E ). Estradiol-induced cleavage of caspase 9 was detected as early as 24 hours after treatment. We did not detect any caspase 3 protein in MCF-7:5C cells (data not shown); however, this finding is consistent with previous reports that indicate a lack of caspase 3 in these cells ( 41 ) . PARP cleavage was observed 48 hours after estradiol treatment, with maximum cleavage between 72 and 96 hours. Interestingly, fulvestrant, the ER antagonist, was able to block the ability of estradiol to induce apoptosis, cytochrome c release, caspase activation, and PARP cleavage (data not shown), which suggests a critical role for the ER in estradiol-induced apoptosis.
Effect of Bcl-x L Overexpression on Estradiol-Induced Apoptosis in MCF-7:5C Cells
Overexpression of Bcl-2/Bcl-x L has been shown to inhibit mitochondrial damage and release of cytochrome c , which are key events in caspase activation and apoptosis ( 42 ) . Therefore, to confirm the role of the mitochondrial pathway in estradiol-induced apoptosis, we investigated whether Bcl-x L overexpression can protect MCF-7:5C cells against estradiol-induced apoptosis in MCF-7:5C cells. Western blot analysis was used to verify Bcl-x L protein overexpression in MCF-7:5C cells stably overexpressing Bcl-x L or the empty vector pBabe ( Fig. 5, A ). For the annexin V study, MCF-7:5C cells stably overexpressing Bcl-x L or the empty vector pBabe were treated with either ethanol vehicle (control) or 1 n M estradiol, and apoptosis was examined by flow cytometry using annexin V–PI staining. Estradiol treatment (for 72 hours) induced marked apoptosis in the pBabe vector control cells but not in the Bcl-x L –overexpressing cells ( Fig. 5, B ). The estradiol-induced reduction in Δψ m , as determined by Rh123 retention assay, was also partially reversed by Bcl-x L overexpression ( Fig. 5, C ). These results provide further evidence that the mitochondrial pathway is required for estradiol-initiated apoptotic signaling in MCF-7:5C cells.
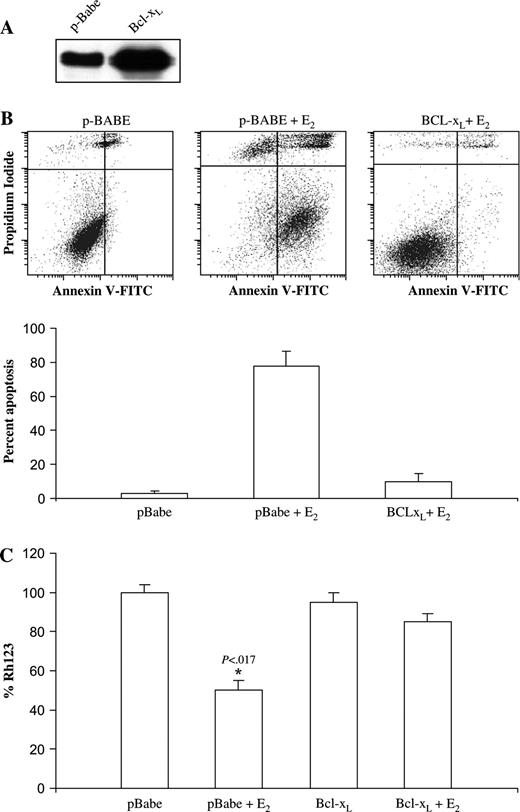
Effect of overexpression of Bcl-x L on estradiol-induced apoptosis of MCF-7:5C cells. Bcl-x L was stably overexpressed in MCF-7:5C cells using the retroviral vector pBabe-Puro-Bcl-x L . A ) Western blot analysis of Bcl-x L expression in Bcl-x L –transfected and pBabe mock-transfected MCF-7:5C cells. B ) Annexin V staining for apoptosis. pBabe vector control and Bcl-x L –overexpressing MCF-7:5C cells were treated with either ethanol vehicle (control) or 1 n M estradiol for 72 hrs. The cells were then double stained with recombinant FITC-conjugated annexin V and propidium iodide and then analyzed by flow cytometry. The percentage of apoptotic cells in each treatment group is shown in the bottom panel . The experiment was repeated three times. The data represent the mean of three independent experiments, and error bars show upper 95% confidence intervals. C ) Loss of mitochondrial transmembrane potential (Δψ m ) was determined by rhodamine-123 (Rh123) retention assay as described in Materials and Methods.
Role of Bax and Bim in Estradiol-Induced Apoptosis in MCF-7:5C Cells
Bax and Bim are important regulators of mitochondrion-mediated apoptosis; hence, we examined the effect of blocking Bax and Bim mRNA expression (using siRNAs) on estradiol-induced apoptosis in MCF-7:5C cells. Transient transfection of MCF-7:5C cells with either Bax or Bim siRNAs almost completely abolished protein levels compared with those in cells transfected with control siRNAs ( Fig. 6, A ). These cells also had 65% [95% CI = 60% to 75%, P <.001] and 85% [95% CI = 80% to 90%, P <.001] reductions in estradiol-induced apoptosis, respectively, compared with cells transfected with the control siRNA ( Fig. 6, B ). Thus, both Bax and Bim appear to be important components of estradiol-induced apoptosis.
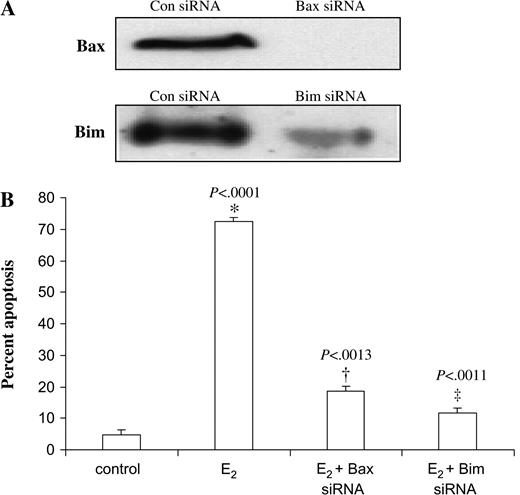
RNA interference and apoptosis in MCF-7:5C cells. A ) MCF-7:5C cells were transfected with a scrambled sequence siRNA (con), Bax siRNA, or Bim siRNA. After 48 hours, lysates were prepared and analyzed by western blotting. Representative blots are shown. B ) Transfected MCF-7:5C cells were treated with ethanol vehicle (control) or 1 n M estradiol (E 2 ) for 72 hours, and the percentage of cells that were apoptotic was assessed by annexin V staining. * P <.001 for the E 2 + Bax siRNA group compared with the E 2 group alone. † P <.001 for the E 2 + Bim siRNA group compared with the E 2 group alone.
Role of p53 in Estradiol-Induced Apoptosis in MCF-7:5C Cells
The p53 tumor suppressor protein is an important apoptosis regulator, and recent evidence suggests that p53-mediated cell death occurs primarily through the intrinsic mitochondrial pathway ( 43 ) . Therefore, we examined the role of p53 in estradiol-induced apoptosis in MCF-7:5C cells. Western blot analysis showed that estradiol markedly increased p53 protein level in MCF-7 cells and MCF-7:5C cells, whereas fulvestrant almost completely decreased p53 protein levels in both cell lines ( Fig. 7, A ). Estradiol treatment also statistically significantly increased p53 mRNA expression in MCF-7:5C cells compared with control ( P = .011) ( Fig. 7, B ). To determine whether p53 was functional in MCF-7:5C cells, we also measured the expression of the p53-regulated proteins p21, Mdm2, Puma, and Noxa. Figure 7, A shows that estradiol treatment markedly increased the protein levels of Puma and Noxa in MCF-7:5C cells but did not alter the levels of p21 or MDM2.
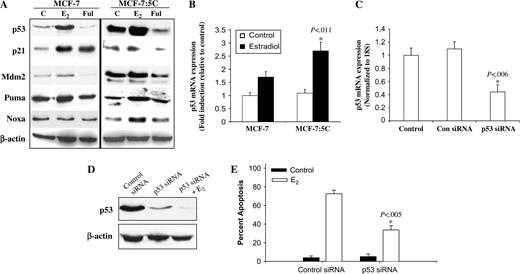
Effect of estradiol and fulvestrant on expression of p53 and p53 target proteins in MCF-7 and MCF-7:5C cells. A ) Cells were treated with ethanol vehicle (C), 1 n M estradiol (E 2 ), or 1 μ M fulvestrant (Ful) for 48 hours. Lysates were analyzed by western blotting with antibodies against p53, p21, MDM2, Puma, and Noxa (Santa Cruz Biotechnology). β-Actin was used as a loading control. B ) Reverse transcriptase polymerase chain reaction (RT-PCR) of p53 expression in MCF-7 and MCF-7:5C cells following estradiol treatment for 48 hours. Ribosomal 18S mRNA was used as a loading control. C ) Inhibition of p53 mRNA expression in MCF-7:5C cells using small siRNA. Ribosomal 18S mRNA was used as a loading control. * P = .006 for difference in p53 expression between p53 siRNA and control siRNA. D ) Western blot analysis of cells transfected with either control siRNA or p53 siRNA. β-Actin was used as a loading control. E ) Percent apoptotic cells determined by annexin V staining of MCF-7:5C cells transfected with control siRNA or p53-specific siRNA and treated with 1 n M E 2 for 72 hours. * P = .005 for E 2 -induced apoptosis in p53-transfected cells compared with cells transfected with the control siRNA. Results are typical of three independent experiments.
We then examined whether p53 induction was required for estradiol-induced apoptosis by using siRNA to suppress p53 mRNA expression in MCF-7:5C cells. Cells transiently transfected with a p53-suppressing siRNA demonstrated dramatic reductions in p53 mRNA and dramatic protein levels compared with those in cells transfected with the scrambled control siRNA ( Fig. 7, C and D ). In addition, cells transfected with the p53 siRNA showed a strong decrease in estradiol-induced apoptosis compared with cells transfected with the control siRNA ( Fig. 7, E ). Overall, these results indicate that p53 induction is important for estradiol-induced apoptosis in MCF-7:5C cells.
Growth of LTED MCF-7:5C Cells In Vivo
The ability of estradiol to induce apoptosis in vitro raised the possibility that this agent might induce tumor regression in vivo. MCF-7:5C cells were bilaterally injected into the mammary fat pads of athymic mice (N = 30), and within approximately 4 weeks, measurable tumors were detected with a mean tumor cross-sectional area of 0.23 cm 2 [95% CI = 0.22 to 0.24]. The mice were then randomly assigned to a treatment group [i.e., estradiol (n = 10), fulvestrant (n = 10), and vehicle control (n = 10)] ( Fig. 8, A ). MCF-7:5C tumors in the vehicle control group continued to grow, reaching a mean cross-sectional area of 0.49 cm 2 [95% CI = 0.47 to 0.51] by week 8. By contrast, MCF-7:5C tumors in mice treated with postmenopausal doses of estradiol (0.3-cm silastic capsule) regressed in a time-dependent manner and were undetectable by week 8 ( Fig. 8, A ). Estradiol-induced tumor regression was observed as early as 1 week after treatment and became more pronounced by 2 weeks ( Fig. 8, A ). Fulvestrant also reduced MCF-7:5C tumor growth relative to control ( P = .007); however, the reduction was statistically significantly less than that of estradiol ( P <.001).
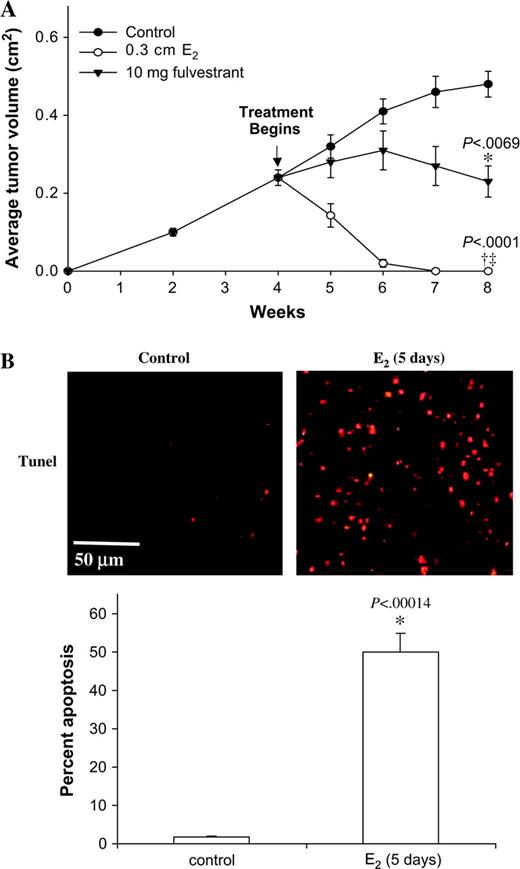
Effect of estradiol and fulvestrant on the growth of MCF-7:5C tumors. A ) Thirty ovariectomized athymic mice were injected bilaterally with MCF-7:5C cells. Tumors were grown in the absence of treatment to a mean cross-sectional area of 0.23 cm 2 . Mice were then randomly assigned into three groups of 10 mice each and treated as follows: 1) vehicle (control), 2) fulvestrant, or 3) estradiol (E 2 ). E 2 was given as a 0.3-cm silastic capsule subcutaneously, and fulvestrant was given subcutaneously as a 5-mg dose twice a week. Tumor size was measured weekly. *Cross-sectional area at 8 weeks of fulvestrant-treated tumors was statistically significantly ( P = .007) different from that of tumors treated with control vehicle. †Cross-sectional area of tumors at 8 weeks treated with E 2 was statistically significantly ( P <.001) different from that of tumors treated with vehicle control. ‡Cross-sectional area of estradiol-treated tumors at 8 weeks was statistically significantly ( P <.001) different from that of fulvestrant-treated tumors. Three separate experiments were performed (on separate occasions), yielding similar results. For each experiment, 30 mice (10 per group) were used. Data are from a representative experiment. Error bars = 95% confidence intervals. B ) Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling (TUNEL) analysis of apoptosis in tumor sections. On day 5, tumors from control and E 2 -treated mice bearing MCF-7:5C tumors were fixed, sectioned, and then placed on slides for detection of apoptosis. Six independent tumors (each from a different mouse) were taken from the control group ( left panel ) or E 2 -treated group ( right panel ) for apoptosis detection. Slides were examined under 40× magnification with a fluorescence microscope. The number of apoptotic cells in four random fields was measured from each tumor section, and representative fields are shown. Scale bars = 50 μm. Bottom panel , percent apoptosis was calculated by dividing the total number of TUNEL-positive cells in each field by the total number of epithelial cells and multiplying by 100. Data are the mean and 95% confidence intervals of four experiments. * P <.001 for the estradiol-treated group versus control group.
To determine whether apoptosis was involved in estradiol-induced regression of MCF-7:5C tumors, we used the TUNEL assay to quantitate apoptosis in tumor sections from control and estradiol treatment groups at day 5 after the commencement of treatment ( Fig. 8, B ). We found that 49.9% [95% CI = 45.9% to 53.9%] of cells in the estradiol-treated MCF-7:5C tumors were apoptotic, whereas only 0.7% [95% CI = 1.6% to 1.8%] of cells in the vehicle control tumors were apoptotic ( P <.001).
D ISCUSSION
In the present study, we investigated the mechanism of estradiol-induced apoptosis in MCF-7:5C cells and found that the mitochondrial (i.e., intrinsic) pathway plays a predominant role in this process. The expression of several proapoptotic proteins—including Bax, Bak, Bim, Noxa, Puma, and p53—was markedly increased with estradiol treatment, and experiments using siRNAs demonstrated that the induction of Bax, Bim, and p53 was necessary for estradiol-induced apoptosis in MCF-7:5C cells. Estradiol treatment also led to a loss of mitochondrial potential and a dramatic increase in the release of cytochrome c from the mitochondria, which resulted in activation of caspases and cleavage of PARP. Furthermore, overexpression of antiapoptotic Bcl-x L was able to protect MCF-7:5C cells from estradiol-induced apoptosis. Estradiol also caused complete and rapid tumor regression in vivo, which was partially reversed by fulvestrant. To our knowledge, this study is the first to show a link between estradiol-induced cell death and activation of the mitochondrial apoptotic pathway in a breast cancer cell model resistant to estrogen withdrawal.
Song et al. ( 19 ) have previously reported a link between estradiol-induced apoptosis and activation of the Fas/FasL signaling pathway in LTED breast cancer cells. However, we found that activation of neither FasR nor FasL was critical for estradiol-induced apoptosis in MCF-7:5C cells. Inhibition of FasR and FasL protein expression using neutralizing antibodies and siRNAs did not prevent estradiol-induced apoptosis in MCF-7:5C cells, and estradiol treatment did not alter caspase 8 activation or Bid processing, which are important events in Fas-mediated apoptosis ( 44 ) . However, there is evidence indicating that Bid cleavage through caspase 8 activation plays a role in mediating cross-talk between the extrinsic and intrinsic apoptosis pathways ( 45 ) . For example, a recent model of Fas-induced apoptosis in breast epithelial cells suggests that proapoptotic Bid, on cleavage and activation by caspase 8, translocates from the cytosol to the mitochondria and causes a conformational change in the N terminus of Bax, resulting in mitochondrial cytochrome c release and amplification of apoptosis ( 46 ) . There is also evidence suggesting that p53 may play a linker role in connecting the Fas death receptor to the mitochondria through Bid translocation and cytochrome c release ( 47 ) . The absence of estradiol-induced caspase 8 activation and Bid cleavage in MCF-7:5C cells suggests, however, that Bid cleavage–mediated cross-talk between the extrinsic and intrinsic pathways may not be a critical factor in estradiol-induced apoptosis in MCF-7:5C cells.
We postulate that estradiol induces mitochondrion-mediated apoptosis, in part, by increasing the expression of proapoptotic Bcl-2 family proteins, disrupting mitochondrial membrane integrity, and facilitating the translocation of cytochrome c from the mitochondria into the cytosol. In the current study, we found that estradiol treatment markedly increased the levels of proapoptotic Bax, Bak, and Bim proteins in MCF-7:5C cells and that these increases coincided with the loss of mitochondrial membrane integrity, cytochrome c release, caspase 9 activation, and PARP cleavage. Estradiol treatment also increased Bax, Bak, and Bim mRNA expression in MCF-7:5C cells (data not shown), and siRNA targeting of Bax and Bim dramatically reduced the ability of estradiol to induce apoptosis in these cells. We also found evidence of mitochondrial accumulation of Bax and Bak proteins following estradiol treatment (data not shown). Both Bax and Bak play an important role in controlling mitochondrial integrity ( 48 ) . Bax is normally found as a monomer in the cytosol of nonapoptotic cells, and it oligomerizes and translocates to the outer mitochondrial membrane in response to apoptotic stimuli ( 49 ) and induces mitochondrial membrane permeabilization ( 50 ) and cytochrome c release ( 51 ) . Bak resides in the mitochondrial outer membrane and has been shown to contain a homologous BH3 binding pocket critical for interactions with the Bak-activating, BH3-only protein Bid ( 52 ) . Once activated, Bak oligomerizes in the mitochondrial outer membrane and induces defects in membrane integrity and the release of apoptogenic factors, including cytochrome c ( 53 ) . Cytochrome c is normally located in the intermembrane space of the mitochondrion, loosely bound to the inner membrane. Once cytochrome c is released into the cytosol, it interacts with apoptotic protease activating factor-1 (Apaf-1) and procaspase 9, leading to the generation of active caspase 9, which is capable of proteolytically activating downstream caspases that then initiate the apoptotic degradation phase ( 54 ) . Most notably, we found substantial release of cytochrome c in the cytosol of estradiol-treated MCF-7:5C cells compared with vehicle-treated control, which was completely blocked by Bcl-x L overexpression (data not shown). Activation of caspase 9 and caspase 7 and cleavage of PARP were also observed in estradiol-treated cells, and these changes were associated with the levels of cytochrome c in the estradiol-treated cells but not in the control cells. Overall, these findings indicate that the mitochondrion is a key target of estradiol-induced apoptosis and that the induction of proapoptotic Bax and Bak may play an important role in facilitating this process.
Although the induction of proapoptotic Bax and Bak disrupts mitochondrial membrane integrity, the overexpression of antiapoptotic proteins such as Bcl-2 and Bcl-x L has been found to stabilize the outer mitochondrial membrane and prevent the release of cytochrome c following a variety of insults, including treatment with chemotherapeutic agents ( 55 , 56 ) . Heterodimers of Bcl-2/Bax have been shown to prevent pore formation and inhibit cytochrome c release and ultimately apoptosis ( 57 ) . In our study, we found that, although estradiol treatment dramatically increased the levels of proapoptotic proteins, it did not statistically significantly alter the levels of antiapoptotic Bcl-2 and Bcl-x L proteins in MCF-7:5C cells. This finding is important because there is evidence that suggests that it is the ratio rather than the amount of antiapoptotic versus proapoptotic proteins that determines whether apoptosis will proceed ( 58 ) . Consistent with this notion, we have found that, in wild-type MCF-7 cells, which are resistant to estradiol-induced apoptosis, estradiol treatment dramatically increased the protein level of antiapoptotic Bcl-2 but did not alter the level of proapoptotic Bax protein. Thus, it is reasonable to suggest that the apoptotic potential of estradiol is directly related to its ability to alter the ratio between proapoptotic and antiapoptotic proteins in target cells.
The tumor suppressor protein p53 also plays a role in mitochondrion-mediated apoptotic cell death ( 43 ) . p53 can mediate intrinsic apoptosis by transcriptional activation of genes that encode proapoptotic proteins such as the BH3-only proteins Noxa and Puma, Bax, p53, and Apaf-1 and by transcriptional repression of Bcl-2 and IAPs ( 59 ) . p53 can also mediate apoptosis via transcription-independent mechanisms. In some cell types, p53-dependent apoptosis occurs in the absence of any gene transcription or translation ( 60 ) . Moreover, transcriptionally inactive p53 mutants have been shown to induce death in tumor cells ( 61 ) . In our study, estradiol treatment markedly increased p53 protein and mRNA and Puma and Noxa protein in MCF-7:5C cells. We also found that blockade of p53 mRNA expression using siRNA reduced estradiol-induced apoptosis in these cells. Although we did not sequence p53 to determine its mutational status, we did observe marked increases in the expression of the p53 target genes Noxa and Puma in estradiol-treated MCF-7:5C cells, which suggests that p53 might be functional in MCF-7:5C cells. Furthermore, blockade of p53 mRNA expression reduced estradiol-induced apoptosis in MCF-7:5C cells. Interestingly, estradiol also increased p53 protein expression in parental MCF-7 cells; however, this induction was observed in the cytosol and was not associated with apoptosis. Estradiol-induced wild-type p53 expression in MCF-7 cells has previously been reported, and it has been suggested that the induction is an ERα-mediated event and is due to ERα interaction with p53, which enhances its stability ( 62 ) .
A direct interaction between ERα and p53 is demonstrated by our finding that fulvestrant, which is known to degrade the ERα, was able to completely block estradiol induction of p53 and reduce the basal level of p53 in MCF-7:5C cells. Furthermore, measurable levels of p53 protein were found in the mitochondrial fraction of estradiol-treated MCF-7:5C cells. Marchenko et al. ( 63 ) have previously reported that the p53 protein localizes to the mitochondria in tumor cells undergoing p53-dependent apoptosis but not in cells undergoing p53-dependent growth arrest or p53-independent cell death. Although the exact mechanism by which mitochondrial p53 triggers apoptosis remains unclear, it has been proposed that p53 interacts directly with antiapoptotic Bcl-2 and Bcl-x L , thereby liberating proapoptotic Bax and Bak to induce changes in the mitochondrial membrane that lead in turn to cytochrome c release, caspase activation, and cell death ( 64 , 65 ) . Overall, these results demonstrate that p53 plays an important role in estradiol-induced apoptosis in MCF-7:5C cells and that p53 acts in concert with the Bcl-2 family protein to activate the mitochondrial pathway.
Although the results in this study strongly support the involvement of the mitochondrial pathway in estradiol-induced apoptosis in MCF-7:5C cells, it is important to note that our study was essentially completed in a single MCF-7 variant. Hence, we cannot discount the importance of the FasR/FasL signaling pathway in estradiol-induced apoptosis in other breast cancer cell models that are resistant to estrogen withdrawal. As mentioned before, Song et al. ( 19 ) have previously reported a statistically significant link between estradiol-induced apoptosis and activation of the FasR/FasL death signaling pathway in an LTED breast cancer cell model. The LTED cells used in their study, however, were derived from a whole cell population, whereas the MCF-7:5C cells used in our study were derived from clonal selection. We chose to focus primarily on MCF-7:5C cells because they represent a pure cell model (i.e., estradiol causes rapid and complete apoptosis both in vitro and in vivo) and because doing so provided the opportunity to fully elucidate the mechanism of estradiol-induced apoptosis. Our laboratory also has developed two other LTED breast cancer cell variants, MCF-7:ED and MCF-7:2A; however, these cells are less sensitive to estradiol-induced apoptosis than are MCF-7:5C cells.
In conclusion, our study provides a mechanistic basis for estradiol-induced apoptosis in a human breast cancer cell model resistant to LTED. Estradiol induced activation of the mitochondrial apoptotic pathway in MCF-7:5C cells by increasing proapoptotic Bax, Bak, Bim, Noxa, Puma, and p53 protein expression and increasing the loss of mitochondrial transmembrane potential and cytochrome c release. Of note, the pure antiestrogen fulvestrant caused growth arrest and tumor stasis but not apoptosis, whereas estradiol caused complete and rapid tumor regression. These laboratory data have important clinical implications, particularly for the use of aromatase inhibitors as long-term therapy, and they suggest that, if and when resistance to aromatase inhibition occurs, a strategy of treatment with estrogen (either exogenous estrogen or the woman's endogenous estrogen) may be sufficient to kill the cancer and control disease progression. We have previously observed that, once the estradiol-induced tumoricidal action is complete in tamoxifen-stimulated tumors, the remaining tumor tissue is again responsive to antihormonal therapy ( 20 ) . A clinical strategy of reducing tumor burden with low-dose, short-term estrogen therapy followed by optimal antihormonal therapy with fulvestrant or aromatase inhibitor should be tested in clinical trials.
Supported by the Avon Foundation, a Specialized Programs of Research Excellence (SPORE) grant CA89018 in breast cancer research (to V. C. Jordan) from the National Cancer Institute, National Institutes of Health, Department of Health and Human Services, and the Lynn Sage Breast Cancer Research Foundation. J. S. Lewis was funded by a Department of Defense Training Grant in Breast Neoplasia DAMD17–00–5671–0386 and Signal Transduction Training Grant #T32 CA70085–06. C. Osipo was funded by a Judy Dlugie Memorial Fund Fellowship for Breast Cancer Research. The sponsors were not involved in the design, conduct, or reporting of the study.
References
McKnight JJ, Gray SB, O'Kane HF, Johnston SR, Williamson KE. Apoptosis and chemotherapy for bladder cancer.
Rupnow BA, Knox SJ. The role of radiation-induced apoptosis as a determinant of tumor responses to radiation therapy.
Dowsett M, Archer C, Assersohn L, Gregory RK, Ellis PA, Salter J, et al. Clinical studies of apoptosis and proliferation in breast cancer.
Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond.
Tsujimoto Y. Bcl-2 family of proteins: life-or-death switch in mitochondria.
Thiantanawat A, Long BJ, Brodie AM. Signaling pathways of apoptosis activated by aromatase inhibitors and antiestrogens.
Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study.
Coombes RC, Hall E, Gibson LJ, Paridaens R, Jassem J, Delozier T, et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer.
Johnston S. Fulvestrant and the sequential endocrine cascade for advanced breast cancer.
Zhang W, Couldwell WT, Song H, Takano T, Lin JH, Nedergaard M. Tamoxifen-induced enhancement of calcium signaling in glioma and MCF-7 breast cancer cells.
Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, et al. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells.
Choi KC, Kang SK, Tai CJ, Auersperg N, Leung PC. Estradiol up-regulates antiapoptotic Bcl-2 messenger ribonucleic acid and protein in tumorigenic ovarian surface epithelium cells.
Kyprianou N, English HF, Davidson NE, Isaacs JT. Programmed cell death during regression of the MCF-7 human breast cancer following estrogen ablation.
Teixeira C, Reed JC, Pratt MA. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl-2 proto-oncogene expression in human breast cancer cells.
Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta.
Jordan VC. Selective estrogen receptor modulation: concept and consequences in cancer.
Lewis JS, Cheng D, Jordan VC. Targeting oestrogen to kill the cancer but not the patient.
Osipo C, Liu H, Meeke K, Jordan VC. The consequences of exhaustive antiestrogen therapy in breast cancer: estrogen-induced tumor cell death.
Song RX, Mor G, Naftolin F, McPherson RA, Song J, Zhang Z, et al. Effect of long-term estrogen deprivation on apoptotic responses of breast cancer cells to 17beta-estradiol.
Yao K, Lee ES, Bentrem DJ, England G, Schafer JI, O'Regan RM, et al. Antitumor action of physiological estradiol on tamoxifen-stimulated breast tumors grown in athymic mice.
Haddow A, Watkinson JM, Paterson E. Influence of synthetic oestrogens upon advanced malignant disease.
Lewis JS, Osipo C, Meeke K, Jordan VC. Estrogen-induced apoptosis in a breast cancer cell model resistant to long-term estrogen withdrawal.
Dodwell D, Vergote I. A comparison of fulvestrant and the third-generation aromatase inhibitors in the second-line treatment of postmenopausal women with advanced breast cancer.
Osipo C, Gajdos C, Liu H, Chen B, Jordan VC. Paradoxical action of fulvestrant in estradiol-induced regression of tamoxifen-stimulated breast cancer.
Liu H, Lee ES, Gajdos C, Pearce ST, Chen B, Osipo C, et al. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo.
Jiang SY, Wolf DM, Yingling JM, Chang C, Jordan VC. An estrogen receptor positive MCF-7 clone that is resistant to antiestrogens and estradiol.
Labarca C, Paigen K. A simple, rapid, and sensitive DNA assay procedure.
MacGregor Schafer J, Liu H, Bentrem DJ, Zapf JW, Jordan VC. Allosteric silencing of activating function 1 in the 4-hydroxytamoxifen estrogen receptor complex is induced by substituting glycine for aspartate at amino acid 351.
Patel T, Gores GJ, Kaufmann SH. The role of proteases during apoptosis.
Park IC, Woo SH, Park MJ, Lee HC, Lee SJ, Hong YJ, et al. Ionizing radiation and nitric oxide donor sensitize Fas-induced apoptosis via up-regulation of Fas in human cervical cancer cells.
Gottardis MM, Jordan VC. Development of tamoxifen-stimulated growth of MCF-7 tumors in athymic mice after long-term antiestrogen administration.
O'Regan RM, Cisneros A, England GM, MacGregor JI, Muenzner HD, Assikis VJ, et al. Effects of the antiestrogens tamoxifen, toremifene, and ICI 182 780 on endometrial cancer growth.
Zeger SL, Liang KY. Longitudinal data analysis for discrete and continuous outcomes.
Soule HD, Vazguez J, Long A, Albert S, Brennan M. A human cell line from a pleural effusion derived from a breast carcinoma.
Lippman M, Bolan G, Huff K. The effects of estrogens and antiestrogens on hormone-responsive human breast cancer in long-term tissue culture.
Lippman ME, Bolan G. Oestrogen-responsive human breast cancer in long term tissue culture.
Jiang SY, Jordan VC. Growth regulation of estrogen receptor-negative breast cancer cells transfected with complementary DNAs for estrogen receptor.
Berry M, Nunez AM, Chambon P. Estrogen-responsive element of the human pS2 gene is an imperfectly palindromic sequence.
Nunez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway.
Hadjiloucas I, Gilmore AP, Bundred NJ, Streuli CH. Assessment of apoptosis in human breast tissue using an antibody against the active form of caspase 3: relation to tumour histopathological characteristics.
Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria.
Schuler M, Green DR. Mechanisms of p53-dependent apoptosis.
Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death.
Murphy KM, Streips UN, Lock RB. Bax membrane insertion during Fas(CD95)-induced apoptosis precedes cytochrome c release and is inhibited by Bcl-2.
Thiery J, Abouzahr S, Dorothee G, Jalil A, Richon C, Vergnon I, et al. p53 potentiation of tumor cell susceptibility to CTL involves Fas and mitochondrial pathways.
Korsmeyer SJ. BCL-2 gene family and the regulation of programmed cell death.
Goping IS, Gross A, Lavoie JN, Nguyen M, Jemmerson R, Roth K, et al. Regulated targeting of BAX to mitochondria.
Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death.
Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c.
Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c.
Green D, Kroemer G. The central executioners of apoptosis: caspases or mitochondria?
Nunez G, Clarke MF. The Bcl-2 family of proteins: regulators of cell death and survival.
Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death.
Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death.
Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy.
Gao C, Tsuchida N. Activation of caspases in p53-induced transactivation-independent apoptosis.
Kokontis JM, Wagner AJ, O'Leary M, Liao S, Hay N. A transcriptional activation function of p53 is dispensable for and inhibitory of its apoptotic function.
Okumura N, Saji S, Eguchi H, Hayashi S, Nakashima S. Estradiol stabilizes p53 protein in breast cancer cell line, MCF-7.
Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling.
Mihara M, Moll UM. Detection of mitochondrial localization of p53.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}