





Abstract
Extensive gene rearrangement is reported in the mitochondrial genomes of lungless salamanders (Plethodontidae). In each genome with a novel gene order, there is evidence that the rearrangement was mediated by duplication of part of the mitochondrial genome, including the presence of both pseudogenes and additional, presumably functional, copies of duplicated genes. All rearrangement-mediating duplications include either the origin of light-strand replication and the nearby tRNA genes or the regions flanking the origin of heavy-strand replication. The latter regions comprise nad6, trnE, cob, trnT, an intergenic spacer between trnT and trnP and, in some genomes, trnP, the control region, trnF, rrnS, trnV, rrnL, trnL1, and nad1. In some cases, two copies of duplicated genes, presumptive regulatory regions, and/or sequences with no assignable function have been retained in the genome following the initial duplication; in other genomes, only one of the duplicated copies has been retained. Both tandem and nontandem duplications are present in these genomes, suggesting different duplication mechanisms. In some of these mitochondrial DNAs, up to 25% of the total length is composed of tandem duplications of noncoding sequence that includes putative regulatory regions and/or pseudogenes of tRNAs and protein-coding genes along with the otherwise unassignable sequences. These data indicate that imprecise initiation and termination of replication, slipped-strand mispairing, and intramolecular recombination may all have played a role in generating repeats during the evolutionary history of plethodontid mitochondrial genomes.
Introduction
In contrast to the conclusions drawn from early and limited sampling, animal mitochondrial genomes possess unexpected diversity both in gene order and in the presence, extent, and distribution of noncoding sequence (Inoue et al. 2003; Boore, Medina, and Rosenberg 2004; Miller et al. 2004; Yokobori et al. 2004). Groups of organisms with highly differing gene orders are appropriate model systems for studying the factors that effect mitochondrial gene rearrangement (Dowton and Campbell 2001). Such groups have been identified among invertebrates (Yamazaki et al. 1997; Dowton and Austin 1999; Shao et al. 2001), and testable hypotheses for the causes of frequent gene rearrangement have been generated; for example, highly rearranged parasitic wasp mitochondrial genomes may result from oxidative stress imposed by the host immune response (Dowton and Campbell 2001). Within the largest salamander family, Plethodontidae, 6 of the 22 mitochondrial genomes examined have rearrangements of independent phylogenetic origin, and 12 of these 22, plus 1 from the family Rhyacotritonidae, have tandem repeats in noncoding sequences. This high instance of gene rearrangement makes plethodontid salamanders an excellent model system for examining the mechanisms of such vertebrate mitochondrial genome instability.
In the most commonly invoked model of mitochondrial gene rearrangement, a region of the genome is duplicated and the supernumerary genes, no longer maintained by selection, are eliminated; the original gene arrangement is either restored or altered, depending on the pattern of gene loss (Moritz, Dowling, and Brown 1987; Boore 2000). Genomic evidence for this model of rearrangement includes (1) repeated motifs, which can cause duplication via slipped-strand mispairing; (2) stem-loop structures, which can cause duplication via intramolecular recombination (Stanton et al. 1994; Moore, Gudikote, and Van Tuyle 1998); and (3) pseudogenes, which may be ancestral duplications in the process of being eliminated (Arndt and Smith 1998; Macey et al. 1998). This mechanism cannot explain gene inversions, which have also been reported (Smith et al. 1989; Boore 1999; Dowton et al. 2003; Miller et al. 2004). In each of the six rearranged genomes reported here, evidence of at least one historical duplication event has been retained. Plethodontid rearrangements involve regions of the genome that are independently rearranged or duplicated in other vertebrate taxa (Moritz and Brown 1986; Desjardins and Morais 1990; Moritz 1991; Pääbo et al. 1991; Quinn and Mindell 1996). The molecular mechanisms of gene rearrangement at work in plethodontids may therefore be the same mechanisms acting across vertebrates.
Organismal-, cellular-, and genomic-level properties may be linked to mitochondrial genome instability, both in these salamanders and more generally. In this paper, we (1) describe mitochondrial genome rearrangements and expansions of noncoding DNA in plethodontid and related salamanders, (2) infer possible molecular mechanisms by which these rearrangements and expansions originated, and (3) outline possible explanations for the extensive mitochondrial genome instability in this clade.
Materials and Methods
Twenty-four complete mitochondrial genomes of plethodontid and related salamanders were sequenced, assembled, and annotated as described elsewhere (Mueller et al. 2004) (GenBank accession numbers AY728212–AY728235). Annotated genomes were examined for novel gene orders and the presence of unassignable sequences. Putative pseudogene sequences in each rearranged genome were identified by position and aligned with the corresponding functional gene sequences using ClustalW (gap parameters set to default: open = 10, extend = 5) and manual adjustment. Two other noncoding regions of the genome were also examined: the control region (CR hereafter), and an intergenic spacer between trnT and trnP (IGS hereafter) present in diverse salamander clades that may be a remnant of at least one older duplication (McKnight and Shaffer 1997; Zardoya et al. 2003; Zhang et al. 2003a,b). Because these regions are highly variable with only a few, or no, short conserved sequences (Shadel and Clayton 1997), we defined these two regions by their genome position. All noncoding regions were tested for the presence of tandem repeat elements and for shared repeats among the different regions by the construction of Pustell DNA-DNA matrices using MacVector (window size = 30, minimum percent score = 80, hash = 6) (Accelrys, San Diego, Calif.). The genome sequence of Hydromantes italicus is incomplete (trnT, the IGS, trnP, the CR, and trnF are missing) and therefore was not examined for repeats. Finally, each genome was tested for possible heteroplasmy in the numbers of repeated regions by examining the sequences of individual clones for their ability to produce assemblies with identical end sequences but different numbers of repeats from the primary assembly.
Results and Discussion
Duplications Have Mediated Most Rearrangements
Six of the 22 plethodontid genomes have novel gene orders and arrangements of noncoding sequences that are consistent with the duplication-random loss model of gene rearrangement (Boore 2000). In this model, a portion of the genome containing at least two genes is duplicated. One of the two copies of each duplicated gene eventually loses function, becomes a pseudogene, and is excised from the genome by mutational processes because it is not maintained by selection. Which gene copy is ultimately lost is determined by the first loss-of-function mutation, with some patterns restoring the original order and others leading to rearrangement. In this study, two such rearrangements include the origin of light-strand replication (OL), two include the IGS, and two include both the origin of heavy-strand replication (OH, contained within the CR) and the IGS. In some cases, there are evident pseudogenes that signal historical duplications. In others, these pseudogenes have decayed beyond ∼60% identity to the functional copy and are inferred based solely on their genome position. In all cases, the exact boundaries of the duplicated fragments have likely been obscured by deletion, and the extent of the original duplication may therefore be underestimated. The phylogenetic positions of the taxa with these rearranged genomes indicate separate rearrangement events, with the possible exception of one rearrangement shared by Aneides flavipunctatus and Aneides hardii, although the genome of A. hardii also appears to have undergone a second duplication-mediated rearrangement (fig. 1). For each rearranged mitochondrial genome, we infer the minimum set of contiguous genes that, when duplicated, creates an intermediate sequence from which gene losses could have established the observed gene arrangement.
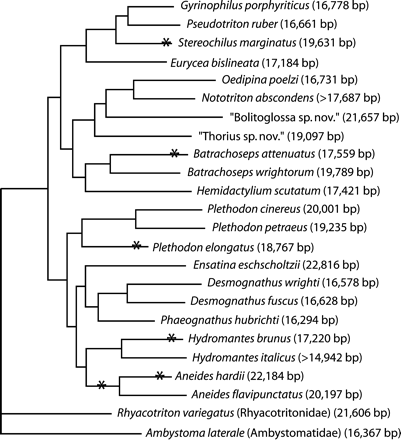
Phylogenetic relationships among plethodontid salamanders and two out-groups from other families as indicated (from Mueller et al. 2004). Asterisks indicate lineages that have experienced a duplication-mediated rearrangement. Aneides hardii and Aneides flavipunctatus may share one synapomorphic rearrangement; however, A. hardii underwent a second duplication-mediated rearrangement, not present in A. flavipunctatus. Numbers to the right of species names are mitochondrial genome sizes. The sequences of two species' genomes, Nototriton abscondens and Hydromantes italicus, are incomplete.
Rearrangements Including the OL
Batrachoseps attenuatus
The hypothesized duplicated region in this species' mitochondrial DNA (mtDNA) includes a small fragment of trnW and all of trnA, trnN, OL, trnC, and trnY (∼300 bp in total) (fig. 2). For the purposes of these analyses, OL refers to the entire noncoding region normally found between trnN and trnC that has the potential to form a large stem-loop structure known to function as an origin of replication in some systems. We refer to a copy of OL by pseudogene annotation if it is greatly deficient in potential for forming this structure relative to the other copy, although no experimental data are available to infer which, if either, actually functions as an origin of replication. Deletions in the pseudogenes have reduced their sizes to the following lengths and percentages of original length: partial ψtrnW + ψtrnA, 77 bp (the percentage of original length cannot be determined because the amount of trnW included in the initial duplication is unknown); ψtrnN, 65 bp (93%); ψOL, 8 bp (27%); ψtrnC, 56 bp (86%); and ψtrnY, 28 bp (amount of trnY duplicated is unknown).
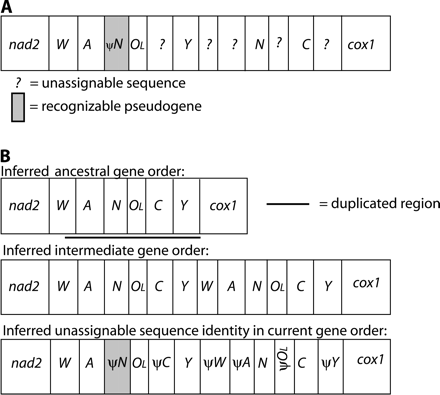
Mitochondrial gene rearrangements in Batrachoseps attenuatus. OL = origin of light-strand replication. Single letters refer to tRNA genes for the corresponding amino acid. Shaded boxes represent recognizable pseudogenes and question marks indicate stretches of sequence that cannot be assigned based on sequence similarity. Lengths of genes and pseudogenes are not to scale. (A) Current gene order. (B) Hypothesized duplication-random loss model for deriving this gene order. ψtrnN retains 60% identity to the functional copy overall, not including two deletions (1 and 3 bp) in the pseudogene; the last 39 bp retain 74% to the functional copy. The remaining pseudogenes have decayed beyond recognition.
Hydromantes brunus
The hypothesized duplicated region in this species' mtDNA is slightly larger than in B. attenuatus, encompassing the end of nad2 and all of trnW in addition to trnA, trnN, OL, trnC, and trnY (∼525 bp) (fig. 3). Deletions in the pseudogenes have reduced their sizes to the following lengths and percentages of original length: partial ψnad2, ∼127 bp (amount of nad2 duplicated is unknown); ψtrnW, ∼60 bp (88%); ψtrnA, 55 bp (81%); ψtrnN, 65 bp (94%); ψOL, 27 bp (73%); ψtrnC, 57 bp (86%); and ψtrnY, 39 bp (58%). Although the initial duplications of B. attenuatus and H. brunus involved similar regions of the genome, different copies of the redundant genes were subsequently lost; this is consistent with random loss, in which the copy of the gene that sustains the first disabling mutation continues to decay.
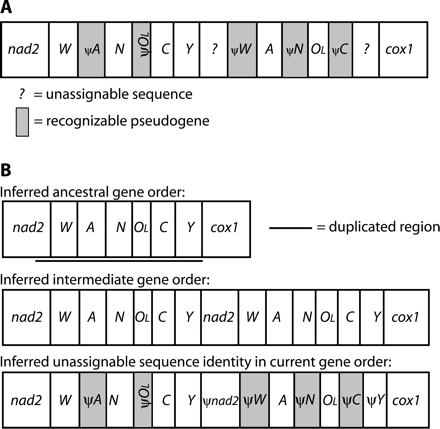
Mitochondrial gene rearrangements in Hydromantes brunus. Designations are as in figure 2. (A) Current gene order. (B) Hypothesized duplication-random loss model for deriving this gene order. ψtrnW retains 69% identity to the functional copy, not including two deletions (3 and 5 bp) in the pseudogene. ψtrnA retains 56% identity to the functional copy overall, not including two deletions (3 and 5 bp) in the pseudogene; the last 44 bp retain 66% identity. ψtrnN retains 82% identity to the functional copy, not including one 5-bp deletion in the pseudogene. ψOL retains 70% identity to the functional copy overall, not including 10 bp missing from its end; the first 18 bp retain 94% identity. ψtrnC retains 87% identity to the functional copy, not including three deletions (2, 1, and 2 bp) in the pseudogene. ψtrnY and ψnad2 have decayed beyond recognition.
Rearrangements Including the CR and/or the IGS
Stereochilus marginatus
The hypothesized duplication in this species' mtDNA includes nad6, trnE, cob, trnT, and the IGS for a total of 1,790 bp plus the unknown length of the IGS (fig. 4). Deletions in the pseudogenes have reduced their sizes to the following lengths and percentages of original lengths: ψnad6, 186 bp (36%); ψtrnE, 66 bp (98.5%); and ψcob, 42 bp (3.7%). ψtrnT, if it still exists, cannot be identified. Two different sets of repeats exist in the two regions of the genome inferred to be copies of the IGS.
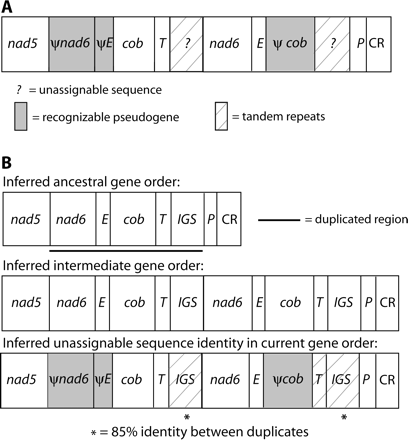
Mitochondrial gene rearrangements in Stereochilus marginatus. Designations are as in figure 2 except that IGS = intergenic spacer between trnT and trnP and CR = control region. (A) Current gene order. (B) Hypothesized duplication-random loss model for deriving this gene order. ψnad6 retains 52% identity to the inferred corresponding 186-bp portion of the functional copy, not including two insertions (2 and 1 bp) in the pseudogene; base pairs 55–132 retain 74% identity. ψtrnE retains 85% identity to the functional copy, not including one 1-bp deletion in the pseudogene. The 42-bp ψcob retains 79% identity to the first 44 bp of the functional copy, not including one 2-bp deletion in the pseudogene. The first putative copy of the IGS, between trnT and nad6, is 1,982 bp in length and contains three complete, and one partial, >95% identical tandem repeats of 437 bp each. The sequence between ψcob and trnP contains five >99% identical copies of a different 110-bp tandem repeat sequence and a sixth 87% identical copy. Notably, following an intervening 102-bp stretch of nonrepetitive sequence, there is a complete copy of the 437-bp repeat unit found in the putative IGS between trnT and nad6. This repeat unit is ∼85% identical to copies in the other putative IGS, despite being separated from them by 1,630 bp. No recognizable ψtrnT remains.
Plethodon elongatus
This species' mitochondrial genome has a gene order and pattern of noncoding sequence consistent with two separate duplications. The initial hypothesized duplication spans the region of the genome including nad6, trnE, cob, trnT, the IGS, trnP, and the CR for a total of 2,811 bp plus the unknown preduplication length of the IGS (fig. 5). Different copies of the redundant genes subsequently decayed to pseudogenes in P. elongatus than in S. marginatus. Deletions in the inferred redundant genes have reduced their sizes to the following lengths and percentages of original lengths: ψnad6 + ψtrnE + ψcob, 47 bp (2.7%); ψtrnP, length unknown because the boundary between the IGS and ψtrnP is unassignable; and ψtrnT + IGS, 107 bp (original length of the IGS is unknown). Notably, two 959-bp 97% identical copies of the putative CR are retained in the genome, indicating that the two may be undergoing concerted evolution as has been reported for several other taxa (Arndt and Smith 1998; Kumazawa et al. 1998; Lee et al. 2001; Inoue et al. 2003).
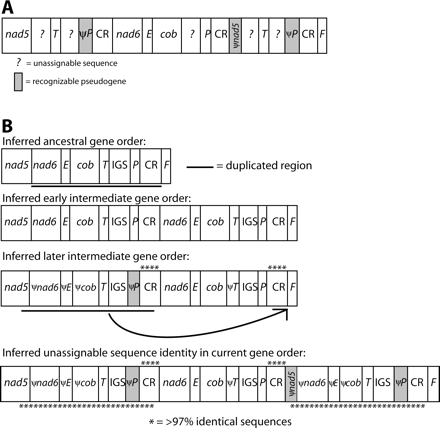
Mitochondrial gene rearrangements in Plethodon elongatus. Designations are as in figure 4. (A) Current gene order. (B) Two hypothesized duplication events mediating rearrangement in the P. elongatus mitochondrial genome. The first duplication resulted in tandem repeats of the region spanning from nad6 to the CR. ψtrnP retains 61% identity to the functional copy overall, not including two insertions (3 bp each) in the pseudogene; the last 44 bp retain 70% identity. ψnad6, ψtrnE, ψcob, and ψtrnT have all decayed beyond recognition. In contrast, the two copies of the CR are 97% identical. Later, a duplicate copy of the fragment comprising the end of nad5, ψnad6, ψtrnE, ψcob, trnT, the IGS, ψtrnP, and a portion of the CR was inserted between the second CR and trnF, likely by intramolecular recombination. The two copies of this fragment are 99% identical.
Following this initial rearrangement by duplication-random loss, we hypothesize that a second duplication gave rise to two copies of the region including the last 57 bp of nad5, ψnad6 + ψtrnE + ψcob, trnT, the IGS, ψtrnP, and the first 761 bp of one CR (1,150 bp in total). These two copies are not adjacent to one another; rather, they are separated by 3,054 bp. The two copies are >99% identical in sequence, implying either (1) very recent duplication or (2) concerted evolution, which is also operating to maintain the sequence identity of the two full-length CRs.
Aneides flavipunctatus
The hypothesized duplication in this species' mtDNA includes nad6, trnE, cob, trnT, the IGS, and trnP for a total of 1,860 bp plus the unknown length of the IGS (fig. 6). The same copies of the redundant genes subsequently decayed to pseudogenes in A. flavipunctatus as in S. marginatus. Deletions in ψnad6 and ψtrnE have completely excised them from the genome. ψcob and ψtrnT, if they still exist, cannot be identified. As seen in the S. marginatus genome, two different sets of repeats exist in the two regions of the genome inferred to contain copies of the IGS.
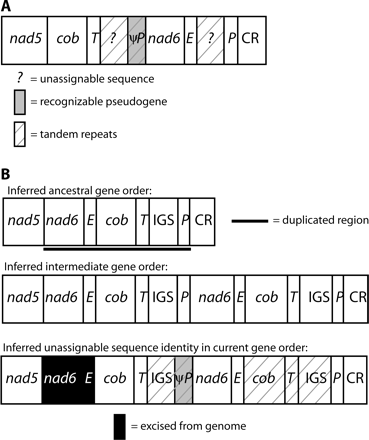
Mitochondrial gene rearrangements in Aneides flavipunctatus. Designations are as in figure 4. (A) Current gene order. (B) Hypothesized duplication-random loss model for deriving this gene order. ψnad6 and ψtrnE have been excised from the genome. The region between trnT and nad6, which comprises a putative IGS and ψtrnP, is 3,050 bp in length and contains seven complete, and one partial, >96% identical 388-bp tandem repeats. Each tandem repeat contains a 72-bp ψtrnP with 54% overall sequence identity to trnP, not including two insertions in the pseudogene (2 and 1 bp); the last 51 bp are 63% identical to the functional trnP. The region between trnE and trnP contains ten >92% identical copies of a 66-bp tandem repeat, a 357-bp stretch of variable numbers of short repeats (5–10 bp), and 71 bp of nonrepetitive sequence. No recognizable ψcob or ψtrnT exists. The two repetitive regions of the genome resemble neither one another nor the CR.
Aneides hardii
This mitochondrial genome has a gene order and pattern of noncoding sequence consistent with two separate tandem duplications (fig. 7). The initial hypothesized duplication includes a near-identical region of the genome duplicated in A. flavipunctatus (from nad6 through the IGS), suggesting that this duplication may be a synapomorphy of the Aneides clade; however, unlike in A. flavipunctatus, no evidence remains that trnP was duplicated in A. hardii. The absence of an identifiable ψtrnP in this duplicated region of the A. hardii genome suggests that (1) the ψtrnP identified in A. flavipunctatus is false, (2) ψtrnP has been excised or completely degraded from A. hardii, or (3) a different duplication-mediated rearrangement occurred independently in each lineage. The second hypothesized duplication in A. hardii includes nad6—the IGS, plus a second IGS, trnP, the CR, trnF, rrnS, trnV, rrnL, trnL1, and nad1 for a total of 6,227 bp and the unknown lengths of the two IGS regions. The copies of nad6 and trnE involved in the second round of duplication must have been functional at the time of reduplication, based on the current position of functional copies in the genome. The ψnad6 and ψtrnE between nad5and cob, which were not involved in the second round of duplication, have been reduced to 11 bp (2%).
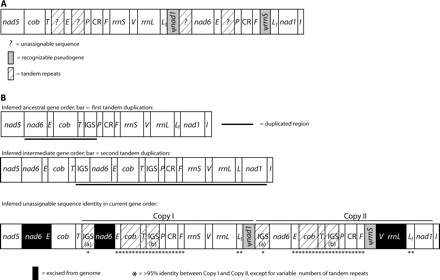
Mitochondrial gene rearrangements in Aneides hardii. Designations are as in figure 4. (A) Current gene order. (B) Two hypothesized duplication events in the history of the A. hardii mitochondrial genome. The first duplication comprised the region spanning from nad6 to the IGS; this duplication may be a synapomorphy shared by A. hardii and Aneides flavipunctatus. The second duplication comprised the region spanning from IGS to nad1. The putative IGS (IGSa) that bounds the two copies of this duplication fragment (Copy I and Copy II) contains varying numbers of a 343-bp tandem repeat. Copy I has one complete and one partial tandem repeat units, and Copy II has four complete >95% identical tandem repeat units and one partial tandem repeat unit. Adjacent to these IGSa sequences, Copy II contains a functional nad6, whereas Copy I possesses neither a functional nad6 nor a recognizable ψnad6. The two copies of trnE, adjacent to nad6 (Copy II) and IGSa (Copy I), are 97% identical to one another excluding a 3-bp deletion in the D-stem of Copy II. ψcob, ψtrnT, and an additional IGS (IGSb), if retained in the genome, should be found in each fragment between trnE and trnP. There is no recognizable ψcob or ψtrnT in this region in either fragment, nor is there any sequence similarity with the 343-bp repeat unit found in IGSa. Rather, this region of each fragment contains 67-bp tandem repeats, followed by ∼300 bp of nonrepetitive sequence. Copy I has six complete tandem repeats; Copy II has four complete repeats and a partial fifth repeat. Not including this indel, the two copies of this region are 96% identical. Both Copy I and Copy II contain trnP, CR, and trnF; the two copies of this region are 96% identical overall, and both copies of both tRNAs have viable secondary structures. Adjacent to trnF, Copy I has a complete rrnS, trnV, rrnL, and trnL1. In contrast, Copy II contains 111 bp that are 86% identical to the first 111 bp of the functional rrnS, followed by a trnL1 that is 97% identical to the trnL1 in Copy I; both trnL1s have viable secondary structure. Adjacent to trnL1, Copy I has a 672-bp ψnad1 that is 92% identical to the first 672 bp of the functional copy but contains multiple stop codons; Copy II contains the functional nad1.
The two large, duplicated fragments resulting from the second hypothesized round of duplication (Copy I and Copy II; see fig. 7) share high levels of sequence similarity over much of their length, although ψnad6, much of ψrrnS, and all of ψtrnV and ψrrnL have been excised from the genome and ψnad1 has accumulated multiple stop codons. In contrast, two very similar, presumably functional, copies of trnP, trnF, trnL1, the CR, and possibly trnE have been retained in this genome. There are several possible explanations for this pattern. Consistent with the duplication-random loss model, this duplication event may be sufficiently recent that mutations have not yet disrupted trnP, trnF, trnL1, the CR, or trnE. Alternatively, these copies may have been maintained by selection or may have had their mutations corrected by gene conversion (Eberhard, Wright, and Bermingham 2001). Selection to retain duplicate copies of these genes or the CR may act either on the production of their gene products or their ability to form secondary structures necessary for message processing (Kumazawa et al. 1998) or on regulatory function, respectively. However, the presence of tandem repeats highly conserved between Copy I and Copy II is difficult to explain by selection in the absence of a clear function assignable to these sequences. Rather, it suggests either a recent duplication event or a purely mechanistic cause of concerted evolution between portions of these two fragments (Moritz and Brown 1987).
Possible Mechanisms Yielding Tandem Duplications
Several mechanisms may produce duplications in a circular molecule: recombination, slipped-strand mispairing, errors in synchronizing the points of initiation and termination of replication, or some combination of these processes (Macey et al. 1997a,b; Boore and Brown 1998; Boore 2000; Dowton and Campbell 2001). At least two such mechanisms are plausible explanations for the patterns observed in the order of genes and noncoding DNA in most plethodontid rearrangements. In the genomes of H. brunus, B. attenuatus, and A. hardii (second duplication), the replication origin is in the center of the duplicated region, indicating that both initiation and termination of replication occurred at alternative sites; alternate initiation or termination alone would duplicate only the region of the genome upstream or downstream, respectively, of the replication origin. A presumably functional OL with viable potential secondary structure (Pääbo et al. 1991; Macey, Schulte, and Larson 2000) exists in both H. brunus and B. attenuatus. This suggests that light-strand replication initiation has not been completely transferred to alternate structures in these genomes and that alternate initiation may be causally linked to imprecise termination. In the genomes of S. marginatus, A. flavipunctatus, and A. hardii (first duplication), no evidence remains that a replication origin was involved in the duplication. The patterns in these genomes are still consistent with initiation and termination of replication at alternate sites, but both sites are located downstream of the OH. The IGS, which is the putative alternative initiation site for S. marginatus and A. hardii, may be an ancient duplicated CR based on its position in the genome (McKnight and Shaffer 1997) and thus may retain limited initiation capability. Imprecise termination alone is also a plausible duplication mechanism for S. marginatus, A. flavipunctatus, and A. hardii based on the proximity of these duplicated regions to the CR and the possible erosion of the ends of the duplicated region. The first duplication in the P. elongatus genome is bounded by the CR; depending on the location of the OH within the CR, imprecise termination alone, or imprecise termination with initiation at an alternate site, is a possible duplication mechanism. We cannot eliminate the alternative possibility that either intramolecular recombination or slipped-strand mispairing caused any of these duplications, nor can we rule them out as mechanisms for excision of redundant genes following these duplications (Holt, Dunbar, and Jacobs 1997; Lunt and Hyman 1997; Kajander et al. 2000; Tang et al. 2000a; Miller et al. 2004).
Possible Mechanisms Yielding Nontandem Duplications
The second duplication event in P. elongatus produced nontandem repeat fragments separated from one another by 3,054 bp, which nonetheless have 99.5% sequence identity. This pattern cannot be easily explained by slipped-strand mispairing, errors in initiation or termination of replication, or any combination of these processes. However, this pattern is consistent with the action of intramolecular recombination. One copy of the region comprising the end of nad5, ψnad6 + ψtrnE + ψcob, trnT, the putative IGS, ψtrnP, and part of one of the two CRs may have been excised from one genome, forming a separate minicircle that was then reintegrated into another genome between the second CR and trnF. Such intramolecular recombination has been reported in the mitochondrial genomes of both healthy and diseased tissue in several taxa (Holt, Dunbar, and Jacobs 1997; Lunt and Hyman 1997; Kajander et al. 2000; Tang et al. 2000a; Miller et al. 2004; Yokobori et al. 2004). In addition to this nontandem repeat in P. elongatus, the pattern of widely separated repeat units in S. marginatus is consistent with the action of intramolecular recombination. However, the pattern in S. marginatus is also consistent with retention of these widely separated repeat units since the original duplication event, coupled with slower accumulation of mutations in the repeat units relative to the remainder of the duplicated fragment.
Tandem Repeats in the CR and IGS That Do Not Effect Gene Rearrangement
Only 4 of the 24 genomes analyzed for this study contain a duplication-mediated rearrangement involving the CR or nearby regions of the genome. Thirteen of the remaining 20 genomes contain tandem repeats of noncoding sequence in the CR and/or the IGS, as reported in other taxa (Wallis 1987; Delarbre et al. 2001). These results are summarized in table 1. The length, number, and sequence of the repeat units vary within and among genomes, although in two cases, the same repeats are present in both the IGS and the CR. In the case of nontandem repeats, intramolecular recombination is a more plausible generation mechanism. In the case of tandem repeats, we cannot discriminate between slipped-strand mispairing and intramolecular recombination. The tandem repeats in the Rhyacotriton variegatus genome are unusual and are discussed in more detail below.
Repeat Elements in the IGS between trnT and trnP and the CR Between trnP and trnF in 13 Species' Genomes
Taxon | IGS Length (bp) | IGS Repeat Motifs, Number × Length (bp) | CR Length (bp) | CR Repeat Motifs, Number × Length (bp) | Sequence in Common Between IGS and CR? |
|---|---|---|---|---|---|
| Gyrinophilus porphyriticus | 666 | 2 × 86, 9 × 7 | 766 | None | No |
| Eurycea bislineata | 1,113 | 3 × 60 | 729 | None | No |
| Nototriton abscondens | >1,198a | 27 × 12 | 1,173 | 22 × 10 | No |
| “Bolitoglossa sp. nov.” | 11 | None | 6,373 | 12 × 325, 6 × 200 | No |
| “Thorius sp. nov.” | 262 | None | 3,539 | 3–36 × ∼10b | No |
| Batrachoseps attenuatus | 652 | 21 × 6–9 | 1,336 | None | No |
| Batrachoseps wrightorum | 183 | None | 4,297 | 13 × 225 | No |
| Hemidactylium scutatum | 1,211 | 8 × 26, 21 × 15 | 930 | 16 × 11 | No |
| Plethodon cinereus | 798 | 1 × 798c | 3,694 | 2 × 1,250 | Yesd |
| Plethodon petraeus | 754 | None | 3,251 | 6 × 54, 9 × 200 | No |
| Desmognathus fuscus | 564 | 2 × 77, 8 × 9, 6 × 10 | 735 | None | No |
| Ensatina eschscholtzii | 1,119 | 1 × 525,e 2 × 71 | 5,004 | 3 × 1,500 | Yesd |
| Rhyacotriton variegatus | 5,527 | 6 × 880f | 755 | None | No |
Taxon | IGS Length (bp) | IGS Repeat Motifs, Number × Length (bp) | CR Length (bp) | CR Repeat Motifs, Number × Length (bp) | Sequence in Common Between IGS and CR? |
|---|---|---|---|---|---|
| Gyrinophilus porphyriticus | 666 | 2 × 86, 9 × 7 | 766 | None | No |
| Eurycea bislineata | 1,113 | 3 × 60 | 729 | None | No |
| Nototriton abscondens | >1,198a | 27 × 12 | 1,173 | 22 × 10 | No |
| “Bolitoglossa sp. nov.” | 11 | None | 6,373 | 12 × 325, 6 × 200 | No |
| “Thorius sp. nov.” | 262 | None | 3,539 | 3–36 × ∼10b | No |
| Batrachoseps attenuatus | 652 | 21 × 6–9 | 1,336 | None | No |
| Batrachoseps wrightorum | 183 | None | 4,297 | 13 × 225 | No |
| Hemidactylium scutatum | 1,211 | 8 × 26, 21 × 15 | 930 | 16 × 11 | No |
| Plethodon cinereus | 798 | 1 × 798c | 3,694 | 2 × 1,250 | Yesd |
| Plethodon petraeus | 754 | None | 3,251 | 6 × 54, 9 × 200 | No |
| Desmognathus fuscus | 564 | 2 × 77, 8 × 9, 6 × 10 | 735 | None | No |
| Ensatina eschscholtzii | 1,119 | 1 × 525,e 2 × 71 | 5,004 | 3 × 1,500 | Yesd |
| Rhyacotriton variegatus | 5,527 | 6 × 880f | 755 | None | No |
The IGS is incompletely sequenced in N. abscondens.
“Thorius sp. nov.” CR contains ≥14 different short (9–11) repeat units repeated between 3 and 36 times.
In the P. cinereus genome, the entire 798-bp IGS is repeated 1.85 times within the CR.
In the mtDNAs of P. cinereus and E. eschscholtzii, the IGS and CR contain similar (∼80%–90% identical) sequences.
In the E. eschscholtzii mtDNA, 525 bp of the IGS are repeated five times within the CR—once in each 1,500-bp CR repeat unit and two additional times. Each 1,500-bp CR repeat unit also contains a ψtrnP that is 91% identical to the functional copy but has a 1-bp deletion in the anticodon.
The IGS repeat unit in the R. variegatus genome is composed of a partial ψcob and additional trnT and is described further in the text.
Repeat Elements in the IGS between trnT and trnP and the CR Between trnP and trnF in 13 Species' Genomes
Taxon | IGS Length (bp) | IGS Repeat Motifs, Number × Length (bp) | CR Length (bp) | CR Repeat Motifs, Number × Length (bp) | Sequence in Common Between IGS and CR? |
|---|---|---|---|---|---|
| Gyrinophilus porphyriticus | 666 | 2 × 86, 9 × 7 | 766 | None | No |
| Eurycea bislineata | 1,113 | 3 × 60 | 729 | None | No |
| Nototriton abscondens | >1,198a | 27 × 12 | 1,173 | 22 × 10 | No |
| “Bolitoglossa sp. nov.” | 11 | None | 6,373 | 12 × 325, 6 × 200 | No |
| “Thorius sp. nov.” | 262 | None | 3,539 | 3–36 × ∼10b | No |
| Batrachoseps attenuatus | 652 | 21 × 6–9 | 1,336 | None | No |
| Batrachoseps wrightorum | 183 | None | 4,297 | 13 × 225 | No |
| Hemidactylium scutatum | 1,211 | 8 × 26, 21 × 15 | 930 | 16 × 11 | No |
| Plethodon cinereus | 798 | 1 × 798c | 3,694 | 2 × 1,250 | Yesd |
| Plethodon petraeus | 754 | None | 3,251 | 6 × 54, 9 × 200 | No |
| Desmognathus fuscus | 564 | 2 × 77, 8 × 9, 6 × 10 | 735 | None | No |
| Ensatina eschscholtzii | 1,119 | 1 × 525,e 2 × 71 | 5,004 | 3 × 1,500 | Yesd |
| Rhyacotriton variegatus | 5,527 | 6 × 880f | 755 | None | No |
Taxon | IGS Length (bp) | IGS Repeat Motifs, Number × Length (bp) | CR Length (bp) | CR Repeat Motifs, Number × Length (bp) | Sequence in Common Between IGS and CR? |
|---|---|---|---|---|---|
| Gyrinophilus porphyriticus | 666 | 2 × 86, 9 × 7 | 766 | None | No |
| Eurycea bislineata | 1,113 | 3 × 60 | 729 | None | No |
| Nototriton abscondens | >1,198a | 27 × 12 | 1,173 | 22 × 10 | No |
| “Bolitoglossa sp. nov.” | 11 | None | 6,373 | 12 × 325, 6 × 200 | No |
| “Thorius sp. nov.” | 262 | None | 3,539 | 3–36 × ∼10b | No |
| Batrachoseps attenuatus | 652 | 21 × 6–9 | 1,336 | None | No |
| Batrachoseps wrightorum | 183 | None | 4,297 | 13 × 225 | No |
| Hemidactylium scutatum | 1,211 | 8 × 26, 21 × 15 | 930 | 16 × 11 | No |
| Plethodon cinereus | 798 | 1 × 798c | 3,694 | 2 × 1,250 | Yesd |
| Plethodon petraeus | 754 | None | 3,251 | 6 × 54, 9 × 200 | No |
| Desmognathus fuscus | 564 | 2 × 77, 8 × 9, 6 × 10 | 735 | None | No |
| Ensatina eschscholtzii | 1,119 | 1 × 525,e 2 × 71 | 5,004 | 3 × 1,500 | Yesd |
| Rhyacotriton variegatus | 5,527 | 6 × 880f | 755 | None | No |
The IGS is incompletely sequenced in N. abscondens.
“Thorius sp. nov.” CR contains ≥14 different short (9–11) repeat units repeated between 3 and 36 times.
In the P. cinereus genome, the entire 798-bp IGS is repeated 1.85 times within the CR.
In the mtDNAs of P. cinereus and E. eschscholtzii, the IGS and CR contain similar (∼80%–90% identical) sequences.
In the E. eschscholtzii mtDNA, 525 bp of the IGS are repeated five times within the CR—once in each 1,500-bp CR repeat unit and two additional times. Each 1,500-bp CR repeat unit also contains a ψtrnP that is 91% identical to the functional copy but has a 1-bp deletion in the anticodon.
The IGS repeat unit in the R. variegatus genome is composed of a partial ψcob and additional trnT and is described further in the text.
In the R. variegatus genome, the 5,527-bp IGS contains six copies of an ∼880-bp tandem repeat. The first ∼820 bp of this repeat are a ψcob, and the last 62 bp are an additional copy of trnT. ψcob is lacking the first 260 bp of the gene, but each of the six copies is >80% identical to the corresponding portion of the functional copy, not including ∼20 bp of deletions. All copies of the pseudogene contain numerous stop codons when translated in all three reading frames. The copies of trnT are ∼90% identical to the inferred original copy but, in contrast to ψcob, all but one may remain functional; their secondary structures appear viable. Following the sixth complete repeat unit, there are 247 bp which appear unrelated to the repeat sequence. In contrast, there are no repeats in the 755-bp CR, nor do the CR and IGS regions share any common sequence.
Heteroplasmy has been reported in some individuals whose mitochondrial genomes contain tandem repeats (Densmore, Wright, and Brown 1985; Wallis 1987; Townsend and Rand 2004). Similarly, patterns in the assembly of individual clone sequences into contigs may suggest heteroplasmy in seven plethodontid genome sequences: Eurycea bislineata, “Bolitoglossa sp. nov.,” Batrachoseps wrightorum, Hemidactylium scutatum, Plethodon petraeus, Ensatina eschscholtzii, and Aneides flavipunctatus. However, we note that polymerase chain reaction (PCR)–based evidence for the presence or absence of heteroplasmy is imperfect. A nonheteroplasmic sequence may indicate that multiple mitochondrial genome haplotypes are absent or that, although present, the additional copy or copies were not amplified by PCR. Furthermore, products that appear heteroplasmic may be generated by strand switching during PCR, analogous to slipped-strand mispairing. Finally, heteroplasmy is predicted in genomes with high numbers of tandem repeats, and the assembly of sequences of individual clones into one contig may prove problematic for such genomes even in the absence of heteroplasmy.
Possible Explanations for Extensive Gene Rearrangement in Plethodontids
A high incidence of rearranged mitochondrial genomes may result from any or some combination of four factors: (1) a high rate of mitochondrial genome partial duplication; (2) a low rate of duplication excision; (3) a low mitochondrial genome effective population size, particularly a bottleneck in the number of mitochondrial genomes transmitted to offspring via maternal oocytes; and (4) selection for, or absence of selection against, rearranged/expanded mitochondrial genomes at either the cell or the organism level. No data addressing any of these four factors are currently available for plethodontids. However, studies of selection on mitochondrial genomes containing duplications have been carried out in other systems. Here, we briefly discuss their possible relevance to the high levels of plethodontid mitochondrial instability. We note, however, that inferences drawn from such studies should be considered as hypotheses directing further research and that studies explicitly measuring all four of these factors in plethodontids and other clades with high levels of mitochondrial genome rearrangement are required to draw any firm conclusions.
The fixation of structurally altered mitochondrial genomes in a population depends in part on whether they are selectively advantageous, neutral, or disadvantageous to the organism. Unlike deletions, large duplications of portions of the mitochondrial genome are generally not pathogenic in humans (Tang et al. 2000b; DiMauro and Schon 2003), suggesting that there may not be strong organism-level selection against the duplications in plethodontid mitochondrial genomes. However, a link between compact mitochondrial genomes and metabolic efficiency has been proposed (Selosse, Albert, and Godelle 2001), and high levels of mitochondrial duplications such as those seen in plethodontids lead to measurable reduction in respiratory chain efficiency in human cell lines (Holt, Dunbar, and Jacobs 1997). Salamanders have extremely low aerobic metabolic requirements compared to other ectotherms (Feder 1976). Organism-level selection against the reduction in aerobic efficiency that may result from a mitochondrial duplication is therefore unlikely to be as strong in plethodontids as in other, more highly aerobic tetrapods. Low energetic costs are also broadly correlated with the accumulation of noncoding DNA in the nuclear genome (Gregory 2003). If relatively weak organism-level selection against mitochondrial duplications is contributing to their high incidence in plethodontids, we would predict (1) high mitochondrial genome instability in other organisms with similarly low aerobic metabolic requirements and (2) small fitness differences between plethodontids with and without mitochondrial duplications relative to differences between other, more highly aerobic animals with and without mitochondrial duplications.
Phylogenetic Applications
The analyses presented in this study describe major genome-level mitochondrial mutations for one representative individual from each included lineage. Therefore, they cannot address the extent to which these rearrangements characterize populations, species, or more inclusive phylogenetic groups, with the possible exception of the two Aneides (Moritz and Brown 1987). Aneides flavipunctatus and A. hardii may share a synapomorphic rearrangement, although high levels of such rearrangements within plethodontids necessitate further sampling from this clade to eliminate the possibility of homoplasy. Additional sampling within all rearranged lineages and their close relatives will enable dating of the appearance and persistence of individual rearrangements, thereby addressing the rate at which pseudogenes decay and/or are excised from the genome. Similarly, it will allow dating of the appearance and persistence of tandem duplications within and around the CR and IGS. Finally, it will enable more accurate identification of the ends of duplicated fragments, allowing characterization of secondary structures, repeats, or other genomic sequences that may facilitate duplication both in plethodontids and more generally. In addition to providing further insight into the evolutionary history of vertebrate mitochondrial genomes, this understanding of molecular- and population-level dynamics of mitochondrial genome instability is critical for evaluating the strength of using mitochondrial genome rearrangements as a genome-level character for phylogenetic analysis (Sankoff et al. 1992; Boore and Brown 1998; Macey, Schulte, and Larson 2000).
Present address: Department of Organismal Biology and Anatomy, University of Chicago.
Scott Edwards, Associate Editor
We thank M. Phillips, R. Zardoya, M. Fujita, R. Gillespie, C. Moritz, D. B. Wake, and M. H. Wake for comments on the manuscript. Part of this work was performed by the University of California Lawrence Berkeley National Laboratory under the auspices of the U.S. Department of Energy, Office of Biological and Environmental Research, contract number DE-AC03-76SF00098. R.L.M. was supported by a National Science Foundation (NSF) predoctoral fellowship and National Institutes of Health training grant. Additional funds came from an NSF doctoral dissertation improvement grant to R.L.M. and David B. Wake (0105824) and the AmphibiaTree Project (NSF EF-0334939).
References
Arndt, A., and M. J. Smith.
———.
Boore, J. L., and W. M. Brown.
Boore, J. L., M. Medina, and L. A. Rosenberg.
Delarbre, C., A.-S. Rasmussen, U. Arnason, and G. Gachelin.
Densmore, L. D., J. W. Wright, and W. M. Brown.
Desjardins, P., and R. Morais.
DiMauro, S., and E. A. Schon.
Dowton, M., and A. D. Austin.
Dowton, M., and N. J. H. Campbell.
Dowton, M., L. R. Castro, S. L. Campbell, S. D. Bargon, and A. D. Austin.
Eberhard, J. R., T. F. Wright, and E. Bermingham.
Feder, M. E.
Gregory, T. R.
Holt, I. J., D. R. Dunbar, and H. T. Jacobs.
Inoue, J. G., M. Miya, K. Tsukamoto, and M. Nishida.
Kajander, O. A., A. T. Rovio, K. Majamaa, J. Poulton, J. N. Spelbrink, I. J. Holt, P. J. Karhunen, and H. T. Jacobs.
Kumazawa, Y., H. Ota, M. Nishida, and T. Ozawa.
Lee, J.-S., M. Miya, Y.-S. Lee, C. G. Kim, E.-H. Park, Y. Aoki, and M. Nishida.
Macey, J. R., A. Larson, N. B. Ananjeva, Z. Fang, and T. J. Papenfuss.
Macey, J. R., A. Larson, N. B. Ananjeva, and T. J. Papenfuss.
Macey, J. R., J. A. Schulte II, and A. Larson.
Macey, J. R., J. A. I. Schulte, A. Larson, and T. J. Papenfuss.
McKnight, M. L., and H. B. Shaffer.
Miller, A. D., T. T. T. Nguyen, C. P. Burridge, and C. M. Austin.
Moore, C. A., J. Gudikote, and G. C. Van Tuyle.
Moritz, C.
Moritz, C., T. E. Dowling, and W. M. Brown.
Moritz, C., and W. M. Brown.
———.
Mueller, R. L., J. R. Macey, M. Jaekel, D. B. Wake, and J. L. Boore.
Pääbo, S., W. K. Thomas, K. M. Whitfield, Y. Kumazawa, and A. C. Wilson.
Quinn, T. W., and D. P. Mindell.
Sankoff, D., G. Leduc, N. Antoine, B. Paquin, B. F. Lang, and R. Cedergren.
Selosse, M.-A., B. Albert, and B. Godelle.
Shadel, G. S., and D. A. Clayton.
Shao, R., N. J. H. Campbell, E. R. Schmidt, and S. C. Barker.
Smith, M. J., D. K. Banfield, K. Doteval, S. Gorski, and D. J. Kowbel.
Stanton, D. J., L. L. Daehler, C. C. Moritz, and W. M. Brown.
Tang, Y., G. Manfredi, M. Hirano, and E. A. Schon.
Tang, Y., E. A. Schon, E. Wilichowski, M. E. Vazquez-Memije, E. Davidson, and M. P. King.
Townsend, J. P., and D. M. Rand.
Wallis, G. P.
Yamazaki, N., R. Ueshima, J. A. Terrett et al. (12 co-authors).
Yokobori, S.-I., N. Fukuda, M. Nakamura, T. Aoyama, and T. Oshima.
Zardoya, R., E. Malaga-Trillo, M. Veith, and A. Meyer.
Zhang, P., Y.-Q. Chen, Y.-F. Liu, H. Zhou, and L.-H. Qu.
Author notes
*Museum of Vertebrate Zoology, University of California, Berkeley; †Evolutionary Genomics Department, Department of Energy Joint Genome Institute and University of California Lawrence Berkeley National Laboratory, Walnut Creek, California; and ‡Department of Integrative Biology, University of California, Berkeley

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}