





Abstract
Inference of evolutionary relationships between nematodes is severely hampered by their conserved morphology, the high frequency of homoplasy, and the scarcity of phylum-wide molecular data. To study the origin of nematode radiation and to unravel the phylogenetic relationships between distantly related species, 339 nearly full-length small-subunit rDNA sequences were analyzed from a diverse range of nematodes. Bayesian inference revealed a backbone comprising 12 consecutive dichotomies that subdivided the phylum Nematoda into 12 clades. The most basal clade is dominated by the subclass Enoplia, and members of the order Triplonchida occupy positions most close to the common ancestor of the nematodes. Crown Clades 8–12, a group formerly indicated as “Secernentea” that includes Caenorhabditis elegans and virtually all major plant and animal parasites, show significantly higher nucleotide substitution rates than the more basal Clades 1–7. Accelerated substitution rates are associated with parasitic lifestyles (Clades 8 and 12) or short generation times (Clades 9–11). The relatively high substitution rates in the distal clades resulted in numerous autapomorphies that allow in most cases DNA barcode–based species identification. Teratocephalus, a genus comprising terrestrial bacterivores, was shown to be most close to the starting point of Secernentean radiation. Notably, fungal feeding nematodes were exclusively found basal to or as sister taxon next to the 3 groups of plant parasitic nematodes, namely, Trichodoridae, Longidoridae, and Tylenchomorpha. The exclusive common presence of fungivorous and plant parasitic nematodes supports a long-standing hypothesis that states that plant parasitic nematodes arose from fungivorous ancestors.
Introduction
Nematodes constitute one of the largest and most widely distributed groups of animals in marine, freshwater, and terrestrial habitats. Their numerical dominance, exceeding often more than 1 million individuals per square meter and accounting for about 80% of all individual animals on earth (Platt 1994), their diversity in lifestyles, and their presence at various trophic levels point at an important role in many ecosystems. Its most well-known representative is Caenorhabditis elegans: the first animal whose genome was completely sequenced (Herman 2004). Apart from the bacterivorous nematodes such as C. elegans, a wide range of trophic ecologies are displayed, such as fungal feeding, predation, and parasitism of plants, invertebrates, higher animals, and humans. Among plant parasitic nematodes, the cyst (Globodera and Heterodera spp.) and root knot nematodes (Meloidogyne spp.) are most notorious, causing major damage to crops such as soybean, potato, and sugar beet. Human parasitic nematodes include, among others, the pinworm Enterobius vermicularis, a worldwide intestinal parasite of mainly children, the causal agents of elephantiasis—Wuchereria bancrofti and Brugia malayi—and Onchocerca volvulus that, in combination with its endosymbiont Wolbachia, causes river blindness (onchocerciasis) (Saint Andre et al. 2002). Nonparasitic nematodes are valuable indicators for the biological condition of soils as this ecologically highly diverse group shows much variation in sensitivity toward environmental stresses and occupies key positions in the soil food web (Bongers and Ferris 1999).
One of the earliest and most influential classifications of the Nematoda was proposed by Chitwood BG and Chitwood MB (1933) and Chitwood (1937). They introduced a division of the phylum into the Aphasmidia and Phasmidia, later renamed “Adenophorea” (gland bearers) and “Secernentea” (secretors), respectively (Chitwood 1958). This division was based on the fact that the Secernentea share several characteristics including the presence of phasmids, a pair of sensory organs located in the lateral posterior region. This division was adhered to in many later classifications even though it was realized that the Adenophorea were not a uniform group (Maggenti 1963; De Coninck 1965). On the basis of an unweighted count of shared morphological features, Andrássy (1976) proposed a tripartite system by subdividing the former Adenophorea into the Torquentia and Penetrantia. The first taxonomic system based on cladistic principles was introduced by Lorenzen (1981). His analysis made clear that there was no support for the Adenophorea as a monophyletic group. Moreover, he showed that the number of informative morphological characters was too low to come up with a plausible alternative.
Only 2 small-subunit (SSU) rDNA–based trees have been constructed so far that attempted to span the entire phylum (Aleshin, Kedrova et al. 1998; Blaxter et al. 1998) to provide a template for phylogenetic studies. Blaxter et al. (1998) defined 5 major clades and confirmed the paraphyly of the Adenophorea. Interestingly, the authors clearly showed that trophic ecologies such as animal and plant parasitism arose several times independently. However, mainly due to the relatively small data set used, namely, 53 taxa, the relationships among the major clades remained unresolved. Comparable results were acquired by Aleshin, Kedrova et al. (1998) based on 19 nematode sequences. Here, we present a phylogenetic reconstruction of 339 nematode taxa throughout the entire phylum Nematoda, inferred from nearly full-length SSU rDNA sequences. Our results revealed a subdivision of the phylum Nematoda into 12 major clades, where the most basal clade (Clade 1) was dominated by representatives of the subclass Enoplia sensu De Ley and Blaxter (De Ley and Blaxter 2002, 2004). Clade 7 comprised only a single family, the Teratocephalidae, and members of the genus Teratocephalus were shown to be most close to the origin of Secernentean (Clades 8–12) radiation. The remarkable and significant acceleration of SSU rDNA substitution rates in the more distal clades that include most major plant and animal parasites gave—in most cases—rise to resolution till species level. This unforeseen resolution implies that SSU rDNA base sequence signatures can be defined at species level for a wide range of parasitic and nonparasitic nematodes.
Materials and Methods
Taxon Sampling
Nematodes were collected from various soil habitats and extracted using standard techniques. Prior to DNA extraction, individual nematodes were identified using a light microscope (Zeiss Axioscope) equipped with differential interference contrast optics. A CCD camera (CoolSnap, RS Photometrics, Tucson, AZ) was used to take a series of digital images of each nematode to retain the possibility to reevaluate the identity of individual specimens. Series of digital images from individual nematodes are available upon request (from J.H.). For classification at family level and below, the nomenclatural system of the Fauna Europaea was used (http://www.faunaeur.org/). For the classification above family level, we conformed to De Ley and Blaxter (2002, 2004).
DNA Extraction and SSU rDNA Amplification and Sequencing
Single nematodes were transferred to a 0.2-ml polymerase chain reaction (PCR) tube containing 25 μl of sterile water. An equal volume of lysis buffer containing 0.2 M NaCl, 0.2 M Tris–HCl (pH 8.0), 1% (v/v) β-mercaptoethanol, and 800 μg/ml proteinase-K was added. Lysis took place in a Thermomixer (Eppendorf, Hamburg, Germany) at 65 °C and 750 rpm for 2 h, followed by 5 min incubation at 100 °C. Lysate was used immediately or stored at −20 °C. SSU rDNA was amplified as 2 partially overlapping fragments using 3 universal and 1 nematode-specific primer (1912R). The latter was included to avoid amplification of nontarget eukaryotic SSU rDNA, for example, from fungal spores attached to the nematode cuticle. For the first fragment, either the primer 988F (5′-ctcaaagattaagccatgc-3′) or the primer 1096F (5′-ggtaattctggagctaatac-3′) was used in combination with the primer 1912R (5′-tttacggtcagaactaggg-3′). The second fragment was amplified with primers 1813F (5′-ctgcgtgagaggtgaaat-3′) and 2646R (5′-gctaccttgttacgactttt-3′). PCR was performed in a final volume of 25 μl and contained 3 μl of a 100 times–diluted crude DNA extract, 0.1 μM of each PCR primer, and a Ready-To-Go PCR bead (Amersham, Little Chalfont, Buckinghamshire, UK). The following PCR profile was used: 94 °C for 5 min; 5× (94 °C, 30 s; 45 °C, 30 s; 72 °C, 70 s) followed by 35 × (94 °C, 30 s; 54 °C, 30 s; 72 °C, 70 s) and 72 °C, 5 min. Gel-purified amplification products (Marligen Bioscience, Ijamsville, MD) were cloned into a TOPO TA vector (Invitrogen, Carlsbad, CA) and sequenced using standard procedures. Newly generated SSU rDNA sequences were deposited at GenBank under the following accession numbers: AY284581–AY284841 and AY593880 (for corresponding species names see Table S1, Supplementary Material).
To distinguish between the 2 closely related potato cyst nematode species Globodera rostochiensis and Globodera pallida on the basis of a single-nucleotide difference in the SSU rDNA sequences, real-time PCR was performed on a Bio-Rad MyiQ thermal cycler (Bio-Rad, Hercules, CA). In a total reaction volume of 25 μl, 3 μl template (10 times diluted nematode lysate prepared as described above) was mixed with a G. rostochiensis–specific primer GrosR1-650R (5′-ggccaacgccggggaa-3′) and a general SSU rDNA primer 988F (5′-ctcaaagattaagccatgc-3′) (end concentrations for both primers 200 nM) and 12.5 μl iQ SYBR Green supermix (Bio-Rad). After 3 amplification cycles with an annealing temperature of 60 °C, the specificity was increased by lowering the denaturation temperature to 89.5 °C.
Sequence Alignment
Nematode SSU rDNA sequences were supplemented with publicly available sequences (accession numbers given in Table S1, Supplementary Material). The choice of outgroup sequences was based on Aleshin, Milyutina et al. (1998) and consisted of arthropods (3x), priapulids (2x), a kinorhynch (1x), nematomorphs (2x), and tardigrades (2x): Dilta littoralis (AF005457), Podura aquatica (AF005452), and Polydesmus coriaceus (AF005449); Priapulus caudatus (Z38009) and Tubiluchus corallicola (AF119086); Pycnophyes kielensis (U67997); Chordodes morgani (AF036639) and Gordius aquaticus (X80233); and Macrobiotus hufelandi (X81442) and Thulinia stephaniae (AF056023), respectively. Nearly full-length SSU rDNA sequences were aligned using the ClustalW algorithm as implemented in BioEdit 5.0.9 (Hall 1999) and manually improved using arthropod secondary structure information (http://www.psb.ugent.be/rRNA/secmodel/index.html, in accordance with Wuyts et al. (2000). The final alignment included 349 nearly full-length SSU rDNA sequences and contained 2,471 aligned positions including gaps.
Phylogenetic Analyses
Bayesian inference (BI), maximum parsimony (MP), and Neighbor-Joining (NJ) were used to reconstruct the phylogeny within the phylum Nematoda. Modeltest 3.06 (Posada and Crandall 1998) identified the general time reversible (GTR) model with invariable sites and a Γ-shaped distribution of substitution rates as the best substitution model. The Bayesian tree was constructed using the program MrBayes 3.0 (Ronquist and Huelsenbeck 2003). The alignment was divided into a stem and a loop partition according to SSU rDNA secondary structure. For both partitions, the GTR model with invariable sites was used with the default flat priors unlinked between partitions. A gamma parameter could not be included due to computing memory limitations. The program was run on the TERAS computer cluster (SARA Computing and Networking Services, Amsterdam, The Netherlands). Each chain was run on a separate processor. Four independent computations with random starting trees and 4 Markov chains were run for 8,000,000 generations with a sampling frequency of 200 generations. The burnins of 1, 3, 3.5, and 1 million generations were discarded. Sampled trees were combined in a 50% majority-rule consensus tree. Nodes with a posterior probability (PP) lower than 95 (Erixon et al. 2003) or a bootstrap support lower than 65% were considered unresolved (Hillis and Bull 1993).
The MP tree was constructed using PAUP* 4.0b10 (Swofford 1998). Default parameters were used with gaps treated as a fifth character state. A total of 16,887 equally parsimonious trees were saved, and a 50% majority-rule consensus tree was bootstrapped 1,000 times, not saving multiple trees during branch swapping. The Neighbor-Joining (NJ) tree was constructed using PAUP* applying the model (GTR + I + Γ) and parameter values determined by Modeltest. The resulting tree was bootstrapped 1,000 times.
The program RRTree (Robinson-Rechavi and Huchon 2000) was used to compare SSU rDNA substitution rates between clades. Significance of relative rate differences was tested using a Bonferroni correction.
Results and Discussion
Representative and balanced taxon sampling is a prerequisite for the reconstruction of phylogenetic relationships within the widespread and speciose phylum Nematoda (Moreira and Philippe 2000). So far, Rhabditidae, relatives of the bacterivorous model organism C. elegans; the suborder Spirurina, which consists exclusively of zooparasites; and Tylenchina, a suborder that includes numerous plant parasites, are relatively overrepresented (Blaxter et al. 2000; Sudhaus and Fitch 2001; Baldwin et al. 2004). Molecular information is scarce for the majority of bacterivorous, fungivorous, carnivorous, and omnivorous nematodes. Here, we present 260 newly generated full-length SSU rDNA sequences mainly from representatives of basal clades, and nonparasitic representatives throughout the phylum, and use these data to derive deep phylogenetic relationships, to deduce the evolution of feeding types, and to define its potential for DNA sequence signature–based community analysis.
Although C. elegans and a number of other bacterivorous Rhabditidae can be grown on growth medium agar plates seeded with bacteria to obtain numerous individuals from the same species, the majority of nematodes appear to be nonculturable. Therefore, single nematodes were used as starting material. After taking a series of high-resolution images, individual nematodes were lysed and their SSU rDNA sequences were determined. By doing so, a database was built that robustly links morphological and molecular data. Newly generated data were combined with 176 publicly available sequences. Consensus sequences were generated in case species were represented by multiple sequences. The final alignment consisted of 339 nematode taxa and 10 outgroup sequences.
Phylogenetic Analysis
In 4 independent runs with nearly identical results (only the first run is used here), Bayesian analysis of 349 taxa yielded a phylogenetic tree with a backbone consisting of 12 consecutive dichotomies from the tree root onward (fig. 1). Eight dichotomies are strongly supported (PP of 1.0) and 1 dichotomy is quite robust (PP of 0.95), whereas 3 nodes are weakly supported with PP values between 0.64 and 0.81. MP-based data analysis revealed a similar tree topology, although the number of resolved nodes was lower. NJ analysis resulted in a tree topology comparable with the MP tree, although the resolution within the clades was lower. Figure 2 shows the overall topologies of the BI, the MP, and the NJ trees. Detailed representations including full taxon names are presented as Supplementary Material (Figs. S1, S2, and S3). The lower resolution of MP can be explained by 1) saturation—a consequence of the large number of sequences analyzed—and by 2) a relatively high variation in branch lengths—inherent to the analysis of a phylum-wide data set (Felsenstein 1978; Philippe et al. 2000). The BI criterion is less susceptible to both methodological problems because it includes a mutation model (Moreira and Philippe 2000). In addition, the BI is more sensitive in detecting the phylogenetic signal when taxa differ in few characters (Alfaro et al. 2003). The relatively poor resolution of the NJ tree can be explained by the fact that distance methods, such as NJ, are in fact not suitable for the inference of more distant phylogenetic relationships, especially when the molecular clock assumption is not valid, as is the case with our data (Holder and Lewis 2003).
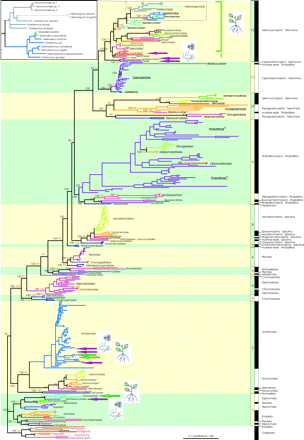
Bayesian tree of the phylum Nematoda. Alternating yellow and green backgrounds define the subdivision of the phylum Nematoda into 12 clades. Within each clade, nematode families have separate colors. Support values are indicated at the deep nodes: the first number (black) is the Bayesian PP and the second number (orange) is the MP bootstrap value. “-” indicates that the node was part of a polytomy in MP. Other nodes down to the family level are marked with a black asterisk if the support from the BI tree is significant (PP ≥ 95) and an orange asterisk if the support from the MP tree is significant (bootstrap ≥ 65). Underlined family names are paraphyletic, family names marked with an asterisk are polyphyletic, and family names in italics are embedded in another family. The black and white bars indicate (sub- and infra-) orders as defined by De Ley and Blaxter (2002, 2004). Plant parasitic and fungivorous taxa are indicated by a pictogram and a purple (fungivores) or green (plant parasites) arrow or bar. The insert shows the most distal part of the tree in more detail.
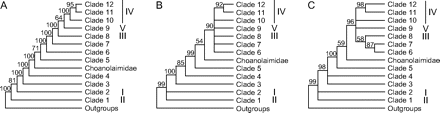
Schematic representations of the division of the phylum Nematoda into 12 clades, according to (A) BI, (B) MP, and (C) NJ. Branches with bootstrap support <50% in MP and NJ are shown as unresolved. The 5 clades defined by Blaxter et al. (1998) are indicated in Roman numerals behind the corresponding clades in our clade division.
The use of full-length SSU rDNA sequences (≈1,700 bp each) and extensive additional taxon sampling give a detailed insight into the deep phylogenetic relationships between all major taxa within the phylum Nematoda for the first time. On the basis of this analysis, we propose a revision of the current clade division (Blaxter et al. 1998) (fig. 2). Instead of a division into 5 clades with many families not placed in a clade at all, we propose a division into 12 clades that—except for the Choanolaimidae—include all sampled nematode families (table 1—NJ clades are omitted as NJ is not suitable for pinpointing distant relationships). Bayesian analysis suggested the family Choanolaimidae to be placed between Clades 4 and 5, whereas in the parsimony tree it was positioned between Clades 5 and 6 (fig. 2). Therefore, we refrained from assigning a clade to this family.
Clade Topology as Reconstructed with BI (fig. 1) and Compared with MP Analysis and Blaxter et al. (1998)
BI Clades (fig. 1) | MP Subclades | Clade Taxa | Blaxter et al. (1998) | Bayesian PP | Bootstrap MP |
|---|---|---|---|---|---|
| 1 | Enoplida, Triplonchida, Bastianiidae*, Rhabdolaimidae* (Plectida) | II | 100 | 70 | |
| 2 | Trichinellida, Mononchida, Mermithida, Dorylaimida | I | 100 | — | |
| 2a | Trichinellida | 100 | 100 | ||
| 2b | Mononchida, Mermithida, Dorylaimida | 100 | 95 | ||
| 3 | Chromadorida, Prodesmodora (Desmodoridae, Desmodorida) | — | 100 | 100 | |
| 4 | Desmodorida, Chromadoridae (Chromadorida) | — | 100 | 100 | |
| Choanolaimidae (Chromadorida) | — | — | — | ||
| 5 | Monhysterida, Areolaimida, Aulolaimidae (Plectida) | — | 96 | 69 | |
| 6 | Plectida | — | 100 | 94 | |
| 7 | Teratocephalidae* (Rhabditida, incertae sedis) | — | 100 | 100 | |
| 8 | Spirurina | III | 100 | 100 | |
| 9 | Myolaimina, Rhabditina | V | 100 | — | |
| 9a | Myolaimus sp. (Myolaimina) | — | — | ||
| 9b | Diplogasteromorpha, Bunonematomorpha, and Rhabditoides inermis (Rhabditomorpha) | 100 | 66 | ||
| 9c | Rhabditoides inermiformis (Rhabditomorpha) | — | — | ||
| 9d | Poikilolaimus spp. (Rhabditomorpha) | 100 | 100 | ||
| 9e | Rhabditomorpha | 100 | 91 | ||
| 10 | Tylenchina, Brevibuccidae (Rhabditida, incertae sedis) | IV | 100 | — | |
| 10a | Steinernema (Panagrolaimorpha) and Brevibucca (Rhabditida, incertae sedis) | — | 57 | ||
| 10b | Panagrolaimidae (Panagrolaimorpha) and Plectonchus (Rhabditida, incertae sedis) | 100 | 99 | ||
| 10d | Rhabditophanes and Strongyloides (Panagrolaimorpha), Aphelenchoides, Bursaphelenchus, and Seinura (Tylenchomorpha) | — | 69 | ||
| 11 | Cephalobomorpha | IV | 100 | 100 | |
| 12 | Tylenchomorpha | IV | 100 | 66 |
BI Clades (fig. 1) | MP Subclades | Clade Taxa | Blaxter et al. (1998) | Bayesian PP | Bootstrap MP |
|---|---|---|---|---|---|
| 1 | Enoplida, Triplonchida, Bastianiidae*, Rhabdolaimidae* (Plectida) | II | 100 | 70 | |
| 2 | Trichinellida, Mononchida, Mermithida, Dorylaimida | I | 100 | — | |
| 2a | Trichinellida | 100 | 100 | ||
| 2b | Mononchida, Mermithida, Dorylaimida | 100 | 95 | ||
| 3 | Chromadorida, Prodesmodora (Desmodoridae, Desmodorida) | — | 100 | 100 | |
| 4 | Desmodorida, Chromadoridae (Chromadorida) | — | 100 | 100 | |
| Choanolaimidae (Chromadorida) | — | — | — | ||
| 5 | Monhysterida, Areolaimida, Aulolaimidae (Plectida) | — | 96 | 69 | |
| 6 | Plectida | — | 100 | 94 | |
| 7 | Teratocephalidae* (Rhabditida, incertae sedis) | — | 100 | 100 | |
| 8 | Spirurina | III | 100 | 100 | |
| 9 | Myolaimina, Rhabditina | V | 100 | — | |
| 9a | Myolaimus sp. (Myolaimina) | — | — | ||
| 9b | Diplogasteromorpha, Bunonematomorpha, and Rhabditoides inermis (Rhabditomorpha) | 100 | 66 | ||
| 9c | Rhabditoides inermiformis (Rhabditomorpha) | — | — | ||
| 9d | Poikilolaimus spp. (Rhabditomorpha) | 100 | 100 | ||
| 9e | Rhabditomorpha | 100 | 91 | ||
| 10 | Tylenchina, Brevibuccidae (Rhabditida, incertae sedis) | IV | 100 | — | |
| 10a | Steinernema (Panagrolaimorpha) and Brevibucca (Rhabditida, incertae sedis) | — | 57 | ||
| 10b | Panagrolaimidae (Panagrolaimorpha) and Plectonchus (Rhabditida, incertae sedis) | 100 | 99 | ||
| 10d | Rhabditophanes and Strongyloides (Panagrolaimorpha), Aphelenchoides, Bursaphelenchus, and Seinura (Tylenchomorpha) | — | 69 | ||
| 11 | Cephalobomorpha | IV | 100 | 100 | |
| 12 | Tylenchomorpha | IV | 100 | 66 |
NOTE.—Dashes indicate not applicable data, and asterisks indicate families discussed in text.
Clade Topology as Reconstructed with BI (fig. 1) and Compared with MP Analysis and Blaxter et al. (1998)
BI Clades (fig. 1) | MP Subclades | Clade Taxa | Blaxter et al. (1998) | Bayesian PP | Bootstrap MP |
|---|---|---|---|---|---|
| 1 | Enoplida, Triplonchida, Bastianiidae*, Rhabdolaimidae* (Plectida) | II | 100 | 70 | |
| 2 | Trichinellida, Mononchida, Mermithida, Dorylaimida | I | 100 | — | |
| 2a | Trichinellida | 100 | 100 | ||
| 2b | Mononchida, Mermithida, Dorylaimida | 100 | 95 | ||
| 3 | Chromadorida, Prodesmodora (Desmodoridae, Desmodorida) | — | 100 | 100 | |
| 4 | Desmodorida, Chromadoridae (Chromadorida) | — | 100 | 100 | |
| Choanolaimidae (Chromadorida) | — | — | — | ||
| 5 | Monhysterida, Areolaimida, Aulolaimidae (Plectida) | — | 96 | 69 | |
| 6 | Plectida | — | 100 | 94 | |
| 7 | Teratocephalidae* (Rhabditida, incertae sedis) | — | 100 | 100 | |
| 8 | Spirurina | III | 100 | 100 | |
| 9 | Myolaimina, Rhabditina | V | 100 | — | |
| 9a | Myolaimus sp. (Myolaimina) | — | — | ||
| 9b | Diplogasteromorpha, Bunonematomorpha, and Rhabditoides inermis (Rhabditomorpha) | 100 | 66 | ||
| 9c | Rhabditoides inermiformis (Rhabditomorpha) | — | — | ||
| 9d | Poikilolaimus spp. (Rhabditomorpha) | 100 | 100 | ||
| 9e | Rhabditomorpha | 100 | 91 | ||
| 10 | Tylenchina, Brevibuccidae (Rhabditida, incertae sedis) | IV | 100 | — | |
| 10a | Steinernema (Panagrolaimorpha) and Brevibucca (Rhabditida, incertae sedis) | — | 57 | ||
| 10b | Panagrolaimidae (Panagrolaimorpha) and Plectonchus (Rhabditida, incertae sedis) | 100 | 99 | ||
| 10d | Rhabditophanes and Strongyloides (Panagrolaimorpha), Aphelenchoides, Bursaphelenchus, and Seinura (Tylenchomorpha) | — | 69 | ||
| 11 | Cephalobomorpha | IV | 100 | 100 | |
| 12 | Tylenchomorpha | IV | 100 | 66 |
BI Clades (fig. 1) | MP Subclades | Clade Taxa | Blaxter et al. (1998) | Bayesian PP | Bootstrap MP |
|---|---|---|---|---|---|
| 1 | Enoplida, Triplonchida, Bastianiidae*, Rhabdolaimidae* (Plectida) | II | 100 | 70 | |
| 2 | Trichinellida, Mononchida, Mermithida, Dorylaimida | I | 100 | — | |
| 2a | Trichinellida | 100 | 100 | ||
| 2b | Mononchida, Mermithida, Dorylaimida | 100 | 95 | ||
| 3 | Chromadorida, Prodesmodora (Desmodoridae, Desmodorida) | — | 100 | 100 | |
| 4 | Desmodorida, Chromadoridae (Chromadorida) | — | 100 | 100 | |
| Choanolaimidae (Chromadorida) | — | — | — | ||
| 5 | Monhysterida, Areolaimida, Aulolaimidae (Plectida) | — | 96 | 69 | |
| 6 | Plectida | — | 100 | 94 | |
| 7 | Teratocephalidae* (Rhabditida, incertae sedis) | — | 100 | 100 | |
| 8 | Spirurina | III | 100 | 100 | |
| 9 | Myolaimina, Rhabditina | V | 100 | — | |
| 9a | Myolaimus sp. (Myolaimina) | — | — | ||
| 9b | Diplogasteromorpha, Bunonematomorpha, and Rhabditoides inermis (Rhabditomorpha) | 100 | 66 | ||
| 9c | Rhabditoides inermiformis (Rhabditomorpha) | — | — | ||
| 9d | Poikilolaimus spp. (Rhabditomorpha) | 100 | 100 | ||
| 9e | Rhabditomorpha | 100 | 91 | ||
| 10 | Tylenchina, Brevibuccidae (Rhabditida, incertae sedis) | IV | 100 | — | |
| 10a | Steinernema (Panagrolaimorpha) and Brevibucca (Rhabditida, incertae sedis) | — | 57 | ||
| 10b | Panagrolaimidae (Panagrolaimorpha) and Plectonchus (Rhabditida, incertae sedis) | 100 | 99 | ||
| 10d | Rhabditophanes and Strongyloides (Panagrolaimorpha), Aphelenchoides, Bursaphelenchus, and Seinura (Tylenchomorpha) | — | 69 | ||
| 11 | Cephalobomorpha | IV | 100 | 100 | |
| 12 | Tylenchomorpha | IV | 100 | 66 |
NOTE.—Dashes indicate not applicable data, and asterisks indicate families discussed in text.
The Origin of Nematode Radiation
Clade 1 is presumably the most basal clade in the BI tree (fig. 2). However, it is not possible to make a strong statement on the basis of the currently presented SSU rDNA sequence data as the node joining Clades 2–12 in the BI-based tree is supported by a relatively low PP value (0.81). BI and maximum likelihood analysis of a limited number of representatives with short branch lengths did not result in a more robust topology at the base of the tree (data not shown). Hence, it was investigated whether the hypothesis of Clade 1, being the most basal clade, was supported by other independent data.
Clade 1 includes representatives of the Enoplia and 2 Plectida families, the Rhabdolaimidae and the Bastianiidae. This subclass Enoplia comprises only 2 orders, Enoplida and Triplonchida, and representatives of the latter, Trischistoma sp. and Tripyla sp. 4, occupy the positions closest to the base of the phylum Nematoda (fig. S1). The basal position of the Enoplia in this SSU rDNA–based phylogenetic tree is supported by patchy embryological and morphological data. Embryo development within this subclass deviates from the standard pattern observed for nematodes as there is no asymmetrically dividing germ line and no bilateral symmetry during early embryogenesis (Malakhov 1994; Voronov et al. 1998; Schierenberg 2005) and they have only a weakly centralized nervous system (Malakhov 1994). In these aspects of embryo development, Enoplia resemble other animals and, thus, these characteristics can be considered as plesiomorphies (Schierenberg 2005). The basal position of the Enoplia is further supported by the retention of the nuclear envelope in the mature spermatozoa, an ancestral character (Baccetti et al. 1983). Spermatozoa from other nematodes outside the Enoplia always lack such an envelope (Justine 2002; Yushin 2003). Taking these additional morphological and embryological data into account, we suggest that Clade 1, as defined in figure 1, is indeed the most basal clade within the phylum Nematoda.
Within Clade 1, members of the family Tripylidae (order Triplonchida) have the shortest branch lengths. In contrast to the (limited number of) nematode taxa investigated so far (including the Enoplida), Tobrilus diversipapillatus, a representative of the Triplonchida, was shown to form a prominent coeloblastula, and gastrulation followed a pattern that is common within the animal kingdom but highly unusual among nematodes (Schierenberg 2005). Hence, currently available embryological data apparently support the very basal position of the Triplonchida in the SSU rDNA–based phylogenetic tree.
At first sight, the firm placement of the Bastianiidae and the Rhabdolaimidae (order Plectida according to De Ley and Blaxter 2002) within the basal orders Triplonchida and Enoplida is remarkable as the Plectida were previously suggested as the origin of Secernentean radiation (Blaxter et al. 1998). However, the order Plectida was suggested to be a mixture of paraphyletic and misplaced families (De Ley and Blaxter 2002). The Bastianiidae strongly resemble the Prismatolaimidae (Triplonchida) (Coomans and Raski 1988; Lorenzen 1994), and this supports its newly established phylogenetic position. Morphological support for the Rhabdolaimidae as a member of Enoplida comes in the shape of the amphids, a pair of sensory organs located on the head of a nematode. These are nonspiral and pocket shaped (Lorenzen 1994), a feature that is characteristic for the Enoplia sensu Lorenzen (newly proposed Clades 1 and 2).
Acceleration of SSU rDNA Substitution Rates
Based on a limited number of SSU rDNA sequences, nematodes were suggested to have a substitution rate 2–3 times greater than those of most other Metazoa (Aguinaldo et al. 1997). The large branch lengths of, for example, crown taxa belonging to Clades 9 (including C. elegans) and 10 (including, e.g., Strongyloides stercoralis) (fig. 1) seemed to support this statement, and a relative rate test (Li and Bousquet 1992) was performed to compare substitution rates of SSU rDNA among the clades. This test compares the weighted distances of the taxa of 2 clades with an outgroup (Robinson et al. 1998) and, in general, basal clades (Clades 1–7; formerly indicated as Adenophorea) evolve significantly slower than distal clades (Clades 8–12; formerly indicated as Secernentea) (table 2). Within Clade 1, sequences of Tripyla sp. (family Tripylidae), Paramphidelus hortensis (family Alaimidae), and Trischistoma sp. (Tripylidae) were closest to the most basal node within the phylogenetic tree.
Pairwise Differences among Clades in Relative Evolutionary Rates as Calculated by RRTree. The Outgroup Containing Other Metazoan Species Is Used as Outgroup in all Pairwise Comparisons
Clade | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | — | 1.249 | 0.932 | 1.065 | 1.133 | 1.177 | 1.171 | 1.274 | 1.399 | 1.847 | 1.381 | 1.464 |
| 2 | 0.034* | — | 0.786 | 0.891 | 0.913 | 0.905 | 1.050 | 1.072 | 1.096 | 1.554 | 1.107 | 1.261 |
| 3 | −0.010 | −0.039* | — | 1.135 | 1.199 | 1.202 | 1.309 | 1.371 | 1.513 | 1.948 | 1.499 | 1.573 |
| 4 | 0.010 | −0.019 | 0.022 | — | 1.088 | 1.092 | 1.167 | 1.245 | 1.333 | 1.737 | 1.370 | 1.402 |
| 5 | 0.020 | −0.015 | 0.028* | 0.014 | — | 1.040 | 1.031 | 1.121 | 1.181 | 1.604 | 1.268 | 1.330 |
| 6 | 0.027 | −0.016 | 0.031 | 0.015 | 0.007 | — | 1.000 | 1.100 | 1.150 | 1.582 | 1.209 | 1.296 |
| 7 | 0.026 | 0.009 | 0.053* | 0.032 | 0.005 | 0.000 | — | 1.072 | 1.173 | 1.490 | 1.223 | 1.260 |
| 8 | 0.042* | 0.013 | 0.059* | 0.042* | 0.021 | 0.018 | 0.015 | — | 1.072 | 1.276 | 1.259 | 1.353 |
| 9 | 0.042* | 0.012 | 0.047* | 0.034* | 0.021 | 0.018* | 0.021* | 0.010 | — | 1.282 | 1.203 | 1.432 |
| 10 | 0.108* | 0.086* | 0.126* | 0.109* | 0.090* | 0.088* | 0.086* | 0.056* | 0.038* | — | 0.784 | 0.825 |
| 11 | 0.056* | 0.019 | 0.074* | 0.060* | 0.046* | 0.038* | 0.041* | 0.048* | 0.028* | −0.053* | — | 1.049 |
| 12 | 0.062* | 0.042* | 0.082* | 0.064* | 0.049* | 0.046* | 0.047* | 0.064* | 0.053* | −0.042* | 0.010 | — |
Clade | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | — | 1.249 | 0.932 | 1.065 | 1.133 | 1.177 | 1.171 | 1.274 | 1.399 | 1.847 | 1.381 | 1.464 |
| 2 | 0.034* | — | 0.786 | 0.891 | 0.913 | 0.905 | 1.050 | 1.072 | 1.096 | 1.554 | 1.107 | 1.261 |
| 3 | −0.010 | −0.039* | — | 1.135 | 1.199 | 1.202 | 1.309 | 1.371 | 1.513 | 1.948 | 1.499 | 1.573 |
| 4 | 0.010 | −0.019 | 0.022 | — | 1.088 | 1.092 | 1.167 | 1.245 | 1.333 | 1.737 | 1.370 | 1.402 |
| 5 | 0.020 | −0.015 | 0.028* | 0.014 | — | 1.040 | 1.031 | 1.121 | 1.181 | 1.604 | 1.268 | 1.330 |
| 6 | 0.027 | −0.016 | 0.031 | 0.015 | 0.007 | — | 1.000 | 1.100 | 1.150 | 1.582 | 1.209 | 1.296 |
| 7 | 0.026 | 0.009 | 0.053* | 0.032 | 0.005 | 0.000 | — | 1.072 | 1.173 | 1.490 | 1.223 | 1.260 |
| 8 | 0.042* | 0.013 | 0.059* | 0.042* | 0.021 | 0.018 | 0.015 | — | 1.072 | 1.276 | 1.259 | 1.353 |
| 9 | 0.042* | 0.012 | 0.047* | 0.034* | 0.021 | 0.018* | 0.021* | 0.010 | — | 1.282 | 1.203 | 1.432 |
| 10 | 0.108* | 0.086* | 0.126* | 0.109* | 0.090* | 0.088* | 0.086* | 0.056* | 0.038* | — | 0.784 | 0.825 |
| 11 | 0.056* | 0.019 | 0.074* | 0.060* | 0.046* | 0.038* | 0.041* | 0.048* | 0.028* | −0.053* | — | 1.049 |
| 12 | 0.062* | 0.042* | 0.082* | 0.064* | 0.049* | 0.046* | 0.047* | 0.064* | 0.053* | −0.042* | 0.010 | — |
NOTE.—Below diagonal: pairwise differences in number of substitutions per site (clade in column − clade in row). Significance level after Bonferroni correction for multiple tests is P < 0.001, significant results are in bold. Above diagonal: relative differences in evolutionary rate calculated as distance to outgroup from clade in column divided by distance to outgroup from clade in row.
Pairwise Differences among Clades in Relative Evolutionary Rates as Calculated by RRTree. The Outgroup Containing Other Metazoan Species Is Used as Outgroup in all Pairwise Comparisons
Clade | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | — | 1.249 | 0.932 | 1.065 | 1.133 | 1.177 | 1.171 | 1.274 | 1.399 | 1.847 | 1.381 | 1.464 |
| 2 | 0.034* | — | 0.786 | 0.891 | 0.913 | 0.905 | 1.050 | 1.072 | 1.096 | 1.554 | 1.107 | 1.261 |
| 3 | −0.010 | −0.039* | — | 1.135 | 1.199 | 1.202 | 1.309 | 1.371 | 1.513 | 1.948 | 1.499 | 1.573 |
| 4 | 0.010 | −0.019 | 0.022 | — | 1.088 | 1.092 | 1.167 | 1.245 | 1.333 | 1.737 | 1.370 | 1.402 |
| 5 | 0.020 | −0.015 | 0.028* | 0.014 | — | 1.040 | 1.031 | 1.121 | 1.181 | 1.604 | 1.268 | 1.330 |
| 6 | 0.027 | −0.016 | 0.031 | 0.015 | 0.007 | — | 1.000 | 1.100 | 1.150 | 1.582 | 1.209 | 1.296 |
| 7 | 0.026 | 0.009 | 0.053* | 0.032 | 0.005 | 0.000 | — | 1.072 | 1.173 | 1.490 | 1.223 | 1.260 |
| 8 | 0.042* | 0.013 | 0.059* | 0.042* | 0.021 | 0.018 | 0.015 | — | 1.072 | 1.276 | 1.259 | 1.353 |
| 9 | 0.042* | 0.012 | 0.047* | 0.034* | 0.021 | 0.018* | 0.021* | 0.010 | — | 1.282 | 1.203 | 1.432 |
| 10 | 0.108* | 0.086* | 0.126* | 0.109* | 0.090* | 0.088* | 0.086* | 0.056* | 0.038* | — | 0.784 | 0.825 |
| 11 | 0.056* | 0.019 | 0.074* | 0.060* | 0.046* | 0.038* | 0.041* | 0.048* | 0.028* | −0.053* | — | 1.049 |
| 12 | 0.062* | 0.042* | 0.082* | 0.064* | 0.049* | 0.046* | 0.047* | 0.064* | 0.053* | −0.042* | 0.010 | — |
Clade | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | — | 1.249 | 0.932 | 1.065 | 1.133 | 1.177 | 1.171 | 1.274 | 1.399 | 1.847 | 1.381 | 1.464 |
| 2 | 0.034* | — | 0.786 | 0.891 | 0.913 | 0.905 | 1.050 | 1.072 | 1.096 | 1.554 | 1.107 | 1.261 |
| 3 | −0.010 | −0.039* | — | 1.135 | 1.199 | 1.202 | 1.309 | 1.371 | 1.513 | 1.948 | 1.499 | 1.573 |
| 4 | 0.010 | −0.019 | 0.022 | — | 1.088 | 1.092 | 1.167 | 1.245 | 1.333 | 1.737 | 1.370 | 1.402 |
| 5 | 0.020 | −0.015 | 0.028* | 0.014 | — | 1.040 | 1.031 | 1.121 | 1.181 | 1.604 | 1.268 | 1.330 |
| 6 | 0.027 | −0.016 | 0.031 | 0.015 | 0.007 | — | 1.000 | 1.100 | 1.150 | 1.582 | 1.209 | 1.296 |
| 7 | 0.026 | 0.009 | 0.053* | 0.032 | 0.005 | 0.000 | — | 1.072 | 1.173 | 1.490 | 1.223 | 1.260 |
| 8 | 0.042* | 0.013 | 0.059* | 0.042* | 0.021 | 0.018 | 0.015 | — | 1.072 | 1.276 | 1.259 | 1.353 |
| 9 | 0.042* | 0.012 | 0.047* | 0.034* | 0.021 | 0.018* | 0.021* | 0.010 | — | 1.282 | 1.203 | 1.432 |
| 10 | 0.108* | 0.086* | 0.126* | 0.109* | 0.090* | 0.088* | 0.086* | 0.056* | 0.038* | — | 0.784 | 0.825 |
| 11 | 0.056* | 0.019 | 0.074* | 0.060* | 0.046* | 0.038* | 0.041* | 0.048* | 0.028* | −0.053* | — | 1.049 |
| 12 | 0.062* | 0.042* | 0.082* | 0.064* | 0.049* | 0.046* | 0.047* | 0.064* | 0.053* | −0.042* | 0.010 | — |
NOTE.—Below diagonal: pairwise differences in number of substitutions per site (clade in column − clade in row). Significance level after Bonferroni correction for multiple tests is P < 0.001, significant results are in bold. Above diagonal: relative differences in evolutionary rate calculated as distance to outgroup from clade in column divided by distance to outgroup from clade in row.
Acceleration of nucleotide substitutions could be attributed to (a combination of) 2 causes: an elevated production of free radicals due to, for example, increased metabolic rates (usually associated with small body size) or an accumulation of DNA replication errors due to shorter generation times (e.g., Gillooly et al. 2005). Both in plant and animal parasitism, infective nematodes are exposed to free radicals (mostly reactive oxygen species) released by hosts as part of their defense response. Clades 8 and 12 are dominated by animal and plant parasites, respectively. We hypothesize that the release of free radicals by plant or animals has contributed to an accelerated evolution of these parasitic nematodes.
Clades 9 and 10 are dominated by bacterial feeding nematode families and contain only a few animal parasitic (Strongyloidea in Clade 9, Strongyloididae in Clade 10) and entomopathogenic nematodes (Heterorhabditidae in Clade 9, Steinernematidae in Clade 10). Clade 11 solely comprises bacterial feeding families. Hence, in Clades 9–11, intimate interactions with other organisms do not explain the observed accelerated substitution rates. Generalized life-history traits, including generation time, are one of the major components that were used by Bongers (1990) to develop an ecological scale for nonparasitic nematode families. Colonizers (c) and persisters (p) are extremes on a scale from 1 to 5, and c − p values of 1 are used to characterize stress-tolerant nematodes. Nematode families with a c − p value 1 have very short life cycles, produce large numbers of small eggs, have voluminous gonads, and are often viviparous. These families show high fluctuations in population densities, and, if present, they are present in huge numbers. Nonparasitic nematode families in Clade 9 have exceptionally low c − p values: Rhabditidae (c − p value 1), Diploscapteridae (1), Neodiplogastridae (1), Diplogastridae (1), Bunonematidae (1), and Myolaimidae (2). Essentially, the same holds for the nonparasitic families in Clade 10: Panagrolaimidae (1), Brevibuccidae (1), and Alloionematidae (1). Notably, Monhysteridae (Clade 5, relatively long branches) is the only family with a c − p value of 1 that is not residing in Clade 9 or 10. Clade 11 consists of Osstellidae and Cephalobidae (the positioning of Brevibucca sp. in this clade is probably an long branch attraction (LBA) artifact) and both families have a c − p value of 2. Hence, the relatively high SSU rDNA substitution rates in Clades 9 and 10 (Clade 11 to some extent) are associated with extremely low c − p values and, by extension, with short generation times.
Nematode Barcoding
The relatively high substitution rates of the SSU rDNA gene in nematodes in Clades 8–12 resulted in a level of sequence diversity that allows, in most cases, nematode identification at species level. Autapomorphies, mostly single-nucleotide polymorphisms (SNP), were found for, for example, the morphologically highly similar potato cyst nematode species Globodera rostochiensis and G. pallida and between the various Helicotylenchus species (see insert in fig. 1). In figure 3, it is shown how a single SNP can be used to (quantitatively) detect G. rostochiensis, whereas equal DNA concentrations of its sibling species G. pallida hardly give rise to any amplification product (ΔCT ≈ 19 [10 juveniles] and ΔCT ≈ 21 [100 juveniles]; CT: threshold cycle). These potato cyst nematode species were chosen to illustrate the potential of SSU rDNA polymorphisms for detection because these 2 species are morphologically nearly indistinguishable (e.g., Jones et al. 1970).
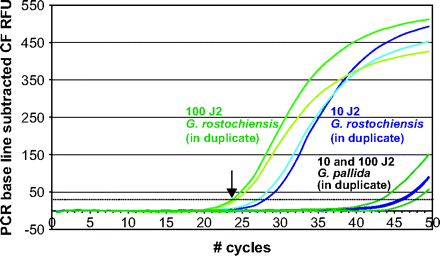
Real-time PCR amplification curves (in duplicate) showing that single-nucleotide differences in SSU rDNA sequences can be used to (quantitatively) detect second-stage juveniles (J2) of the potato cyst nematode species Globodera rostochiensis, whereas equal DNA concentrations of its sibling species Globodera pallida hardly give rise to a product (ΔCT around 20). CT — cycle number at which the fluorescent signal exceeds the threshold value as indicated by the dotted line (foremost left curve indicated by an arrow).
The current phylum-wide data set allows for the identification of individual nematode species within a pool of nontarget taxa, as for instance in case of soil samples (Helder et al. 2004). For nematode biodiversity studies, it has been proposed to define molecular operational taxonomic units on the basis of a defined number of SSU rDNA sequence differences instead of classical species concepts (Floyd et al. 2002). However, different rates of evolution among nematode clades (table 2) implicate that a defined number of nucleotide differences cannot always be linked unequivocally to meaningful biological differences.
The Origin of the Secernentean Radiation
The SSU rDNA sequence data presented here provide a detailed insight into the relationship between Adenophorea (fig. 1; Clades 1–7) and Secernentea (fig. 1; Clades 8–12), a partition of the phylum Nematoda that has dominated nematode systematics since it was proposed by Chitwood BG and Chitwood MB (1933). Secernentea (equivalent to the order Rhabditida with the exception of the Teratocephalidae)—a group that includes virtually all major animal and plant parasites—arose from Adenophorea (Blaxter et al. 1998), and the current SSU rDNA data set suggests that members of the genus Teratocephalus (Clade 7) are the closest living representatives of the common ancestor of the Secernentea. The genus Teratocephalus, the only genus within the family Teratocephalidae, exhibits a mixture of morphological characters of Secernentea and Adenophorea (e.g., Zhang and Baldwin 2001), and the taxonomic position of the family Teratocephalidae is still unclear (De Ley and Blaxter 2002). The family Metateratocephalidae (genera Euteratocephalus and Metateratocephalus) was included in the Teratocephalidae in the past (Lorenzen 1983), but the current data set point at a position in a separate clade, Clade 6, that also includes the Plectidae and Chronogastridae. This observation corresponds with the results from a detailed morphological scanning electron microscopy study on teratocephalids by Boström (1989). He listed 8 morphological characters, all of which call for a taxonomic separation of Teratocephalus and Metateratocephalus, but he found no phasmids—small organs in the tail region that are characteristic for most Secernentea (in some Secernentean taxa, they are secondarily lacking)—in any of the Teratocephalus species under investigation. On the basis of SSU rDNA sequence data, members of the genus Teratocephalus can be considered as the immediate sister group of the Secernentea.
Did Plant Parasites Evolve from Fungivorous Ancestors?
A long-standing hypothesis on the evolution of feeding types among nematodes suggests that plant parasitic nematodes arose from fungivorous ancestors (Maggenti 1971). Analysis of full-length SSU rDNA data reveals the presence of fungivorous nematodes (Yeates et al. 1993) in 3 clades: in Clade 1, representatives of the genus Diphtherophora, Clade 2, members of the Tylencholaimidae (Dorylaimida), and Clade 12, various representatives of the Tylenchomorpha. The tree suggests the presence of a fourth group of fungivorous nematode in Clade 10 (not marked in fig. 1 as fungivorous), namely, representatives of the Aphelenchoididae (various Aphelenchoides species) and Parasitaphelenchidae (i.e., Bursaphelenchus spp.). However, the position of the Aphelenchoididae, Parasitaphelenchidae, and Seinuridae (Tylenchomorpha) within Clade 10 is most likely an LBA artifact as their GC content (≈46%) and that of the Panagrolaimorpha (≈43%) are relatively low and they exhibit long branch lengths as compared with the Aphelenchidae and Paraphelenchidae (GC content ≈ 48%) with whom they are normally associated. Similar concerns were raised by De Ley and Blaxter (2002). It is noted that an additional analysis of Clades 10, 11, and 12 excluding the Panagrolaimorpha did not unite the Paraphelenchidae and Aphelenchidae with the Aphelenchoididae, Parasitaphelenchidae, and Seinuridae (results not shown). Remarkably, fungivorous nematodes are only observed in clades that contain plant parasitic nematodes as well. In Clades 1 and 12, SSU rDNA shows sufficient variation to determine the phylogenetic relationships between fungivorous and plant parasitic nematodes. The SSU rDNA of the Dorylaimida within Clade 2 is remarkably conserved (possibly an indication for rapid speciation), and consequently, the position of fungivores within this clade is unresolved. In figure 1, we show the SSU rDNA–based phylogenetic relationships between fungivorous and plant parasitic nematodes. In Clade 12, fungivorous nematodes occupy a basal position as compared with their plant parasitic relatives. This finding implies a first molecular support for the hypothesis stating that plant parasitic nematodes arose from fungivorous ancestors. In Clade 1, the fungivores and plant parasites are sister groups, and it is impossible to predict the feeding type of their last common ancestor.
Supplementary Material
Table S1 showing all SSU rDNA sequences used in this paper (scientific names and corresponding GenBank accession numbers) and Figures S1, S2, and S3 showing the complete phylogenetic trees of nematodes with full species names and support values (Fig. S1: Bayesian inference; Fig. S2: maximum parsimony; and Fig. S3: Neighbor Joining) see:(Supplementary Material).
These authors contributed equally to this work.
Diethard Tautz, Associate Editor
We would like to thank Freek Bakker of the Biosystematics group, Wageningen University, for the useful discussions. This work was supported by the Dutch Technology Foundation (STW) grant WBI4725. A.v.d.W. was funded by the Netherlands Organization for Scientific Research (NWO-SSEO) grant 0123.060/061. Work at the SARA Computing and Networking Services in Amsterdam, The Netherlands, was sponsored by the National Computing Facilities Foundation with financial support from NWO-SSEO.
References
Aguinaldo AMA, Turbeville JM, Linford LS, Rivera MC, Garey JR, Raff RA, Lake JA.
Aleshin VV, Kedrova OS, Milyutina IA, Vladychenskaya NS, Petrov NB.
Aleshin VV, Milyutina IA, Kedrova OS, Vladychenskaya NS, Petrov NB.
Alfaro ME, Zoller S, Lutzoni F.
Andrássy I.
Baccetti B, Dallai R, Dezio SG, Marinari A.
Baldwin JG, Nadler SA, Adams BJ.
Blaxter M, Dorris M, De Ley P.
Blaxter ML, De Ley P, Garey JR, et al.
Bongers T.
Bongers T, Ferris H.
Boström S.
Chitwood BG.
Chitwood BG.
Coomans AV, Raski DJ.
De Coninck LAP.
De Ley P, Blaxter ML.
De Ley P, Blaxter ML.
Erixon P, Svennblad B, Britton T, Oxelman B.
Felsenstein J.
Floyd R, Abebe E, Papert A, Blaxter M.
Gillooly JF, Allen AP, West GB, Brown JH.
Hall TA.
Helder J, Van den Elsen SJJ, Bongers AMT, Van der Wurff AWG, Bakker J, Kammenga JE, inventors.
Hillis DM, Bull JJ.
Holder M, Lewis PO.
Jones FGW, Carpente JM, Parrott DM, Stone AR, Trudgill DL.
Justine J-L.
Li P, Bousquet J.
Lorenzen S.
Lorenzen S.
Maggenti AR.
Maggenti AR.
Malakhov VV.
Philippe H, Germot A, Moreira D.
Platt HM.
Posada D, Crandall KA.
Robinson M, Gouy M, Gautier C, Mouchiroud D.
Robinson-Rechavi M, Huchon D.
Ronquist F, Huelsenbeck JP.
Saint Andre AV, Blackwell NM, Hall LR, et al.
Schierenberg E.
Sudhaus W, Fitch D.
Swofford DL.
Wuyts J, De Rijk P, Van de Peer Y, Pison G, Rousseeuw P, De Wachter R.
Yeates GW, Bongers T, De Goede RGM, Freckman DW, Georgieva SS.
Yushin VV.
Author notes
*Laboratory of Nematology, Department of Plant Sciences, Wageningen University, Wageningen, The Netherlands; and †Department of Zoology, Biology Faculty, Ivan Franko National University, Lviv, Ukraine

{kind=link}
{kind=link}
{kind=link}