





Abstract
In the Neisseria spp., natural competence for transformation and homologous recombination generate antigenic variants through creation of mosaic genes (such as opas) and through recombination with silent cassettes (such as pilE/pilS) and gene-complement diversity through the horizontal exchange of whole genes or groups of genes, in minimal mobile elements (MMEs). An MME is a region encompassing 2 conserved genes between which different whole-gene cassettes are found in different strains, which are chromosomally incorporated solely through the action of homologous recombination. Comparative analyses of the neisserial genome sequences identified 39 potential MME sites, the contents of which were investigated in 11 neisserial strains. One hundred and eight different MME regions were identified, 20 of which contain novel sequences and these contain 12 newly identified neisserial coding sequences. Neisserial uptake signal sequences are associated with 38 of the 40 MMEs studied. In some sites, divergent dinucleotide signatures of the sequences between the flanking genes suggest relatively recent horizontal acquisition of some cassettes. The neisserial MMEs were used to interrogate all of the other available bacterial genome sequences, revealing frequent conservation of the flanking genes combined with the presence of different gene cassettes between them. In some cases, these sites can definitively be classified as MMEs in these other genera. These findings provide additional evidence for the MME model, indicate that MME-directed investigations are a good basis for the identification of novel strain-specific genes and differences within bacterial populations and demonstrate that these elements are probably ubiquitously involved in genetic exchange, particularly in naturally competent bacteria.
Introduction
The horizontal exchange of DNA is instrumental in the evolution of bacteria, including the acquisition of virulence factors. The movement of DNA between bacteria has been extensively investigated, with particular focuses on self-mobilizing and integrating elements and genes associated with virulence and antibiotic resistance (Ochman et al. 2000; Dutta and Pan 2002; Gogarten et al. 2002; Frank et al. 2005; Zuniga et al. 2005; Gophna et al. 2006; Becq et al. 2007). Additionally, the incorporation of foreign DNA into the chromosome without the involvement of specific mobilization machinery or homologous recombination is possible, although this is a comparatively infrequent event (Hill and Grant 2002; Lawrence 2002).
The presence of different cassettes of genes in different strains of the Neisseria spp. has been described for several chromosomal loci (Zhu et al. 1999; Claus et al. 2000; Klee et al. 2000; Kahler et al. 2001; Saunders and Snyder 2002; Snyder et al. 2004). Nine genetic islands have been reported that distinguish Neisseria meningitidis from Neisseria gonorrhoeae (Klee et al. 2000), including regions that are flanked by rfaE and rfaD, fpr and dinP, pheS and pheT, and tex and galE. The later region contains the meningococcal capsule and transport gene locus, which is absent in N. gonorrhoeae and some strains of N. meningitidis. The locus containing the meningococcal outer membrane protein gene opc, which is also absent in N. gonorrhoeae and some N. meningitidis strains, shows variation in the presence of genes between glyA and dedA (Seiler et al. 1996; Zhu et al. 1999, 2003). Different genes that can include restriction modification systems are found between hrpA and a hypothetical protein gene (Bart et al. 2001) and between trpE and purK (Zhu et al. 1999). Other instances of exchangeable gene cassette systems and regions of pathogen- or strain-specific genes have also been reported (Klee et al. 2000; Kahler et al. 2001; Perrin et al. 2002; Snyder and Saunders 2006).
The model of minimal mobile elements (MMEs) and their role in horizontal gene transfer and integration (fig. 1) have been developed (Saunders and Snyder 2002), in which DNA transfer and homologous recombination acts to mobilize whole-gene cassettes that are flanked by conserved protein-encoding genes. It is believed that these gene cassettes are initially inserted between the conserved genes as a rare event, using little or no homologous sequence. Once in place, however, further mobilization of the element proceeds more efficiently by virtue of the flanking homologous sequences. Especially in naturally transformable species, the chromosomal incorporation of MMEs can be a powerful mechanism for the mobilization of strain-specific genes. The different sequences (containing genes or not) that are present between the same conserved flanking genes subsequently behave as mutually exclusive exchangeable cassettes.
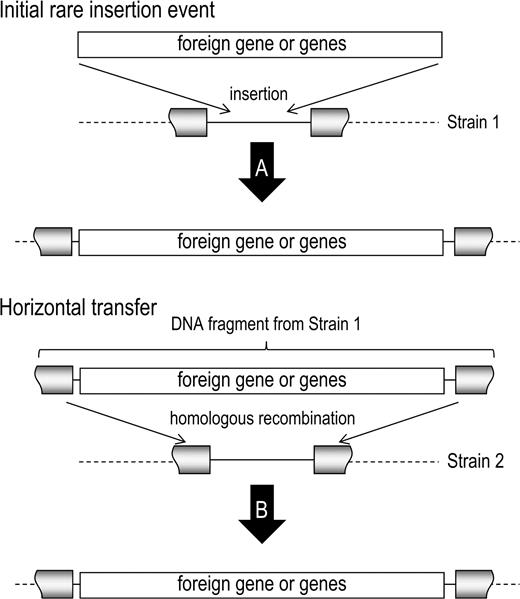
The MME model. By a rare insertion event, a foreign gene or group of genes is inserted into the genome of a bacterial strain (here designated Strain 1). The gene or genes are then present in the chromosome of Strain 1 (A). This segment of DNA can then be horizontally transferred to other bacterial strains, such as that designated here as Strain 2. Due to the conserved nature of the flanking genes (gray) between Strain 1 and Strain 2, the gene becomes integrated into the genome of Strain 2 through homologous recombination. Strain 2 now also has the new gene in this MME site (B) and will have replaced any other sequences located at this site.
To date, MME sites have been identified in the Neisseria spp. (Saunders and Snyder 2002; Snyder et al. 2004, 2005; Snyder and Saunders 2006), Group B Streptococci (Herbert et al. 2005), Helicobacter pylori (Salaün L and Saunders NJ, unpublished data), Escherichia coli (Sibley and Raleigh 2004), and Vibrio cholerae (Miller et al. 2007). They have also been described in H. pylori (Chanto et al. 2002) and Streptococcus suis (Sekizaki et al. 2005), although these authors did not describe them as such. In E. coli, the immigration control region (ICR) is one such MME site. It contains sets of genes that can be exchanged through homologous recombination with conserved flanking sequences. This generates a cassette-like system with the result that different strains contain a variety of different genes within the ICR. The majority of these encode restriction modification systems, although strains containing a penicillin acylase within the ICR and strains without any genes in the ICR have been identified (Sibley and Raleigh 2004).
One neisserial MME site, located between the pheS and pheT genes (MMEpheST), has been previously investigated in detail in 69 strains. In this MME site, 9 different MME regions have been identified, 6 of these encoding 6 different restriction modification systems (Saunders and Snyder 2002). Specific MME sites have also been investigated in N. gonorrhoeae strains MS11 and FA19 pursuing differences in hybridization of these strains to the pan-Neisseria microarray, compared with the sequenced strain FA1090 (Snyder et al. 2004). In a subsequent study, differences in 6 MMEs were found to contribute to the definition of gene-complement combinations strongly associated with clinical isolates causing disseminated gonococcal infections (Snyder LAS and Saunders NJ, unpublished data). At least 3 MMEs contain virulence traits that differentiate the pathogenic and nonpathogenic Neisseria (Snyder and Saunders 2006).
In the present study, the repertoire of MMEs in Neisseria has been further investigated using a genome-wide analysis of 4 strains in which 38 potential MMEs were identified in the pathogenic Neisseria spp. The contents of these regions, MMEpheST and MMEleuDB, were then investigated in a collection of the 11 neisserial strains that showed the greatest diversity in the previous investigation of MMEpheST (Saunders and Snyder 2002), including strains of N. meningitidis, N. gonorrhoeae, Neisseria lactamica, and Neisseria polysaccharea. Several new sites were defined, and the different combinations of MME-associated coding sequences (CDSs) present in these 11 strains were determined, showing that these elements occur in different combinations clearly indicative of their independent movement within these populations. The locations of the neisserial sites were then used to investigate the presence of common MME sites containing strain- and species-specific CDSs in all the other available bacterial genome sequences.
It was discovered that unrelated genome sequences frequently share the conserved flanking gene organizations seen in the Neisseria. CDSs were identified between the previously identified flanking genes that suggest the presence of a similarly located candidate MME (cMME) in 24 of the 40 assessed neisserial MMEs and cMMEs in these unrelated species. In most cases, only a single example of an intervening gene cassette is currently identifiable in these other species. However, further comparisons between the genome sequences of related strains of these other species indicate that 5 of 24 identified cMMEs have more than 1 identifiable intervening cassette and are also MMEs within these other populations. These common sites and flanking genes suggest the potential for these shared MMEs to facilitate the horizontal transfer of genes across genera as well as between strains and closely related species.
Materials and Methods
Comparative Genome Analyses
The genome sequences of N. meningitidis strain MC58 (Tettelin et al. 2000), N. meningitidis strain Z2491 (Parkhill et al. 2000), N. meningitidis strain FAM18 (Bentley et al. 2007), and N. gonorrhoeae strain FA1090 (AE004969) were assessed using the whole-genome database graphical interface ACEDB (Durbin and Thierry-Mieg 1991) as described previously (Snyder et al. 2001; Saunders and Snyder 2002) to identify regions where conserved genes flank strain-specific CDSs. Regions that contained recognizable mobilizing elements, such as transposases, bacteriophage insertion, or chromosome rearrangements, were excluded unless other evidence indicated an MME in other strains at the same site in which no such elements were present.
Bacterial Strains
A set of 11 strains (N. meningitidis strains A22, 860800, 00/240794, 97/282675, 00/240868, 01/241825, and NG E30; N. gonorrhoeae strains 28539 and 26593; N. lactamica; and N. polysaccharea) were selected for polymerase chain reaction (PCR) amplification of the MMEs. These strains showed the greatest differences in the previous study of MMEpheST, in which this single site was investigated in 69 strains (Saunders and Snyder 2002). These 11 strains include representatives of meningococcal serogroups B, D, 29E, H, Y, and Z, as well as blood and joint isolates of N. gonorrhoeae. A control PCR was included, using N. meningitidis strain MC58 and/or N. gonorrhoeae strain FA1090.
Assessment of CDSs within the MME Sites
Primers were designed within the flanking conserved genes of each MME site complementary to sequences that were conserved in all 4 genome-sequenced neisserial strains and that lay outside of any apparent region of recombination, thereby selecting primer sites likely to be conserved in other neisserial strains (listed in supplementary table, Supplementary Material online).
PCR was performed using Invitrogen (Paisley, UK) Taq polymerase, Bioline (London, UK) Bio-X-Act Long Polymerase, or Qiagen (Crawley, UK) HotStarTaq polymerase, according to the manufacturers’ instructions. PCR products were visualized on ethidium bromide-containing agarose gels, and product sizes were determined using Quantity One image analysis software (Biorad, Hemel Hempstead, UK). Product lengths were compared with those from the 4 sequenced strains used for region selection. PCR products that differed in length from those expected from known regions were identified. Those PCR products that were the same size as those from known regions were digested with DpnII (New England Biolabs, Hitchin, UK, 2 units of enzyme were used for 5 μl of PCR product), and the digest patterns were compared with the expected patterns in the sequenced strains.
Those products that were either different in length and/or restriction digest pattern were sequenced using ABI Prism BigDye Terminator Cycle Sequencing version 3.0 (Applied Biosystems, Warrington, UK) and resolved on an ABI Prism 3100 DNA Sequencer (Applied Biosystems). Sequence traces were analyzed using Trev (Bonfield et al. 2002). Sequences were aligned and assembled using the Wisconsin Package from GCG (Accelrys, Cambridge, UK).
Dinucleotide Signature Analysis of Neisserial MMEs
The dinucleotide signature for each of the MME regions was calculated using the sequence located between the coding regions of the flanking conserved genes, provided that this intervening sequence was 600 bp or more in length. The use of shorter sequences introduces sampling effects such that abnormal signatures cannot be reliably attributed to untypical sequence composition. Divergence (termed δ* difference; Karlin et al. [1997]; Karlin [2001]) was determined on the basis of comparison with the dinucleotide signature of the whole-genome sequences.
Presence of Neisserial Uptake Signal Sequences
The presence of neisserial uptake signal sequences (NUSSs) within the MME and the conserved flanking genes was assessed using fuzznuc (Rice et al. 2000) to search for occurrences of the recognized NUSS (GCCGTCTGAA) in the genome sequences of N. gonorrhoeae strain FA1090 and N. meningitidis strains MC58 and Z2491, allowing for 1 mismatch. Single mismatches in otherwise conserved sequence regions have been observed in the commensal Neisseria spp., and such mismatches are present in the pathogenic neisserial genome sequences (Qvarnstrom and Swedberg 2006).
Interrogation of the Available Genome Sequences to Identify MMEs Shared with the Neisseria spp.
Orthologues of the neisserial MME-flanking genes were searched for in the other available bacterial genome sequences using Blast versus translated annotated CDSs from bacterial genomes and major plasmids (downloaded from GenBank, ftp.ncbi.nih.gov/genomes/Bacteria; 569 entries in total). The Blast program BlastP was used to compare each of the translated MME-flanking genes with the translated annotated CDSs from bacterial genomes, using a cutoff for the expectation value (e) of 0.001. This value was chosen to increase the likelihood of identification of the orthologous flanking genes. All other parameters were set at their default values. Annotated CDSs that started or ended in the MME or that encompassed the MME in its entirety were identified.
GenBank accession numbers for the neisserial sequences determined as part of this study are DQ115757, DQ115758, DQ115759, DQ115760, DQ115761, DQ115762, DQ115763, DQ115764, DQ115765, DQ115766, DQ115767, DQ115768, DQ115769, DQ115770, DQ115771, DQ115772, DQ115773, DQ115774, DQ115775, DQ115776, DQ115777, and DQ117942.
Results
Comparative Genome Sequence Analysis
Once all the strain-divergent regions that were too large to be amplified or which contained evidence of other sources of horizontal gene transfer or degeneration were excluded, the comparative genome analyses of N. meningitidis strain MC58, Z2491, and FAM18 and N. gonorrhoeae strain FA1090 identified 37 new potential MME sites as suitable for further investigation (table 1). The previously identified MMEnuoLM was also included in the subsequent investigation of the 11 neisserial strains. In some cases, the MME site contains no CDSs between the conserved flanking regions. These instances in some genome sequences are classified as “empty” MME sites and are likely to represent the locus before the initial insertion event (fig. 2).
Content of MME and cMME Sites in 11 Strains of Neisseria spp.
| Site | Nm Strain A22 | Nm Strain 860 800 | Nm Strain 00/240 794 | Nm Strain 97/282 675 | Nm Strain 00/240 868 | Nm Strain 01/241 825 | Nm Strain NG E30 | Ng Strain 28 539 | Ng Strain 26 593 | Neisseria lactamica | Neisseria polysaccharea | New Regions | New Genes | MME Regions | Present in Other Species l |
| MMEpheSTa | b | b | b | b | b | a | a | c | NDe | c | a | a | a | 8a | Yes |
| MMErpoCrpsL | d | d | d | d | d | d | d | c | c | NPf | g | 0 | 0 | 3 | Yes |
| MMEvalSlysR | b | d | d | c | c | d | d | c | c | NPf | 57k | 1 | 1 | 4 | — |
| cMMEdadAvalS | d | d | d | d | d | d | d | c | c | c | NPf | 0 | 0 | 2 | — |
| cMMEfabZlpxA | g | d | g | g | g | d | d | c | c | b | d | 0 | 0 | 2 | Yes |
| MMEfrr | d | NPf | g | d | NPf | d | d | c | c | 58k | NPf | 1 | 2 | 3 | — |
| cMMEnuoG | c | c | c | c | c | d | c | c | c | NPf | NPf | 0 | 0 | 2 | — |
| cMMEnuoIJ | c | g | c | d | d | c | c | c | g | c | 0 | 0 | 2 | — | |
| cMMEnuoKL | d | d | g | d | d | b | d | c | c | c | d | 0 | 0 | 2 | — |
| MMEnuoLM | d | c | d | d | d | d | d | c | c | d | d | 0 | 0 | 4h | — |
| cMMEpdxJacpS | d | d | d | d | d | d | d | c | c | c | c | 0 | 0 | 3 | Yes |
| cMMEamiC | b | b | g | g | g | b | d | c | c | d | d | 0 | 0 | 3 | — |
| cMMESMC | d | d | d | d | d | d | d | c | c | c | d | 0 | 0 | 2 | — |
| cMMEgcvTH | d | d | NPf | NPf | d | d | d | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMEksgA | d | d | d | d | d | d | d | c | c | c | c | 0 | 0 | 2 | Yes |
| cMMErpoH | d | d | d | d | d | d | d | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMENMB0775-6 | b | g | b | b | b | b | b | c | c | NPf | NPf | 0 | 0 | 3 | — |
| cMMErfaED | NPf | NPf | d | NPf | NPf | NPf | d | c | c | NPf | c | 0 | 0 | 2 | Yes |
| cMMEdld | d | d | c | d | d | d | d | c | d | c | c | 0 | 0 | 2 | Yes |
| MMEtrpEpurK | d | d | d | d | d | c | d | c | c | d | 61k | 1 | 0 | 3 | — |
| cMMEaspA | d | d | 62m | d | d | d | d | NPf | NPf | 62k | d | 1 | 0 | 3 | Yes |
| MMEglyAdedA | g | NPf | g | b | NPf | g | NPf | c | NPf | g | NPf | 0 | 0 | 5j | Yes |
| cMMEglnDguaB | c | c | NPf | NPf | d | c | d | c | c | c | d | 0 | 0 | 2 | Yes |
| cMMEzurcobW | c | NPf | NPf | b | b | NPf | b | c | c | c | c | 0 | 0 | 2 | — |
| MMEnrdAB | 64k | d | g | NPf | d | g | d | c | c | 65k | c | 2 | 2 | 5 | Yes |
| MMEuvrB | NPf | NPf | 66k | NPf | NPf | i | i | c | c | NPf | NPf | 2 | 2 | 6 | Yes |
| cMMEpdhBlpdA | b | b | b | b | b | b | d | c | c | b | b | 0 | 0 | 2 | Yes |
| cMMEfmu | b | b | b | b | b | c | b | c | c | NPf | b | 0 | 0 | 2 | — |
| cMMErbfAtruB | d | d | b | d | d | b | d | c | c | b | b | 0 | 0 | 2 | Yes |
| MMEnifSU | d | d | 67k | 68k | 68m | d | 68m | c | c | 69k | d | 3 | 4 | 5 | Yes |
| MMEfprdinP | NPf | c | g | 70k | 70m | g | g | c | c | NPf | c | 1 | 0 | 5 | Yes |
| MMEhesA | c | c | 71k | 72k | 72m | c | c | c | c | NPf | 73k | 3 | 1 | 5 | Yes |
| cMMEasmAfolP | d | d | d | d | d | d | 77k | NPf | NPf | NPf | NPf | 1 | 0 | 3 | — |
| cMMEgrx | NPf | 74k | d | d | d | d | d | c | c | c | c | 1 | 0 | 3 | — |
| cMMEglyQS | b | b | b | c | c | b | b | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMEpurLhagH | d | d | d | d | d | d | d | 75k | 75m | d | d | 1 | 0 | 3 | Yes |
| cMMEhagHmgtE | d | d | c | NPf | NPf | NPf | c | c | c | c | c | 0 | 0 | 2 | Yes |
| cMMEmetRrpsI | d | NPf | d | d | d | d | d | c | c | 42k | c | 1 | 0 | 3 | Yes |
| MMEobgcysS | NPf | d | d | d | d | c | d | c | c | d | c | 0 | 0 | 2 | — |
| Total | 19 | 12 | 118 | — | |||||||||||
| Site | Nm Strain A22 | Nm Strain 860 800 | Nm Strain 00/240 794 | Nm Strain 97/282 675 | Nm Strain 00/240 868 | Nm Strain 01/241 825 | Nm Strain NG E30 | Ng Strain 28 539 | Ng Strain 26 593 | Neisseria lactamica | Neisseria polysaccharea | New Regions | New Genes | MME Regions | Present in Other Species l |
| MMEpheSTa | b | b | b | b | b | a | a | c | NDe | c | a | a | a | 8a | Yes |
| MMErpoCrpsL | d | d | d | d | d | d | d | c | c | NPf | g | 0 | 0 | 3 | Yes |
| MMEvalSlysR | b | d | d | c | c | d | d | c | c | NPf | 57k | 1 | 1 | 4 | — |
| cMMEdadAvalS | d | d | d | d | d | d | d | c | c | c | NPf | 0 | 0 | 2 | — |
| cMMEfabZlpxA | g | d | g | g | g | d | d | c | c | b | d | 0 | 0 | 2 | Yes |
| MMEfrr | d | NPf | g | d | NPf | d | d | c | c | 58k | NPf | 1 | 2 | 3 | — |
| cMMEnuoG | c | c | c | c | c | d | c | c | c | NPf | NPf | 0 | 0 | 2 | — |
| cMMEnuoIJ | c | g | c | d | d | c | c | c | g | c | 0 | 0 | 2 | — | |
| cMMEnuoKL | d | d | g | d | d | b | d | c | c | c | d | 0 | 0 | 2 | — |
| MMEnuoLM | d | c | d | d | d | d | d | c | c | d | d | 0 | 0 | 4h | — |
| cMMEpdxJacpS | d | d | d | d | d | d | d | c | c | c | c | 0 | 0 | 3 | Yes |
| cMMEamiC | b | b | g | g | g | b | d | c | c | d | d | 0 | 0 | 3 | — |
| cMMESMC | d | d | d | d | d | d | d | c | c | c | d | 0 | 0 | 2 | — |
| cMMEgcvTH | d | d | NPf | NPf | d | d | d | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMEksgA | d | d | d | d | d | d | d | c | c | c | c | 0 | 0 | 2 | Yes |
| cMMErpoH | d | d | d | d | d | d | d | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMENMB0775-6 | b | g | b | b | b | b | b | c | c | NPf | NPf | 0 | 0 | 3 | — |
| cMMErfaED | NPf | NPf | d | NPf | NPf | NPf | d | c | c | NPf | c | 0 | 0 | 2 | Yes |
| cMMEdld | d | d | c | d | d | d | d | c | d | c | c | 0 | 0 | 2 | Yes |
| MMEtrpEpurK | d | d | d | d | d | c | d | c | c | d | 61k | 1 | 0 | 3 | — |
| cMMEaspA | d | d | 62m | d | d | d | d | NPf | NPf | 62k | d | 1 | 0 | 3 | Yes |
| MMEglyAdedA | g | NPf | g | b | NPf | g | NPf | c | NPf | g | NPf | 0 | 0 | 5j | Yes |
| cMMEglnDguaB | c | c | NPf | NPf | d | c | d | c | c | c | d | 0 | 0 | 2 | Yes |
| cMMEzurcobW | c | NPf | NPf | b | b | NPf | b | c | c | c | c | 0 | 0 | 2 | — |
| MMEnrdAB | 64k | d | g | NPf | d | g | d | c | c | 65k | c | 2 | 2 | 5 | Yes |
| MMEuvrB | NPf | NPf | 66k | NPf | NPf | i | i | c | c | NPf | NPf | 2 | 2 | 6 | Yes |
| cMMEpdhBlpdA | b | b | b | b | b | b | d | c | c | b | b | 0 | 0 | 2 | Yes |
| cMMEfmu | b | b | b | b | b | c | b | c | c | NPf | b | 0 | 0 | 2 | — |
| cMMErbfAtruB | d | d | b | d | d | b | d | c | c | b | b | 0 | 0 | 2 | Yes |
| MMEnifSU | d | d | 67k | 68k | 68m | d | 68m | c | c | 69k | d | 3 | 4 | 5 | Yes |
| MMEfprdinP | NPf | c | g | 70k | 70m | g | g | c | c | NPf | c | 1 | 0 | 5 | Yes |
| MMEhesA | c | c | 71k | 72k | 72m | c | c | c | c | NPf | 73k | 3 | 1 | 5 | Yes |
| cMMEasmAfolP | d | d | d | d | d | d | 77k | NPf | NPf | NPf | NPf | 1 | 0 | 3 | — |
| cMMEgrx | NPf | 74k | d | d | d | d | d | c | c | c | c | 1 | 0 | 3 | — |
| cMMEglyQS | b | b | b | c | c | b | b | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMEpurLhagH | d | d | d | d | d | d | d | 75k | 75m | d | d | 1 | 0 | 3 | Yes |
| cMMEhagHmgtE | d | d | c | NPf | NPf | NPf | c | c | c | c | c | 0 | 0 | 2 | Yes |
| cMMEmetRrpsI | d | NPf | d | d | d | d | d | c | c | 42k | c | 1 | 0 | 3 | Yes |
| MMEobgcysS | NPf | d | d | d | d | c | d | c | c | d | c | 0 | 0 | 2 | — |
| Total | 19 | 12 | 118 | — | |||||||||||
NOTE.—Nm, Neisseria meningitidis and Ng, Neisseria gonorrhoeae.
Data for this MME is from the investigation described in Saunders and Snyder (2002). Neisseria polysaccharea is AF542178, Nm strain 01/241825 is similar to AJ311178, and Nm strain NG E30 is similar to AF542173.
PCR product size and digest pattern indicate this region is similar to that in Nm strain Z2491.
PCR product size and digest pattern indicate this region is similar to that in Ng strain FA1090.
PCR product size and digest pattern indicate this region is similar to that in Nm strain MC58.
Not done.
No PCR product.
PCR product size and digest pattern indicate this region is similar to that in Nm strain FAM18.
The MME region from N. gonorrhoeae strain MS 11 (AY386266) was sequenced previously (Snyder et al. 2004).
Region is like that seen in Nm strain 00/240794 (DQ115766), but with an IS1301 inserted between bases 1130 and 1131.
The MME region from N. polysaccharea strain 89357 (AY072809) was sequenced previously (Zhu et al. 2003).
GenBank accession numbers—42: DQ117942; 57: DQ115757; 58: DQ115758; 61: DQ115761; 62: DQ115762; 64: DQ115764; 65: DQ115765; 66: DQ115766; 67: DQ115767; 68: DQ115768; 69: DQ115769; 70: DQ115770; 71: DQ115771; 72: DQ115772; 73: DQ115773; 74: DQ115774; 75: DQ115775; and 77: DQ115777.
Neisseria spp., MME, and cMME sites that are present with gene cassettes between the flanking genes in other bacterial genera.
PCR product size and digest pattern indicate this region is similar to that in the accession number indicated.
Content of MME and cMME Sites in 11 Strains of Neisseria spp.
| Site | Nm Strain A22 | Nm Strain 860 800 | Nm Strain 00/240 794 | Nm Strain 97/282 675 | Nm Strain 00/240 868 | Nm Strain 01/241 825 | Nm Strain NG E30 | Ng Strain 28 539 | Ng Strain 26 593 | Neisseria lactamica | Neisseria polysaccharea | New Regions | New Genes | MME Regions | Present in Other Species l |
| MMEpheSTa | b | b | b | b | b | a | a | c | NDe | c | a | a | a | 8a | Yes |
| MMErpoCrpsL | d | d | d | d | d | d | d | c | c | NPf | g | 0 | 0 | 3 | Yes |
| MMEvalSlysR | b | d | d | c | c | d | d | c | c | NPf | 57k | 1 | 1 | 4 | — |
| cMMEdadAvalS | d | d | d | d | d | d | d | c | c | c | NPf | 0 | 0 | 2 | — |
| cMMEfabZlpxA | g | d | g | g | g | d | d | c | c | b | d | 0 | 0 | 2 | Yes |
| MMEfrr | d | NPf | g | d | NPf | d | d | c | c | 58k | NPf | 1 | 2 | 3 | — |
| cMMEnuoG | c | c | c | c | c | d | c | c | c | NPf | NPf | 0 | 0 | 2 | — |
| cMMEnuoIJ | c | g | c | d | d | c | c | c | g | c | 0 | 0 | 2 | — | |
| cMMEnuoKL | d | d | g | d | d | b | d | c | c | c | d | 0 | 0 | 2 | — |
| MMEnuoLM | d | c | d | d | d | d | d | c | c | d | d | 0 | 0 | 4h | — |
| cMMEpdxJacpS | d | d | d | d | d | d | d | c | c | c | c | 0 | 0 | 3 | Yes |
| cMMEamiC | b | b | g | g | g | b | d | c | c | d | d | 0 | 0 | 3 | — |
| cMMESMC | d | d | d | d | d | d | d | c | c | c | d | 0 | 0 | 2 | — |
| cMMEgcvTH | d | d | NPf | NPf | d | d | d | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMEksgA | d | d | d | d | d | d | d | c | c | c | c | 0 | 0 | 2 | Yes |
| cMMErpoH | d | d | d | d | d | d | d | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMENMB0775-6 | b | g | b | b | b | b | b | c | c | NPf | NPf | 0 | 0 | 3 | — |
| cMMErfaED | NPf | NPf | d | NPf | NPf | NPf | d | c | c | NPf | c | 0 | 0 | 2 | Yes |
| cMMEdld | d | d | c | d | d | d | d | c | d | c | c | 0 | 0 | 2 | Yes |
| MMEtrpEpurK | d | d | d | d | d | c | d | c | c | d | 61k | 1 | 0 | 3 | — |
| cMMEaspA | d | d | 62m | d | d | d | d | NPf | NPf | 62k | d | 1 | 0 | 3 | Yes |
| MMEglyAdedA | g | NPf | g | b | NPf | g | NPf | c | NPf | g | NPf | 0 | 0 | 5j | Yes |
| cMMEglnDguaB | c | c | NPf | NPf | d | c | d | c | c | c | d | 0 | 0 | 2 | Yes |
| cMMEzurcobW | c | NPf | NPf | b | b | NPf | b | c | c | c | c | 0 | 0 | 2 | — |
| MMEnrdAB | 64k | d | g | NPf | d | g | d | c | c | 65k | c | 2 | 2 | 5 | Yes |
| MMEuvrB | NPf | NPf | 66k | NPf | NPf | i | i | c | c | NPf | NPf | 2 | 2 | 6 | Yes |
| cMMEpdhBlpdA | b | b | b | b | b | b | d | c | c | b | b | 0 | 0 | 2 | Yes |
| cMMEfmu | b | b | b | b | b | c | b | c | c | NPf | b | 0 | 0 | 2 | — |
| cMMErbfAtruB | d | d | b | d | d | b | d | c | c | b | b | 0 | 0 | 2 | Yes |
| MMEnifSU | d | d | 67k | 68k | 68m | d | 68m | c | c | 69k | d | 3 | 4 | 5 | Yes |
| MMEfprdinP | NPf | c | g | 70k | 70m | g | g | c | c | NPf | c | 1 | 0 | 5 | Yes |
| MMEhesA | c | c | 71k | 72k | 72m | c | c | c | c | NPf | 73k | 3 | 1 | 5 | Yes |
| cMMEasmAfolP | d | d | d | d | d | d | 77k | NPf | NPf | NPf | NPf | 1 | 0 | 3 | — |
| cMMEgrx | NPf | 74k | d | d | d | d | d | c | c | c | c | 1 | 0 | 3 | — |
| cMMEglyQS | b | b | b | c | c | b | b | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMEpurLhagH | d | d | d | d | d | d | d | 75k | 75m | d | d | 1 | 0 | 3 | Yes |
| cMMEhagHmgtE | d | d | c | NPf | NPf | NPf | c | c | c | c | c | 0 | 0 | 2 | Yes |
| cMMEmetRrpsI | d | NPf | d | d | d | d | d | c | c | 42k | c | 1 | 0 | 3 | Yes |
| MMEobgcysS | NPf | d | d | d | d | c | d | c | c | d | c | 0 | 0 | 2 | — |
| Total | 19 | 12 | 118 | — | |||||||||||
| Site | Nm Strain A22 | Nm Strain 860 800 | Nm Strain 00/240 794 | Nm Strain 97/282 675 | Nm Strain 00/240 868 | Nm Strain 01/241 825 | Nm Strain NG E30 | Ng Strain 28 539 | Ng Strain 26 593 | Neisseria lactamica | Neisseria polysaccharea | New Regions | New Genes | MME Regions | Present in Other Species l |
| MMEpheSTa | b | b | b | b | b | a | a | c | NDe | c | a | a | a | 8a | Yes |
| MMErpoCrpsL | d | d | d | d | d | d | d | c | c | NPf | g | 0 | 0 | 3 | Yes |
| MMEvalSlysR | b | d | d | c | c | d | d | c | c | NPf | 57k | 1 | 1 | 4 | — |
| cMMEdadAvalS | d | d | d | d | d | d | d | c | c | c | NPf | 0 | 0 | 2 | — |
| cMMEfabZlpxA | g | d | g | g | g | d | d | c | c | b | d | 0 | 0 | 2 | Yes |
| MMEfrr | d | NPf | g | d | NPf | d | d | c | c | 58k | NPf | 1 | 2 | 3 | — |
| cMMEnuoG | c | c | c | c | c | d | c | c | c | NPf | NPf | 0 | 0 | 2 | — |
| cMMEnuoIJ | c | g | c | d | d | c | c | c | g | c | 0 | 0 | 2 | — | |
| cMMEnuoKL | d | d | g | d | d | b | d | c | c | c | d | 0 | 0 | 2 | — |
| MMEnuoLM | d | c | d | d | d | d | d | c | c | d | d | 0 | 0 | 4h | — |
| cMMEpdxJacpS | d | d | d | d | d | d | d | c | c | c | c | 0 | 0 | 3 | Yes |
| cMMEamiC | b | b | g | g | g | b | d | c | c | d | d | 0 | 0 | 3 | — |
| cMMESMC | d | d | d | d | d | d | d | c | c | c | d | 0 | 0 | 2 | — |
| cMMEgcvTH | d | d | NPf | NPf | d | d | d | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMEksgA | d | d | d | d | d | d | d | c | c | c | c | 0 | 0 | 2 | Yes |
| cMMErpoH | d | d | d | d | d | d | d | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMENMB0775-6 | b | g | b | b | b | b | b | c | c | NPf | NPf | 0 | 0 | 3 | — |
| cMMErfaED | NPf | NPf | d | NPf | NPf | NPf | d | c | c | NPf | c | 0 | 0 | 2 | Yes |
| cMMEdld | d | d | c | d | d | d | d | c | d | c | c | 0 | 0 | 2 | Yes |
| MMEtrpEpurK | d | d | d | d | d | c | d | c | c | d | 61k | 1 | 0 | 3 | — |
| cMMEaspA | d | d | 62m | d | d | d | d | NPf | NPf | 62k | d | 1 | 0 | 3 | Yes |
| MMEglyAdedA | g | NPf | g | b | NPf | g | NPf | c | NPf | g | NPf | 0 | 0 | 5j | Yes |
| cMMEglnDguaB | c | c | NPf | NPf | d | c | d | c | c | c | d | 0 | 0 | 2 | Yes |
| cMMEzurcobW | c | NPf | NPf | b | b | NPf | b | c | c | c | c | 0 | 0 | 2 | — |
| MMEnrdAB | 64k | d | g | NPf | d | g | d | c | c | 65k | c | 2 | 2 | 5 | Yes |
| MMEuvrB | NPf | NPf | 66k | NPf | NPf | i | i | c | c | NPf | NPf | 2 | 2 | 6 | Yes |
| cMMEpdhBlpdA | b | b | b | b | b | b | d | c | c | b | b | 0 | 0 | 2 | Yes |
| cMMEfmu | b | b | b | b | b | c | b | c | c | NPf | b | 0 | 0 | 2 | — |
| cMMErbfAtruB | d | d | b | d | d | b | d | c | c | b | b | 0 | 0 | 2 | Yes |
| MMEnifSU | d | d | 67k | 68k | 68m | d | 68m | c | c | 69k | d | 3 | 4 | 5 | Yes |
| MMEfprdinP | NPf | c | g | 70k | 70m | g | g | c | c | NPf | c | 1 | 0 | 5 | Yes |
| MMEhesA | c | c | 71k | 72k | 72m | c | c | c | c | NPf | 73k | 3 | 1 | 5 | Yes |
| cMMEasmAfolP | d | d | d | d | d | d | 77k | NPf | NPf | NPf | NPf | 1 | 0 | 3 | — |
| cMMEgrx | NPf | 74k | d | d | d | d | d | c | c | c | c | 1 | 0 | 3 | — |
| cMMEglyQS | b | b | b | c | c | b | b | c | c | d | d | 0 | 0 | 2 | Yes |
| cMMEpurLhagH | d | d | d | d | d | d | d | 75k | 75m | d | d | 1 | 0 | 3 | Yes |
| cMMEhagHmgtE | d | d | c | NPf | NPf | NPf | c | c | c | c | c | 0 | 0 | 2 | Yes |
| cMMEmetRrpsI | d | NPf | d | d | d | d | d | c | c | 42k | c | 1 | 0 | 3 | Yes |
| MMEobgcysS | NPf | d | d | d | d | c | d | c | c | d | c | 0 | 0 | 2 | — |
| Total | 19 | 12 | 118 | — | |||||||||||
NOTE.—Nm, Neisseria meningitidis and Ng, Neisseria gonorrhoeae.
Data for this MME is from the investigation described in Saunders and Snyder (2002). Neisseria polysaccharea is AF542178, Nm strain 01/241825 is similar to AJ311178, and Nm strain NG E30 is similar to AF542173.
PCR product size and digest pattern indicate this region is similar to that in Nm strain Z2491.
PCR product size and digest pattern indicate this region is similar to that in Ng strain FA1090.
PCR product size and digest pattern indicate this region is similar to that in Nm strain MC58.
Not done.
No PCR product.
PCR product size and digest pattern indicate this region is similar to that in Nm strain FAM18.
The MME region from N. gonorrhoeae strain MS 11 (AY386266) was sequenced previously (Snyder et al. 2004).
Region is like that seen in Nm strain 00/240794 (DQ115766), but with an IS1301 inserted between bases 1130 and 1131.
The MME region from N. polysaccharea strain 89357 (AY072809) was sequenced previously (Zhu et al. 2003).
GenBank accession numbers—42: DQ117942; 57: DQ115757; 58: DQ115758; 61: DQ115761; 62: DQ115762; 64: DQ115764; 65: DQ115765; 66: DQ115766; 67: DQ115767; 68: DQ115768; 69: DQ115769; 70: DQ115770; 71: DQ115771; 72: DQ115772; 73: DQ115773; 74: DQ115774; 75: DQ115775; and 77: DQ115777.
Neisseria spp., MME, and cMME sites that are present with gene cassettes between the flanking genes in other bacterial genera.
PCR product size and digest pattern indicate this region is similar to that in the accession number indicated.
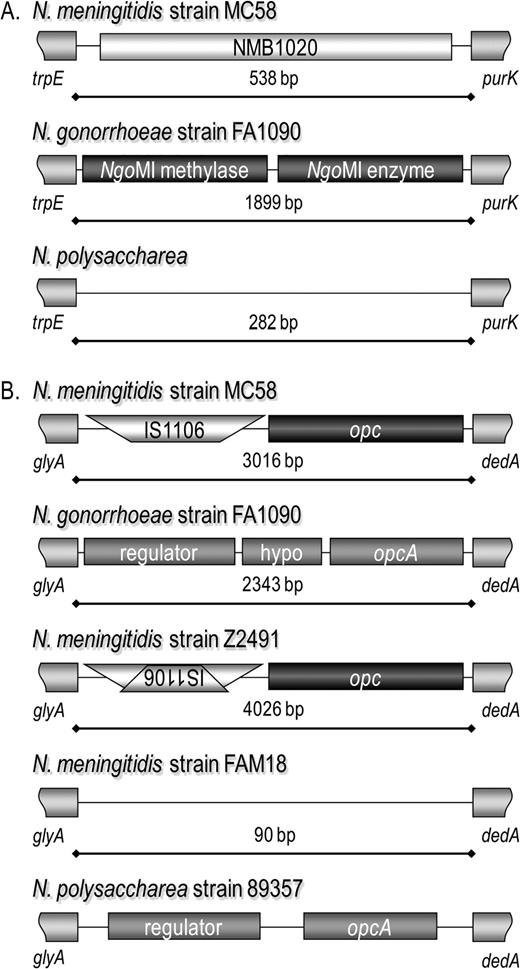
Examples of empty MME sites. In the MME between trpE and purK (A), a hypothetical gene is present in Neisseria meningitidis strain MC58. In Neisseria gonorrhoeae strain FA1090, the NgoMI restriction modification system genes are present in the same location. Sequencing from Neisseria polysaccharea revealed only a short intergenic region between trpE and purK (DQ115761). In the MME between glyA and dedA (B), several different genes and gene combinations have been identified previously (Zhu et al. 1999), including the absence of genes in this region in N. meningitidis strain FAM18. It should be noted that the N. gonorrhoeae and N. polysaccharea OpcA have 53.8% and 54.7% amino acid similarity to the N. meningitidis strain MC58 Opc. The sequence length for N. polysaccharea is not shown because this information is not available from the GenBank sequence (AY072809).
Each site was classified as an MME or a cMME on the basis of the presence of CDS-containing cassettes (fig. 3). In cases where only one CDS-containing cassette was identified between the conserved flanking genes, these sites remain cMMEs until additional different CDS-containing sequences are identified in the same locations. Based on this initial genome sequence-based analysis, the content of 10 MMEs (MMErpoCrpsL, MMEvalSlysR, MMEnuoLM, MMEtrpEpurK, MMEglyAdedA, MMEnrdAB, MMEuvrB, MMEnifSU, MMEfprdinP, and MMEobgcysS) and 28 cMMEs were further pursued in other neisserial strains.
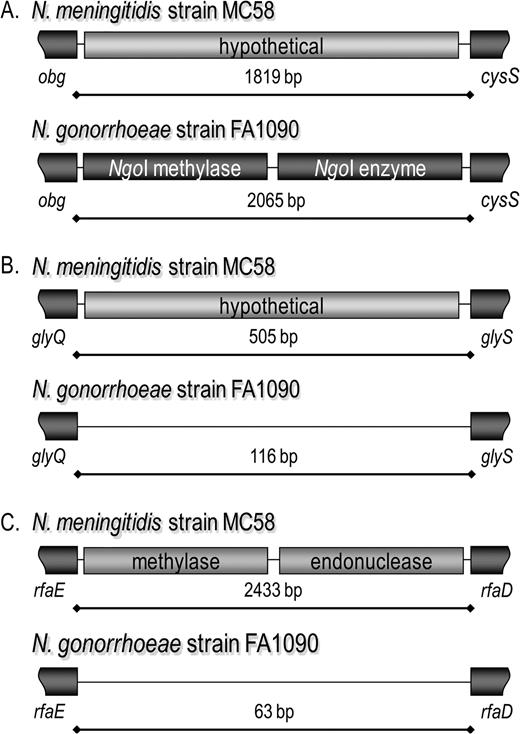
Different gene-containing regions within sites support their identification as MMEs. Only 2 different MME regions have thus far been identified in some sites. When both contain CDSs, as in the MMEobgcysS (A), it is easily identified as an MME. When 1 of the 2 regions within a site is empty, as in the MMEglyQS (B) and MMErfaED (C), the identification of the site as an MME is less clear, and these remain cMMEs.
Investigation of MME and cMME Site Content in 11 Neisserial Strains
The results of the PCR-based investigation of the contents of the 38 identified MME and cMME sites in the 11 neisserial strains are summarized in table 1. The results from MMEpheST shown in table 1 were obtained previously (Saunders and Snyder 2002). A total of 20 new intervening sequences were identified on the basis of size and restriction digest differences, from 9 MME and 5 cMME sites. Three of the different intervening sequences only differ from previously known sequences either by the presence of indels or the insertions of an Insertion Sequence (IS) element, which does not contribute to the reclassification of these cMME sites as MMEs (cMMEasmAfolP strain NG E30; cMMEpurLhagH strain 28539; and cMMEgrx strain 860800). The MME model addresses whole-gene cassettes; therefore, reclassification from cMME to MME requires that there be different whole-gene cassettes at the MME site. Although indels could arise within a strain and could be transferred horizontally, the MME facilitates the exchange of complete genes; thus, an indel does not fulfill the criteria of different gene-containing cassettes at the site. Insertion sequences can be self-mobilized without the homologous recombination of the MME; therefore, the addition of an insertion element does not constitute a different gene cassette at the site, only the secondary insertion of a self-mobilizing sequence. For MMEhesA, 3 new regions were sequenced, 1 with a 183-bp indel (strain 97/282675) compared with strain MC58, 1 with an IS30 and no other features (strain 00/240794), and 1 with a new inserted hypothetical gene (N. polysaccharea), which forms the basis of its classification as an MME. In MMEuvrB, an IS element is inserted into 1 of 2 new hypothetical genes in strain 01/241825 that in strain 00/240794 are present in the absence of the IS element. In one further case, a new hypothetical gene is identified adjacent to an intergenic IS30 in MMEnrdAB strain A22. Four new sequences contained no CDSs between the conserved MME-flanking genes in these strains (MMEtrpEpurK, N. polysaccharea; cMMEmetRrpsI, N. lactamica; MMEfprdinP, strain 97/282675; and cMMEaspA, N. lactamica). These represent new empty MME sites.
In the remaining 7 new MME-associated regions, 10 novel neisserial CDSs are present, in addition to the 2 CDSs present in strain 01/241825 and the CDS sequenced from N. polysaccharea in MMEhesA described above. A single CDS is present between the conserved flanking genes in MMEvalSlysR, N. polysaccharea; MMEnrdAB, N. lactamica; MMEnifSU, strain 00/240794; and MMEnifSU, N. lactamica. Two CDSs are present in MMEfrrN. lactamica, MMEuvrB strain 00/240794, and MMEnifSU strain 97/282675.
Characteristics of the Flanking Regions
The recombinational joints for the 5 MME sites illustrated in figures 2 and 3 are shown in the supplementary figure recombinational joints (Supplementary Material online). These examples are typical of what has been observed at the other neisserial MME sites. For the majority of the sequence of the flanking gene, the DNA sequence has a percent identity that would be expected on average when comparing 2 neisserial gene sequences. Closer to the internal MME sequence, however, single base polymorphisms are observed at the recombinational joint. For example, rfaE from N. meningitidis strain MC58 is 94.6% identical to rfaE from N. gonorrhoeae strain FA1090. When the first three-quarters of the gene sequences are compared (750 bp), these genes are 95.33% identical. The final quarter of the genes’ sequence have 92% identity to one another. Similar trends were observed at other MME sites. In some cases (i.e., glyQ and rfaE), the differences between the flanking gene sequences closest to the MME gene cassette or empty region have altered the location of the initiation or termination codon of the flanking gene (supplementary figure recombinational joints, Supplementary Material online).
An Additional MME Site Was Identified between leuD and leuB
All of the neisserial genome sequences investigated contain the same restriction modification genes between leuD and leuB, which did not lead to the identification of this region on the basis of the general search criteria used. This site was considered to be a potential MME because of the conserved nature of the flanking genes, their encoded central metabolic functions, and because restriction enzyme genes are frequently associated with MMEs. Sequencing from N. meningitidis strain 00/240794 identified a novel hypothetical gene in this location, thus identifying this site as an MME (fig. 4).
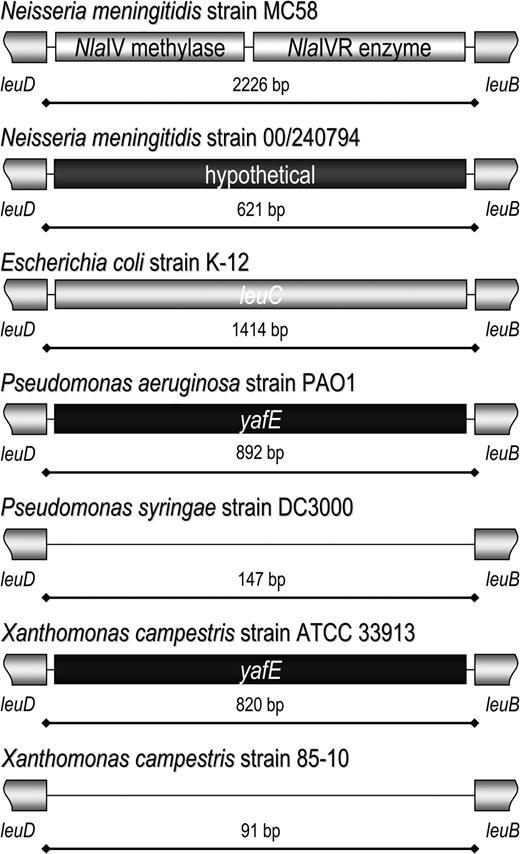
Identification of an MME based on gene content and its presence in other unrelated species. Although all 4 neisserial genome sequences contain the same NlaIV restriction modification system genes, the region between leuD and leuB has features of an MME because of the flanking genes and because restriction modification systems are the most common identifiable gene type found in MMEs. Investigation of this region revealed that Neisseria meningitidis strain 00/240794 contains a hypothetical gene within MMEleuDB. In most bacterial species, leuC is present in this location as it is in Escherichia coli strain K-12. In Pseudomonas aeruginosa strain PAO1 and Xanthomonas campestris strain ATCC33913, yafE is present between leuB and leuD, whereas the region is empty in Pseudomonas syringe strain DC3000 and X. campestris strain 85-10.
The Supplementary Material online contains figures illustrating the neisserial sequences associated with the 37 MMEs and cMMEs identified and investigated on the basis of the sequence analysis, MMEleuDB, and the previously identified MMEpheST and MMEnuoLM, as well as a separate document with figures of examples of degenerate regions that were also investigated and subsequently determined not to be neisserial MMEs.
Dinucleotide Signature Analysis of the MME Sites
The dinucleotide signature of each of the MME regions was determined from the sequences between the coding regions of the conserved flanking genes that were over 600 bp in length. The results are presented at http://www.compbio.ox.ac.uk/data/MME/neiss/Neiss_MMEs.html. Twenty-two MMEs and cMMEs (55%) had no δ* difference signatures of over 120, which is normally considered to represent sequences that are “closely similar,” “moderately similar,” and “weakly similar” (Karlin et al. 1997; Karlin 2001). Nine (22.5%) (MMEpheST, MMEnuoLM, cMMEpdxJacpS, cMMEglnDguaB, MMEnrdAB, cMMEfmu, MMEfprdinP, cMMEgrx, and cMMEmetRrpsI) had at least 1 gene cassette with a δ* difference signature of over 160, which is typically considered to be “distant” (160–200) or “very distant” (over 200). The remaining 9 (22.5%) (MMEvalSlysR, cMMEamiC, cMMESMC, cMMErfaDE, MMEleuDB, cMMEzurcobW, MMEnifSU, MMEhesA, and MMEobgcysS) had at least 1 gene cassette with a δ* difference signature between 120 and 160, which is classed as “distantly similar.” The designations within this scale, originally described by Karlin et al. (1997) and Karlin (2001), are normally used for comparing results between different species. On this scale, sea urchins are weakly similar to mammals and insects are weakly similar to monocot plants. As such, the selection of distantly similar as a threshold provides a fairly conservative estimate of recent horizontal acquisition. That 18 of the MMEs and cMMEs (45%) have dinucleotide signature differences that are above 120 is consistent with the hypothesis that these CDSs are being transferred between, as well as within, unrelated bacterial species.
Association of NUSSs with the MME Sites
The NUSS (GCCGTCTGAA, allowing for 1 mismatch) is present within, or in, the flanking CDSs of 38 (95%) of the 40 MMEs and cMMEs investigated in this study. It is present in both flanking genes and within at least 1 of the cassettes in 7 sites, in 1 flanking gene and within at least 1 of the gene cassettes in a further 20 sites, within at least 1 of the gene cassettes in a further 10 sites, and in both of the flanking genes in an additional 2 sites (table 3). For the remaining 2 cMMEs investigated, cMMErpoH and cMMEmetRrpsI, a sequence similar to the uptake signal sequence is present in 1 of the flanking genes if 2 mismatches are allowed. This suggests that within Neisseria, the presence of an NUSS may be associated with the efficiency of transfer and/or incorporation of MMEs.
MME and cMME Sites That in 1 or More Strains Do Not Contain Genes between the Flanking Genes—Empty Sites
| Sites | Strain Containing an Empty Site |
| MMEpheSTa | Neisseria elongata and Neisseria flavescens |
| MMErpoCrpsL | Nm strain FAM18 |
| cMMEdadAvalS | Ng strain FA1090 |
| cMMEfabZlpxA | Nm strain MC58 |
| MMEfrr | Nm strain MC58 |
| cMMEnuoG | Ng strain FA1090 |
| cMMEnuoIJ | Ng strain FA1090 |
| cMMEnuoKL | Ng strain FA1090 |
| MMEnuoLM | Nm strain MC58 |
| cMMEpdxJacpS | Ng strain FA1090 |
| cMMEamiC | Nm strain MC58 |
| cMMESMC | Nm strain MC58 |
| cMMEgcvTH | Nm strain MC58 |
| cMMEksgA | Nm strain MC58 |
| cMMENMB0775-6 | Nm strain MC58 |
| cMMErfaDE | Ng strain FA1090 |
| cMMEdld | Nm strain MC58 |
| MMEtrpEpurK | Neisseria polysaccharea |
| cMMEaspA | Nm strain MC58 & Neisseria lactamica |
| MMEglyAdedA | Nm strain FAM18 |
| cMMEglnDguaB | Ng strain FA1090 |
| cMMEzurcobW | Ng strain FA1090 |
| MMEnrdAB | Ng strain FA1090 |
| cMMEpdhBlpdA | Nm strain Z2491 |
| cMMEfmu | Ng strain FA1090 |
| cMMErbfAtruB | Nm strain MC58 |
| MMEfprdinP | Nm strain 97/282675 |
| MMEhesA | Ng strain FA1090 |
| cMMEasmAflop | Nm strain Z2491 |
| cMMEgrx | Ng strain FA1090 |
| cMMEglyQS | Ng strain FA1090 |
| cMMEpurLhagH | Nm strain MC58 |
| cMMEhagHmgtE | Ng strain FA1090 |
| cMMEmetRrpsI | N. lactamica |
| Sites | Strain Containing an Empty Site |
| MMEpheSTa | Neisseria elongata and Neisseria flavescens |
| MMErpoCrpsL | Nm strain FAM18 |
| cMMEdadAvalS | Ng strain FA1090 |
| cMMEfabZlpxA | Nm strain MC58 |
| MMEfrr | Nm strain MC58 |
| cMMEnuoG | Ng strain FA1090 |
| cMMEnuoIJ | Ng strain FA1090 |
| cMMEnuoKL | Ng strain FA1090 |
| MMEnuoLM | Nm strain MC58 |
| cMMEpdxJacpS | Ng strain FA1090 |
| cMMEamiC | Nm strain MC58 |
| cMMESMC | Nm strain MC58 |
| cMMEgcvTH | Nm strain MC58 |
| cMMEksgA | Nm strain MC58 |
| cMMENMB0775-6 | Nm strain MC58 |
| cMMErfaDE | Ng strain FA1090 |
| cMMEdld | Nm strain MC58 |
| MMEtrpEpurK | Neisseria polysaccharea |
| cMMEaspA | Nm strain MC58 & Neisseria lactamica |
| MMEglyAdedA | Nm strain FAM18 |
| cMMEglnDguaB | Ng strain FA1090 |
| cMMEzurcobW | Ng strain FA1090 |
| MMEnrdAB | Ng strain FA1090 |
| cMMEpdhBlpdA | Nm strain Z2491 |
| cMMEfmu | Ng strain FA1090 |
| cMMErbfAtruB | Nm strain MC58 |
| MMEfprdinP | Nm strain 97/282675 |
| MMEhesA | Ng strain FA1090 |
| cMMEasmAflop | Nm strain Z2491 |
| cMMEgrx | Ng strain FA1090 |
| cMMEglyQS | Ng strain FA1090 |
| cMMEpurLhagH | Nm strain MC58 |
| cMMEhagHmgtE | Ng strain FA1090 |
| cMMEmetRrpsI | N. lactamica |
Data for this MME is from the investigation described in Saunders and Snyder (2002).
MME and cMME Sites That in 1 or More Strains Do Not Contain Genes between the Flanking Genes—Empty Sites
| Sites | Strain Containing an Empty Site |
| MMEpheSTa | Neisseria elongata and Neisseria flavescens |
| MMErpoCrpsL | Nm strain FAM18 |
| cMMEdadAvalS | Ng strain FA1090 |
| cMMEfabZlpxA | Nm strain MC58 |
| MMEfrr | Nm strain MC58 |
| cMMEnuoG | Ng strain FA1090 |
| cMMEnuoIJ | Ng strain FA1090 |
| cMMEnuoKL | Ng strain FA1090 |
| MMEnuoLM | Nm strain MC58 |
| cMMEpdxJacpS | Ng strain FA1090 |
| cMMEamiC | Nm strain MC58 |
| cMMESMC | Nm strain MC58 |
| cMMEgcvTH | Nm strain MC58 |
| cMMEksgA | Nm strain MC58 |
| cMMENMB0775-6 | Nm strain MC58 |
| cMMErfaDE | Ng strain FA1090 |
| cMMEdld | Nm strain MC58 |
| MMEtrpEpurK | Neisseria polysaccharea |
| cMMEaspA | Nm strain MC58 & Neisseria lactamica |
| MMEglyAdedA | Nm strain FAM18 |
| cMMEglnDguaB | Ng strain FA1090 |
| cMMEzurcobW | Ng strain FA1090 |
| MMEnrdAB | Ng strain FA1090 |
| cMMEpdhBlpdA | Nm strain Z2491 |
| cMMEfmu | Ng strain FA1090 |
| cMMErbfAtruB | Nm strain MC58 |
| MMEfprdinP | Nm strain 97/282675 |
| MMEhesA | Ng strain FA1090 |
| cMMEasmAflop | Nm strain Z2491 |
| cMMEgrx | Ng strain FA1090 |
| cMMEglyQS | Ng strain FA1090 |
| cMMEpurLhagH | Nm strain MC58 |
| cMMEhagHmgtE | Ng strain FA1090 |
| cMMEmetRrpsI | N. lactamica |
| Sites | Strain Containing an Empty Site |
| MMEpheSTa | Neisseria elongata and Neisseria flavescens |
| MMErpoCrpsL | Nm strain FAM18 |
| cMMEdadAvalS | Ng strain FA1090 |
| cMMEfabZlpxA | Nm strain MC58 |
| MMEfrr | Nm strain MC58 |
| cMMEnuoG | Ng strain FA1090 |
| cMMEnuoIJ | Ng strain FA1090 |
| cMMEnuoKL | Ng strain FA1090 |
| MMEnuoLM | Nm strain MC58 |
| cMMEpdxJacpS | Ng strain FA1090 |
| cMMEamiC | Nm strain MC58 |
| cMMESMC | Nm strain MC58 |
| cMMEgcvTH | Nm strain MC58 |
| cMMEksgA | Nm strain MC58 |
| cMMENMB0775-6 | Nm strain MC58 |
| cMMErfaDE | Ng strain FA1090 |
| cMMEdld | Nm strain MC58 |
| MMEtrpEpurK | Neisseria polysaccharea |
| cMMEaspA | Nm strain MC58 & Neisseria lactamica |
| MMEglyAdedA | Nm strain FAM18 |
| cMMEglnDguaB | Ng strain FA1090 |
| cMMEzurcobW | Ng strain FA1090 |
| MMEnrdAB | Ng strain FA1090 |
| cMMEpdhBlpdA | Nm strain Z2491 |
| cMMEfmu | Ng strain FA1090 |
| cMMErbfAtruB | Nm strain MC58 |
| MMEfprdinP | Nm strain 97/282675 |
| MMEhesA | Ng strain FA1090 |
| cMMEasmAflop | Nm strain Z2491 |
| cMMEgrx | Ng strain FA1090 |
| cMMEglyQS | Ng strain FA1090 |
| cMMEpurLhagH | Nm strain MC58 |
| cMMEhagHmgtE | Ng strain FA1090 |
| cMMEmetRrpsI | N. lactamica |
Data for this MME is from the investigation described in Saunders and Snyder (2002).
The Presence of NUSSs Associated with the MME Sites
| Site | Flank A | Internal | Flank B |
| MMEpheST | fmz | fm1 | fmz |
| MMErpoCrpsL | fmz | fmz | |
| MMEvalSlysR | fmz | fmz | |
| cMMEdadAvalS | fmz | fmz | fmz |
| cMMEfabZlpxA | f1m1z1 | ||
| MMEfrr | f | fmz | |
| cMMEnuoG | mz | fmz | |
| cMMEnuoIJ | m1z1 | fm1z1 | |
| cMMEnuoKL | mz | fmz | |
| MMEnuoLM | fmz | f1mz | |
| cMMEpdxJacpS | fm1z | ||
| cMMEamiC | fmz | fmz | |
| cMMESMC | fmz | fmz | |
| cMMEgcvTH | fmz | ||
| cMMEksgA | fmz | fmz | |
| cMMErpoH | |||
| cMMENMB0775-6 | fmz | ||
| cMMErfaED | f | z1 | |
| cMMEdld | fmz | fmz | z |
| MMEtrpEpurK | mz | f1 | fm1z |
| cMMEaspA | fmz | ||
| MMEleuDB | fmz | ||
| MMEglyAdedA | fmz | ||
| cMMEglnDguaB | fmz | fmz | |
| cMMEzurcobW | z | ||
| MMEnrdAB | fmz | m1z1 | |
| MMEuvrB | f1m1z | fmz | fmz |
| cMMEpdhBlpdA | z | ||
| cMMEfmu | fmz | fm1z1 | |
| cMMErbfAtruB | f1m1z | fmz | |
| MMEnifSU | f1mz | ||
| MMEfprdinP | mz | z1 | |
| MMEhesA | fmz | fmz | |
| cMMEasmAfolP | fmz | fmz | f1m1 |
| cMMEgrx | fmz | f1mz | |
| cMMEglyQS | f | fmz | |
| cMMEpurLhagH | fmz | fmz | fmz |
| cMMEhagHmgtE | fmz | fmz | |
| cMMEmetRrpsI | |||
| MMEobgcysS | f | fmz1 |
| Site | Flank A | Internal | Flank B |
| MMEpheST | fmz | fm1 | fmz |
| MMErpoCrpsL | fmz | fmz | |
| MMEvalSlysR | fmz | fmz | |
| cMMEdadAvalS | fmz | fmz | fmz |
| cMMEfabZlpxA | f1m1z1 | ||
| MMEfrr | f | fmz | |
| cMMEnuoG | mz | fmz | |
| cMMEnuoIJ | m1z1 | fm1z1 | |
| cMMEnuoKL | mz | fmz | |
| MMEnuoLM | fmz | f1mz | |
| cMMEpdxJacpS | fm1z | ||
| cMMEamiC | fmz | fmz | |
| cMMESMC | fmz | fmz | |
| cMMEgcvTH | fmz | ||
| cMMEksgA | fmz | fmz | |
| cMMErpoH | |||
| cMMENMB0775-6 | fmz | ||
| cMMErfaED | f | z1 | |
| cMMEdld | fmz | fmz | z |
| MMEtrpEpurK | mz | f1 | fm1z |
| cMMEaspA | fmz | ||
| MMEleuDB | fmz | ||
| MMEglyAdedA | fmz | ||
| cMMEglnDguaB | fmz | fmz | |
| cMMEzurcobW | z | ||
| MMEnrdAB | fmz | m1z1 | |
| MMEuvrB | f1m1z | fmz | fmz |
| cMMEpdhBlpdA | z | ||
| cMMEfmu | fmz | fm1z1 | |
| cMMErbfAtruB | f1m1z | fmz | |
| MMEnifSU | f1mz | ||
| MMEfprdinP | mz | z1 | |
| MMEhesA | fmz | fmz | |
| cMMEasmAfolP | fmz | fmz | f1m1 |
| cMMEgrx | fmz | f1mz | |
| cMMEglyQS | f | fmz | |
| cMMEpurLhagH | fmz | fmz | fmz |
| cMMEhagHmgtE | fmz | fmz | |
| cMMEmetRrpsI | |||
| MMEobgcysS | f | fmz1 |
NOTE.—f, m, and z indicate the presence of 1 or more copies of the NUSS in the flanking genes (Flank A and Flank B) or in the sequence between the flanking genes (Internal) of Neisseria gonorrhoeae strain FA1090, Neisseria meningitidis strain MC58, and N. meningitidis strain Z2491, respectively. f1, m1, and z1 indicates that 1 or more copies of the NUSS could only be detected when allowing for 1 mismatch.
The Presence of NUSSs Associated with the MME Sites
| Site | Flank A | Internal | Flank B |
| MMEpheST | fmz | fm1 | fmz |
| MMErpoCrpsL | fmz | fmz | |
| MMEvalSlysR | fmz | fmz | |
| cMMEdadAvalS | fmz | fmz | fmz |
| cMMEfabZlpxA | f1m1z1 | ||
| MMEfrr | f | fmz | |
| cMMEnuoG | mz | fmz | |
| cMMEnuoIJ | m1z1 | fm1z1 | |
| cMMEnuoKL | mz | fmz | |
| MMEnuoLM | fmz | f1mz | |
| cMMEpdxJacpS | fm1z | ||
| cMMEamiC | fmz | fmz | |
| cMMESMC | fmz | fmz | |
| cMMEgcvTH | fmz | ||
| cMMEksgA | fmz | fmz | |
| cMMErpoH | |||
| cMMENMB0775-6 | fmz | ||
| cMMErfaED | f | z1 | |
| cMMEdld | fmz | fmz | z |
| MMEtrpEpurK | mz | f1 | fm1z |
| cMMEaspA | fmz | ||
| MMEleuDB | fmz | ||
| MMEglyAdedA | fmz | ||
| cMMEglnDguaB | fmz | fmz | |
| cMMEzurcobW | z | ||
| MMEnrdAB | fmz | m1z1 | |
| MMEuvrB | f1m1z | fmz | fmz |
| cMMEpdhBlpdA | z | ||
| cMMEfmu | fmz | fm1z1 | |
| cMMErbfAtruB | f1m1z | fmz | |
| MMEnifSU | f1mz | ||
| MMEfprdinP | mz | z1 | |
| MMEhesA | fmz | fmz | |
| cMMEasmAfolP | fmz | fmz | f1m1 |
| cMMEgrx | fmz | f1mz | |
| cMMEglyQS | f | fmz | |
| cMMEpurLhagH | fmz | fmz | fmz |
| cMMEhagHmgtE | fmz | fmz | |
| cMMEmetRrpsI | |||
| MMEobgcysS | f | fmz1 |
| Site | Flank A | Internal | Flank B |
| MMEpheST | fmz | fm1 | fmz |
| MMErpoCrpsL | fmz | fmz | |
| MMEvalSlysR | fmz | fmz | |
| cMMEdadAvalS | fmz | fmz | fmz |
| cMMEfabZlpxA | f1m1z1 | ||
| MMEfrr | f | fmz | |
| cMMEnuoG | mz | fmz | |
| cMMEnuoIJ | m1z1 | fm1z1 | |
| cMMEnuoKL | mz | fmz | |
| MMEnuoLM | fmz | f1mz | |
| cMMEpdxJacpS | fm1z | ||
| cMMEamiC | fmz | fmz | |
| cMMESMC | fmz | fmz | |
| cMMEgcvTH | fmz | ||
| cMMEksgA | fmz | fmz | |
| cMMErpoH | |||
| cMMENMB0775-6 | fmz | ||
| cMMErfaED | f | z1 | |
| cMMEdld | fmz | fmz | z |
| MMEtrpEpurK | mz | f1 | fm1z |
| cMMEaspA | fmz | ||
| MMEleuDB | fmz | ||
| MMEglyAdedA | fmz | ||
| cMMEglnDguaB | fmz | fmz | |
| cMMEzurcobW | z | ||
| MMEnrdAB | fmz | m1z1 | |
| MMEuvrB | f1m1z | fmz | fmz |
| cMMEpdhBlpdA | z | ||
| cMMEfmu | fmz | fm1z1 | |
| cMMErbfAtruB | f1m1z | fmz | |
| MMEnifSU | f1mz | ||
| MMEfprdinP | mz | z1 | |
| MMEhesA | fmz | fmz | |
| cMMEasmAfolP | fmz | fmz | f1m1 |
| cMMEgrx | fmz | f1mz | |
| cMMEglyQS | f | fmz | |
| cMMEpurLhagH | fmz | fmz | fmz |
| cMMEhagHmgtE | fmz | fmz | |
| cMMEmetRrpsI | |||
| MMEobgcysS | f | fmz1 |
NOTE.—f, m, and z indicate the presence of 1 or more copies of the NUSS in the flanking genes (Flank A and Flank B) or in the sequence between the flanking genes (Internal) of Neisseria gonorrhoeae strain FA1090, Neisseria meningitidis strain MC58, and N. meningitidis strain Z2491, respectively. f1, m1, and z1 indicates that 1 or more copies of the NUSS could only be detected when allowing for 1 mismatch.
The Presence of Neisseria-Associated MME Sites Containing Intervening CDSs in Other Bacterial Species
All 330 of the nonneisserial bacterial genome sequences currently available were interrogated for the presence of potential MMEs flanked by the genes identified in the study of the neisserial MMEs and cMMEs. In each case, homologues of the neisserial flanking genes were sought and the presence of any annotated CDSs between them was noted. The complete results of this interrogation are available at http://www.compbio.ox.ac.uk/data/MME/all/all_genomes_MMEs.html. These data were analyzed to identify all cases in which there were annotated CDSs between the flanking genes and where the flanking genes were not separated by more than 15 CDSs. Identified regions were assessed by viewing the genomic region in the National Center for Biotechnology Information genome sequence databases to determine whether the genomic organization of the site was consistent with it representing an MME in that species. This included an assessment of the conserved organization of the flanking genes and the absence of indicators of horizontal gene transfer by other means, such as the presence of bacteriophage genes or transposons.
In many cases, there is currently only a single genome sequence representing a bacterial species or group of related species currently available, so it is not possible to confirm whether these regions are MMEs facilitating genetic exchange within these other species. On the basis of the presence of additional different CDSs located between these flanking genes and the sequences currently available, 24 of the 40 neisserial MME-associated regions have sequences suggestive of them also being MMEs in other species (table 1 and also MMEleuDB, which is not listed in table 1). Additional sequence information is required in most cases to determine if there are any variations in the intervening CDSs between strains of the same or closely related species indicative of interstrain transfer. However, in some species multiple genome sequences are available, comparison of which revealed strain-specific gene-complement differences in these same chromosomal locations.
MMEs Common to the Neisseria spp. and Rhodopseudomonas, Streptococcus, Pseudomonas, Lactobacillus, Haemophilus, and Mannheimia
MMEpheST contains 2 hypothetical genes in Rhodopseudomonas palustris strain BisB18 and a DUF559 endonuclease in R. palustris strains BisB5, CGA009, and HaA2. An acetyltransferase is present in this location in most Streptococcus species but is absent in Streptococcus pyogenes (fig. 5). A further 13 gene cassettes are present in this location in other species. The presence of so many different gene cassettes between pheS and pheT is suggestive of this being an MME in these other species; however, the lack of related genome sequences for comparative analysis means the MME model cannot currently be conclusively fulfilled except for R. palustris. In total, including the neisserial regions, 26 different genetic elements have now been identified that are located between these 2 highly conserved metabolic housekeeping genes.
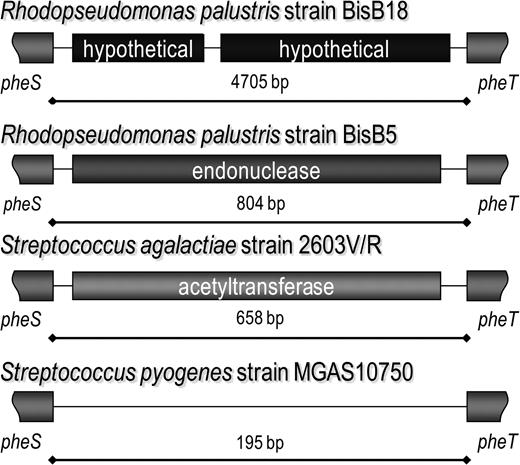
MMEpheST in other bacterial species. Nine different regions have been previously identified between the neisserial pheS and pheT genes. In Rhodopseudomonas palustris, 2 different gene-containing regions are present in MMEpheST. This region is potentially an MME in the streptococci; Streptococcus agalactiae and other streptococci have an acetyltransferase between pheS and pheT, whereas this site in Streptococcus pyogenes is empty.
In Pseudomonas syringe, 3 different gene-containing regions were identified in MMEnrdAB from strains DC3000, 1448A, and B728a, with a different cassette being present in Pseudomonas putida strain KT2440 in this location (fig. 6). In the other genome sequences, 21 different gene cassettes were identified within MMEnrdAB.
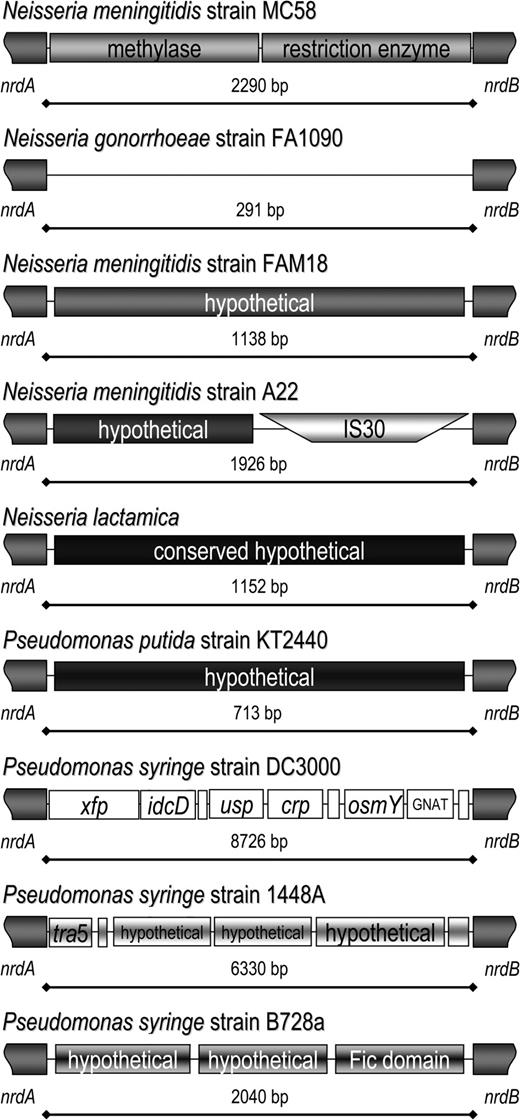
MMEnrdAB contains 5 different regions in Neisseria and 4 different regions in Pseudomonas. Nine different MME regions have been identified between nrdA and nrdB. In addition to the 3 regions identified in the neisserial genome sequences, Neisseria meningitidis strain A22 contains a newly identified hypothetical gene and an IS30 (DQ115764). Neisseria lactamica contains a large conserved hypothetical gene at MMEnrdAB (DQ115765). Pseudomonas putida strain KT2400 contains a different hypothetical gene and 3 different strains, if Pseudomonas syringe contain 3 different gene cassettes.
Fifteen different gene cassettes were identified within MMErbfAtruB, including 2 in different Lactobacillus species. In Lactobacillus plantarum strain WCFS1, the region contains aroJ, aroF, a hypothetical gene, aroE, tyrA, and aroI. In Lactobacillus sakei strain 23K, the same region contains an oxidase, a glycosyltransferase, and 2 hypothetical genes. The other genome sequences of lactobacilli have adjacent rbfA and truB sequences, making this MME empty in those strains (fig. 7).
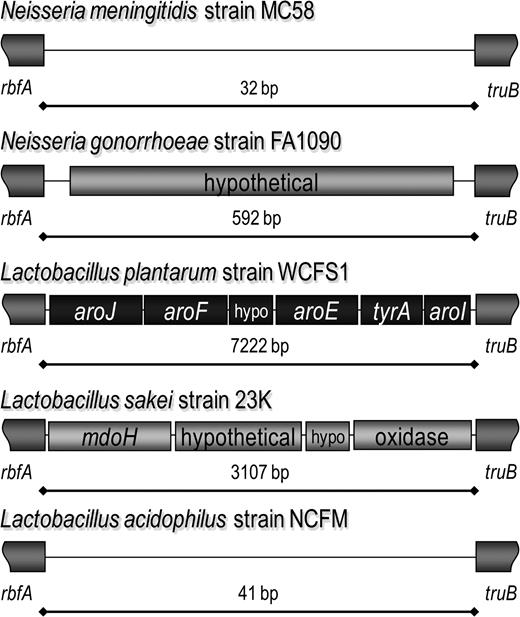
Neisseria and Lactobacillus share MMErbfAtruB. Neisserial strains either have an empty MMErbfAtruB, as in Neisseria meningitidis strain MC58 and FAM18, or the site contains a hypothetical gene homologous to NMA1587, as in Neisseria gonorrhoeae strain FA1090 and N. meningitidis strain Z2491. In lactobacilli, MMErbfAtruB is empty in Lactobacillus acidophilus, but 2 different gene cassettes are present in Lactobacillus plantarum and Lactobacillus sakei.
It was noted that the genes for the NlaIV restriction modification system are located between leuB and leuD in all 4 of the neisserial genome sequences evaluated (fig. 4). In most bacterial species for which homologues are present and have been sequenced, leuC is present between leuB and leuD. This is the case for all of the genome sequences from E. coli, Haemophilus influenzae, Bacillus spp., Staphylococcus spp., and Vibrio spp., for example. Investigations of other genome sequences revealed that in Pseudomonas aeruginosa, Pseudomonas fluorescens, and P. putida, a CDS annotated as yafE is present, which contains the smtA domain. There are no genes between leuB and leuD in P. syringe. A CDS homologous to the PseudomonasyafE is present in Xanthomonas campestris strain ATCC33913 and X. campestris strain 8004, whereas the region is empty in X. campestris strain 85-10 and Xanthomonas citri.
The first conclusive evidence of interspecies MME-mediated horizontal gene transfer is associated with MMEglyQS. These genes are adjacent in bacterial strains such as E. coli strain K-12 and encode the glycyl-tRNA synthetase α and β subunits. In Neisseria, this site is thus far known to contain either a hypothetical protein, as in N. meningitidis strain MC58 and most of the meningococcal strains tested, or an empty site, as in N. gonorrhoeae strain FA1090, most of the gonococcal strains tested, and N. meningitidis strains 00/240868 and 97/282675. There are 11 annotated coding regions between glyQ and glyS in H. pylori strain 26695 (gpsA, acpP, HP0963, HP0964, HP0965, HP0966, vapD, HP0968, czcA, czcB, and HP0971), and 14 other gene-containing regions were identified at this site from other bacterial genome sequences. In H. influenzae strain Rd, a hypothetical gene and a CDS encoding a DUF559 endonuclease is present in this location, whereas in strain 86-28NP, there are 2 different hypothetical genes and the same endonuclease. In Haemophilus ducreyi, MMEglyQS is empty. It is not only interesting to note that, although the other CDSs are not homologues, the DUF559 endonuclease is present in this location in both H. influenzae strains, where they have 100% amino acid identity, but also there is a DUF559 endonuclease present between glyQ and glyS in Mannheimia succiniciproducens that shares 84.3% amino acid identity and 86.8% similarity with the H. influenzae endonuclease. An additional DUF559 endonuclease is present in this location in R. palustris, where the similarity to the H. influenzae protein is 50.9% (fig. 8).
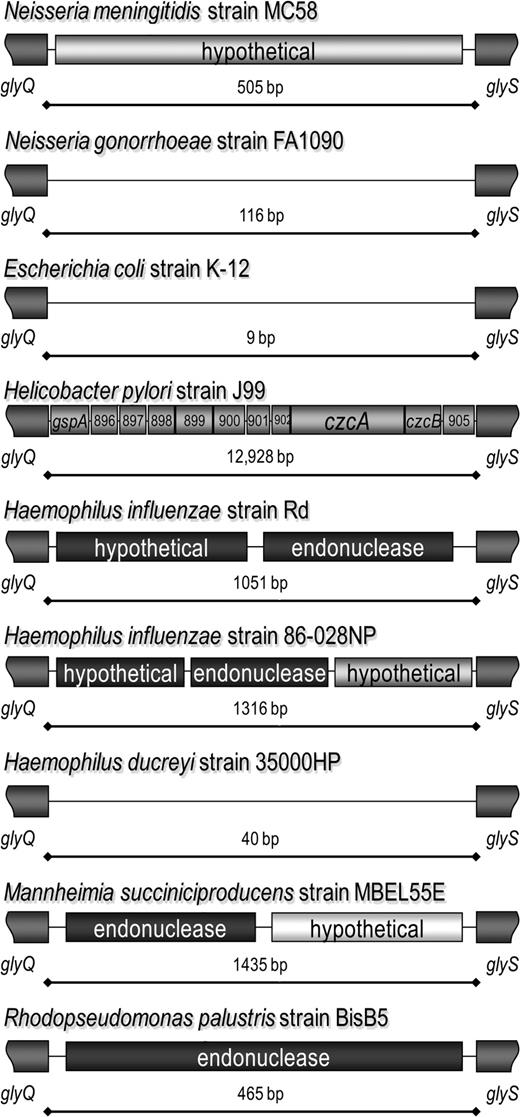
MMEglyQS is present in unrelated species. The glyQ and glyS genes are adjacent in Neisseria gonorrhoeae strain FA1090 and Escherichia coli strain K12 (not pictured), but in other species there are different sets of CDSs in this MME site. Between the Helicobacter pylori strain 26695 glyQ and glyS genes are gpsA, acpP, HP0963, HP0964, HP0965, HP0966, vapD, HP0968, czcA, czcB, and HP0971 (not pictured). More striking are the differences between Haemophilus influenzae strains Rd and 86-028NP and that this site is empty in Haemophilus ducreyi. The endonuclease that the 2 H. influenzae sites share is also found between glyQ and glyS in Mannheimia succiniciproducens and Rhodopseudomonas palustris.
Given the cross-species presence of a DUF559 endonuclease in MMEglyQS and in MMEpheST, the genomic sequence databases were searched for other instances of features that had been annotated as members of this conserved domain group. This identified a region in R. palustris strains BisB18 and HaA2 that contains a DUF559 endonuclease flanked by ugpB (an extracellular solute-binding protein family 1) and ugpA (a binding protein–dependent transport systems inner membrane component), whereas in strain BisB5 these CDSs are adjacent. This indicates that similar CDSs can be associated with different MME sites in different species.
Discussion
The MMEs of the Neisseria spp.
The identification of novel intervening sequences and the presence of the different MME-associated CDSs and empty cassettes in different combinations within the strains tested (table 1) provides clear evidence of the independent mobility of these CDSs within these populations and for the MME model. As can be seen by the results of table 1, the gene cassettes identified in N. meningitidis strain MC58 MME sites (MC58-like) are found combined within the same strain as other MME sites with the gene cassettes identified from N. meningitidis strain Z2491 (Z2491-like), N. meningitidis strain FAM18 (FAM18-like), and N. gonorrhoeae strain FA1090 (FA1090-like), as well as gene cassettes newly identified in this study. This shuffling of MME site CDS constituents in comparison with the genome sequences and between the strains investigated here provides clear evidence of horizontal exchange.
The presence of different cassettes of CDSs in different strains of the Neisseria spp. has been described for several chromosomal loci (Zhu et al. 1999; Claus et al. 2000; Klee et al. 2000; Kahler et al. 2001; Saunders and Snyder 2002; Snyder et al. 2004). This study used comparative genomics to identify sites of gene-complement variation, specifically targeting MME and cMME sites, for further investigation. Of the 9 genetic islands that have been reported previously to distinguish N. meningitidis from N. gonorrhoeae (Klee et al. 2000), 4 can now be classified as MMEs or cMMEs based on the MME model (fig. 1). Using the nomenclature of this previous study, region 4 is MMEpheST, region 5 is cMMErfaED, and region 6 is MMEfprdinP (see table 1). Region 1, the meningococcal capsule locus, is also likely to be an MME (Snyder et al. 2005), although the size and variation of this site is too extensive for it to have been addressed in this study, and this region has already been studied by others (Claus et al. 1997; Klee et al. 2000; Dolan-Livengood et al. 2003). Of the remaining regions previously described (Klee et al. 2000), 4 are associated with self-mobilizing elements (transposons, IS elements, and bacteriophage), whereas region 2 has inserted within a cvaB homologue.
The MME-flanking genes are typically similarly oriented housekeeping genes with related functions, and it is presumed that most MME sites initially contain no intervening genes. As such, empty MMEs represent the native state of the chromosome before the incorporation of the MME-associated genes. There is nothing to suggest that these empty sites represent the consequences of a deletion event; no remnants of deleted genes are observed, the sequence diverges at or close to the beginning/end of the conserved flanking genes, and the presence of different alternative genes at the same site cannot be accounted for simply by deletion of ancestral genes. For example, figure 2 shows the empty MME sites identified in this study in N. polysaccharea between trpE and purK and in N. meningitidis strain FAM18 between glyA and dedA that are likely to represent the native state of these regions, both of which have been previously identified as containing different genes in other neisserial strains (Zhu et al. 1999).
When a site contains more than 1 cassette-containing genes that differ between 2 or more genome sequences, this confirms the classification of that location as an MME site, as in figure 3A. Twenty-six of the 40 sites selected for investigation in this study currently remain classified as cMMEs because only one gene-containing alternative has so far been identified (indicated as cMME on tables 2–4 and throughout the text). Examples of sites that remain classified as cMMEs are shown in figure 3B and C, where a gene-containing cassette is present in one strain and in another the site is empty. Although often the alternative region of a cMME is empty, in some cases there are additional differences at these locations due to the insertion of Correia repeat enclosed elements (Correia et al. 1986, 1988; Liu et al. 2002), which are mobile elements that are probably tolerated at these sites for the same reason that the MME-associated genes can be inserted without disrupting the functions of the surrounding essential genes. (see Supplementary Material online: illustrations for cMMEamiC, cMMErpoH, cMMENMB0775-6, and cMMEmetRrpsI).
In the case of candidate regions in which only one set of CDSs has been identified to date, there are still 2 potentially mutually exclusive cassettes present, which rather than exchanging alternative functions, exchange a coding and a noncoding alternative. If shown to be mobile, these sites could still be encompassed by the MME model because they would be associated with the gain and loss of whole genes, rather than being transferred by other mechanisms, or generating mosaic genes. However, identification of additional alternative gene-containing regions would strengthen their classification as MMEs.
Seven percent of the 378 successful PCRs (27 of 378) contained new regions or new strain-specific variants of previously defined regions and half (20 of 40) of the sites contained new, different strain-specific regions. In N. meningitidis strain 00/240794, 5 new sequences were discovered; 5 were found in N. lactamica; 3 each in N. polysaccharea and N. meningitidis strains NG E30, 97/282675, and 00/240868; and 1 each in N. meningitidis strains A22, 01/241825, and 860800; and N. gonorrhoeae strains 28539 and 26593 (table 1 and fig. 4). As such, the targeting of MMEs has shown itself to be a highly efficient means of identifying new strain-specific differences and genes. However, it should be noted that the MMEs containing larger regions (>5 kb) were excluded from the study and a further 12% of the selected targets (51 of 429) did not generate a PCR product despite the use of more than 1 combination of flanking primers. This is likely to be related to the combined effects of the presence of polymorphisms in the flanking regions (supplementary figure recombinational joints, Supplementary Material online) and alternate MME-associated regions that are too long to amplify. Although the flanking genes are conserved and their homology generates the homologous recombination substrate, the portions closest to the incorporated sequences are more diverse than average due to the recombination process by which MMEs are incorporated (supplementary figure recombinational joints, Supplementary Material online). These polymorphisms are not believed to be so significant as to negatively impact homologous recombination, as most are single nucleotide polymorphisms, but such small changes can severely negatively impact the performance of PCR. As such, the limitations of the methodological approach are likely to underestimate the true diversity associated with these regions.
As mentioned above, there are slight sequence differences between strains observed in the conserved flanking regions at some of the MME sites (supplementary figure recombinational joints, Supplementary Material online). Most often these are single base differences that occur most frequently toward the internal portion of the MME. The remainder of the gene remains highly homologous; therefore, these polymorphisms, although a reflection of the homologous recombination event that has introduced the differences at the MME sites, are not expected to negatively impact the potential for homologous recombination. They may actually provide protection for the gene cassettes within the MME by ensuring that the regions of most homology are within the farthest regions of the flanking genes, thus ensuring that the coding potential of the gene cassettes will not be disrupted through their integration.
The functions of many of the annotated coding regions within MME sites have not been investigated; therefore, the majority remain strain-specific hypothetical genes of unknown function. Of the 121 different hypothetical genes thus far identified within neisserial cMMEs, 76 (63%) are of unknown function. The high percentage of annotated coding regions of unknown function is likely a reflection of the fact that strain-specific genes are generally poorly studied and understood because they are not part of the common metabolic processes, which have been most studied and readily identified on the basis of homology. The most common identifiable function of MME-associated genes is restriction modification; 13 restriction modification systems can be identified within 7 neisserial MME sites: MMEpheST, MMEfrr, cMMENMB0775-6, cMMErfaDE, MMEtrpEpurK, MMEnrdAB, and MMEobgcysS. The prevalence of restriction modification systems within MMEs may simply be the result of their ready identification through homology, whereas most other putative genes in the MME sites remain of unknown function. It is also possible, however, that the MME represents a dynamic system for the shuffling and exchange of restriction modification systems. Certainly in E. coli, its MME has been called the ICR because of the influence that the restriction modification systems themselves have on the survival of horizontally transferred DNA within the cell (Sibley and Raleigh 2004). It may not then be surprising that genes so integrally important in horizontal transfer are themselves horizontally transferred to generate diversity in the population's potential to accept and incorporate foreign DNA.
The efficiency of gene transfer by the MME model in the Neisseria spp. is likely to be related to natural transformation and hence upon the presence of NUSS within or adjacent to the MMEs. The 10-bp NUSS promotes the efficient uptake of exogenous DNA by at least 4 orders of magnitude (Stein 1991). Also, the presence of this relatively neisseria-specific sequence within the exchanged central region would suggest that these cassettes have been present within these species for some considerable time and would increase the efficiency of their transfer between neisserial strains. Of the 40 MME sites investigated, 28 contain NUSSs in 1 or both conserved flanking genes and an additional 10 contain NUSSs between the flanking genes in 1 or more strains (table 3). This means that 95% of the neisserial MMEs investigated have an associated NUSS.
MMEs and Horizontal Transfer between Unrelated Species
As a test of whether the sequence composition features of the CDSs within MMEs was consistent with the model that they are also horizontally transferred between species, the dinucleotide signature of each region over 600 bp was determined and compared with whole-genome sequence (http://www.compbio.ox.ac.uk/data/MME/neiss/Neiss_MMEs.html). For 18 of the 40 MMEs investigated, at least 1 of the regions between the conserved genes displayed a divergence signature over 120, classified as distantly similar (120–160), distant (160–200), and very distant (over 200). Sequences with less divergent dinucleotide signatures may indicate that the gene cassettes analyzed are neisserial in origin, have been acquired from a species with a similar dinucleotide signature, or have been present in the Neisseria for a sufficient time to become ameliorated.
The Neisserial MMEs in Other Bacterial Species
For more than half of the neisserial MMEs and cMMEs investigated here (24 of 40), CDS-containing cassettes can be found between the same flanking genes in other bacterial genome sequences. This strongly suggests that there are common exchange sites being utilized within, and possibly also between, unrelated species, which is most clearly illustrated for those bacteria, where the comparison of genome sequences from different strains reveals that sites that are MMEs in the Neisseria spp. are also MMEs in other species. The best example of potential transfer between species currently is associated glyQ and glyS (fig. 8). The DUF559 endonuclease in MMEglyQS is associated with different hypothetical genes in the 2 H. influenzae strains and is also present between glyQ and glyS in M. succiniciproducens, where the DUF559 endonuclease is 86.8% amino acid similar to H. influenzae, and there is also a different hypothetical gene. A DUF559 endonuclease is also present here in R. palustris, where the similarity to the H. influenzae endonuclease is 50.9%. The changes within the gene cassette may be secondary, due to either deletion of 1 or more genes or the insertion of additional genes. It is clear, however, that this endonuclease in this location is conserved between the distantly related genera Haemophilus, Mannheimia, and Rhodopseudomonas. DUF559 endonucleases are also found in other MME sites, particularly in Rhodopseudomonas. Indeed, the prevalence of neisserial MME and cMME sites in this genus would suggest that it may carry other MME sites and is worthy of a genome-scale comparative analysis focused upon this species to identify and pursue these.
The identification of MME sites shared between different unrelated species potentially favors interspecies, as well as intraspecies, mobilization of whole-gene cassettes. Although no common genes were found between the Neisseria spp. and the other genome sequences investigated, this may simply be due to limitations of the genome sequence data currently available, both from our sequencing of neisserial MMEs and from the whole-genome data. Shared common MME sites between the Neisseria and other bacteria were identified, providing additional support to previous evidence that MMEs are present in species other than the Neisseria spp. and suggesting that these common sites can provide a means of horizontal transfer between unrelated bacterial species. The supplementary figure flanking gene alignments (Supplementary Material online) contains graphical illustrations of the similarity between the nucleotide sequence of N. meningitidis strain MC58-flanking genes and the flanking genes from the other species show in figures 4, 5, and 8. Although the homology between species is understandably lower than between strains of the same species, the potential for cross-species transfer cannot be ignored, particularly if stretches of DNA exist that are likely to facilitate homologous recombination. More data from other species are likely to reveal clear examples of gene cassette transfer to unrelated species sharing patches of homology.
It is most likely that MME horizontal gene exchange and incorporation will be more prevalent in naturally competent species. Uptake of the DNA fragment containing the conserved flanking genes and exchangeable gene cassette by a transformable species, whether facilitated by an uptake signal sequence or not, would permit homologous recombination between the fragment and the chromosome. It is potentially possible for the MME-containing fragment to enter a noncompetent cell by conjugation or transduction. The likelihood of transfer of the MME fragment by these means is not high, but it cannot be discounted as a possibility. However, once within the cell by conjugation or transduction, chromosomal integration would have to be due to homologous recombination to remain true to the MME model. If the fragment were resident on a conjugation plasmid or were chromosomally integrated because of plasmid or phage integration events, the element would not fulfill the MME model of incorporation. Therefore, we believe that MMEs will predominate in the naturally competent species, such as those in which they have already been identified.
MMEs have now been identified from this work and previous studies in Neisseria (Saunders and Snyder 2002; Snyder et al. 2004), Streptococcus (Herbert et al. 2005; Sekizaki et al. 2005), Helicobacter (Chanto et al. 2002; Salaün and Saunders, unpublished data), E. coli (Sibley and Raleigh 2004), V. cholerae (Miller et al. 2007), Haemophilus, Rhodopseudomonas, Pseudomonas, and Lactobacillus. Aside from the Neisseria spp. study described here, no comprehensive genome-wide comparative analyses have been done to find MMEs in these bacteria. The MMEs that have been identified thus far in these species are single sites of strain difference. Further investigations targeted at the identification of MMEs in these genera are therefore likely to reveal additional MME sites.
Conclusions
One hundred and three different MME-associated cassettes have now been recognized within the 40 MME and cMME sites that have been identified within the Neisseria species. MMEs are a substantial component of gene-complement differences between neisserial strains. The cassettes in these 40 sites can probably be readily passed and chromosomally integrated between competent neisserial strains, aided by the presence of NUSSs. As such, they represent a repertoire of alternative cassettes each associated with gain, loss, or change of function that, when exchanged independently, can exist in vast numbers of possible combinations; 40 sites with only 2 alternatives for each MME (which is therefore an underestimate) has the potential to generate more than 1 trillion different gene complements within Neisseria, for example.
The identification and targeting of these sites is an efficient basis for the identification of new strain-specific regions and genes that can be used to study the basis of strain-specific behavioral differences.
In addition, as evidenced by the dinucleotide signature analysis of the associated CDSs, the diversity of CDSs present within these highly conserved regions, and the presence of the same CDSs that have been transferred within MMEs in different bacterial genera, there is now compelling evidence that MMEs are likely to be important in genetic exchange between, as well as within, bacterial species and this is a generally applicable model for horizontal exchange within and between bacterial systems. The degree to which the sites are conserved and contain diverse cassettes generates the potential for far-reaching gene flow between very distant bacterial species.
This work and L.A.S.S. were supported in part by a Wellcome Trust Project grant awarded to N.J.S. Thomas Sleeman, as part of an undergraduate project, contributed to this work but is not listed here as an author because he could not be reached during the preparation of the manuscript. The N. gonorrhoeae strain FA1090 genome sequence is available on GenBank (AE004969). The sequence used in this study was obtained from http://www.genome.ou.edu/gono.html (downloaded in 2000). In comparison with the more recent GenBank submission of this sequence, no differences were noted that would alter the conclusions of this study. The bioinformatics infrastructure and tools were provided and supported by the Sir William Dunn School of Pathology/Weatherall Institute of Molecular Medicine Computational Biology Research Group.
References
Author notes
Present address: Centre for Systems Biology, School of Biosciences, University of Birmingham, Edgbaston, United Kingdom.
William Martin, Associate Editor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}