





Abstract
For a very long time, Type II restriction enzymes (REases) have been a paradigm of ORFans: proteins with no detectable similarity to each other and to any other protein in the database, despite common cellular and biochemical function. Crystallographic analyses published until January 2008 provided high-resolution structures for only 28 of 1637 Type II REase sequences available in the Restriction Enzyme database (REBASE). Among these structures, all but two possess catalytic domains with the common PD-(D/E)XK nuclease fold. Two structures are unrelated to the others: R.BfiI exhibits the phospholipase D (PLD) fold, while R.PabI has a new fold termed ‘half-pipe’. Thus far, bioinformatic studies supported by site-directed mutagenesis have extended the number of tentatively assigned REase folds to five (now including also GIY-YIG and HNH folds identified earlier in homing endonucleases) and provided structural predictions for dozens of REase sequences without experimentally solved structures. Here, we present a comprehensive study of all Type II REase sequences available in REBASE together with their homologs detectable in the nonredundant and environmental samples databases at the NCBI. We present the summary and critical evaluation of structural assignments and predictions reported earlier, new classification of all REase sequences into families, domain architecture analysis and new predictions of three-dimensional folds. Among 289 experimentally characterized (not putative) Type II REases, whose apparently full-length sequences are available in REBASE, we assign 199 (69%) to contain the PD-(D/E)XK domain. The HNH domain is the second most common, with 24 (8%) members. When putative REases are taken into account, the fraction of PD-(D/E)XK and HNH folds changes to 48% and 30%, respectively. Fifty-six characterized (and 521 predicted) REases remain unassigned to any of the five REase folds identified so far, and may exhibit new architectures. These enzymes are proposed as the most interesting targets for structure determination by high-resolution experimental methods. Our analysis provides the first comprehensive map of sequence-structure relationships among Type II REases and will help to focus the efforts of structural and functional genomics of this large and biotechnologically important class of enzymes.
INTRODUCTION
Type II restriction endonucleases (REases) are enzymes that recognize short DNA sequences (usually 4–8-bp long) and cleave the target in both strands at, or in close proximity to the recognition site. Orthodox REases are homodimeric, cleave within palindromic sequences, require Mg 2+ ions and can act on single copies of their targets. Type II enzymes that exhibit structural and functional peculiarities (requirement of more than one target site for cleavage, cleavage at a distance from the asymmetrical target, etc.) have been classified into subtypes [nomenclature reviewed in ref. ( 1 )]. Because of remarkably high specificity in recognizing and cleaving their target sequences, they are of high interest as model systems for analyzing protein-DNA interactions and one of the most frequently used tools for recombinant DNA technology [most recent reviews: ( 2 , 3 ), a comprehensive collection of reviews on REases has been also published as a book ( 4 )]. In nature, Type II REases are found in prokaryotic organisms, where they form so-called Restriction-Modification (RM) systems with DNA methyltransferases (MTases) of the same or very similar substrate specificity. DNA MTases use S -adenosylmethionine (AdoMet) as a methyl group donor to modify specific bases in the target sequence, thereby rendering it resistant to cleavage by the REase. Thus, while the RM system's own DNA (together with the whole DNA of the prokaryotic host) is protected against suicidal degradation by REase, any foreign DNA that invades the host cell and lacks protective methylation (e.g. phages, plasmids, etc.), may be efficiently destroyed ( 5 ). In order to distinguish the components of RM system the names of MTase and REase are preceded with ‘M’. and ‘R.’ prefixes, respectively, (e.g. M.FokI and R.FokI).
Type II REases have a very high specificity and simple substrate requirements, which makes them very popular as tools in biotechnology. There are other classes of REases (Types I, III and IV), multisubunit and complex molecular machines that may combine multiple activities including restriction, methylation and DNA translocation, require additional cofactors (e.g. AdoMet, ATP or GTP), bind more than one target site, and cleave outside the recognition sequence, often at a random distance. Comparative analysis of these enzymes is outside the scope of this article, the reader is referred to recent review articles for a survey and summary of their functional properties ( 4 , 6 , 7 ). A wealth of information about all REases, including sequences, structures and functional annotations, is stored in a dedicated database REBASE ( 8 ).
Since the first genes encoding Type II REases were cloned and sequenced, comparisons have been made, aimed at detecting similarities indicative of common evolutionary history and/or mechanism of action ( 9–11 ). Surprisingly, these analyses revealed very little sequence similarity, usually limited to groups of isoschizomers, i.e. enzymes that exhibit identical DNA recognition sites and cleavage specificities ( 11 , 12 ). Database searches with REase sequences typically revealed either no significant similarity to any protein, or very high similarity (often >90% identity) to a few isoschizomers, and no similarity to other proteins. This strongly biased distribution of similarities and dissimilarity made comparative sequence analysis of all REases impossible with the use of standard tools for sequence alignment and raised a question whether the diversity of amino acid sequences of REases indicates polyphyletic evolution (convergence) or extreme divergence from a common ancestor ( 5 , 13 ).
The first answer to the question whether or not REases are related to each other was provided by crystallographic analyses. Already the first two structures of REases with apparently dissimilar sequences [R.EcoRI ( 14 ) and R.EcoRV ( 15 )] revealed a common three-dimensional fold and similar active sites ( 16 ), which indicates that they are evolutionarily related and that the overall sequence dissimilarity is due to divergent evolution (homology) rather than convergence (analogy). Essentially the same features were repeatedly observed in all crystal structures of Type II REases, at least until 2005, and in many other nucleases involved in a variety of cellular processes, e.g. DNA repair enzyme MutH or Holliday junction resolvases ( 17 , 18 ). Catalytic domains of these proteins share a common structural core, comprising a mixed β-sheet of 4 strands flanked on both sides by α-helices and additional, variable elements of secondary structure ( 16 , 19–21 ). The core serves as a scaffold for a weakly conserved active site, typically comprising two or three acidic residues (Asp or Glu) and one Lys residue, which together form the hallmark bipartite catalytic motif (P) D … Xn …( D/E )X K (where X is any amino acid). This motif has led to naming this superfamily of proteins as ‘PD-(D/E)XK’ ( 22 , 23 ).
It was found that some members of the PD-(D/E)XK superfamily exhibit deviations from the consensus. First, the active sites of Type II REases often contain nonstandard residues at the otherwise conserved positions, e.g. Q or N at the positions occupied by the (D/E)XK half-motif ( 24 , 25 ). Second, catalytic residues have been also found to ‘migrate’ between nonequivalent positions in sequence, preserving the spatial orientation of functional groups in the active site without the correspondence at the level of the sequence alignment ( 26–28 ). These two features have been also reported in some non-REase members of the PD-(D/E)XK superfamily ( 23 , 29 , 30 ), but when combined with the extreme overall sequence divergence characteristic for REases, they essentially prevent the identification of an active site by ‘sequence gazing’. As a result, sequence–function analysis usually requires the aid of three-dimensional structure (ideally—solved experimentally, or obtained by comparative modeling techniques).
Type II REases are notorious for presenting elaborations of the common fold in the form of large insertions and terminal extensions that often contain regular elements of secondary structure, even entire domains. These elaborations form a variable ‘shell’ surrounding the conserved core and are often involved in DNA binding or formation of contacts between protomers in oligomeric structures. They may be responsible for the formation of completely different quaternary structures even by enzymes that are very similar at the level of tertiary structure, e.g. R.EcoRV and R.BglI ( 31 ). In a phylogenetic tree of PD-(D/E)XK enzymes with known structures, Type II REases radiate from all major branches of the superfamily, indicating multiple independent recruitment of the same fold to the process of restriction. The accumulation of a large number of changes suggests higher speed of evolution associated with being involved in restriction, compared to other PD-(D/E)XK enzymes involved in house-keeping processes such as DNA repair ( 20 ). Type II REases are therefore extremely hard targets for protein structure prediction methods, and even detection of the PD-(D/E)XK motif in their sequence remains a formidable challenge ( 20 , 32 ).
Not all REases, however, are members of the PD-(D/E)XK superfamily. In 2000, three groups discovered a few REases that appeared to be members of structurally and evolutionarily unrelated superfamilies: Siksnys and co-workers discovered that R.BfiI belongs to the phospholipase D (PLD) superfamily ( 33 ), the group of Koonin and independently one of the authors of this article (J.M.B.) predicted that a few REases belong to the HNH superfamily ( 34 , 35 ); J.M.B. also predicted that R.Eco29kI and its two nearly identical isoschizomers belong to the GIY-YIG superfamily ( 35 ). Since then, all these theoretical predictions have been confirmed experimentally. The structure of R.BfiI has been solved, revealing a PLD-like dimer of catalytic domains with a single symmetrical active site at the domain interface ( 36 ). Structural models of HNH nuclease domain in R.KpnI ( 37 ) and GIY-YIG nuclease domain in R.Eco29kI ( 38 ) have been supported by mutagenesis and biochemical experiments. Most recently, a newly identified REase R.PabI was predicted to be a candidate for a new fold ( 39 ), which has been validated by X-ray crystallography and mutagenesis, revealing a novel tertiary and quaternary architecture ( 40 ). It must be mentioned that two of these nonstandard enzymes (R.BfiI and R.PabI) exhibit a feature that may be even more unusual than their nonclassical folds: they cleave DNA in the absence of metal ions ( 33 , 40 ). Thus, structurally characterized Type II REases present five unrelated three-dimensional folds, several different variants of active sites and catalytic mechanisms, and a plethora of modes for protein–protein and protein–DNA interactions.
REBASE, the database of restriction enzymes makes available to the public (as of 25 January 2008) 1637 sequences of Type II REases, including 302 experimentally characterized enzymes and 1335 putative ones, inferred from sequence comparisons or genomic analyses. Many REase candidates are ORFans, i.e. proteins that show no similarity to any other protein (or only very high similarity to a few other proteins). Some of them have been predicted only because they are encoded by genes located close to genes encoding true or predicted DNA MTases. The disproportion between the number of known or predicted sequences and the number of experimentally characterized proteins with known three-dimensional structures (>50 to 1) is similar to the average value reported for sequences inferred from genome sequencing projects. Thus, Type II REases can be regarded as a ‘firing range’ for structural genomics projects in a sense that any methodology (theoretical or experimental) developed to narrow down this gap may be broadly applicable to all proteins. Some efforts have been made in this direction. Bioinformatics analyses have been made to assign a fraction of REase sequences to the previously identified folds ( 22 , 23 , 34 , 35 , 41 ) and site-directed mutagenesis has been used, often in connection with the circular dichroism (CD) analysis, to test some of these predictions [e.g. ( 27 , 42–49 )]. Because of the difficulties in predicting variable regions, most of the published alignments and models contain only the catalytic domain, or just the immediate neighborhood of the active site. Nonetheless, these predictions, especially if supported by experimental data, are usually sufficient to provide a confident three-dimensional fold assignment (which implies evolutionary relationship to other members), and provide numerous additional hints regarding the possible mechanism of action (e.g. the mode of DNA binding).
Bioinformatic and low-resolution experimental analyses have aided X-ray crystallography in assigning a number of Type II REases to known folds and superfamilies. However, a large fraction of REases remains without any predictions or experimental data. Moreover, there is no single resource a researcher could use, that indicates whether any structural or evolutionary prediction has been made for a given REase sequence, what the assigned fold is, where the structural model is available, and whether any experimental data support the theoretical analyses. Currently, navigation in a large volume of data and literature concerning different REase structures and families is very difficult not only for newcomers in the REase field, but also for biochemists, who are not necessarily experts in molecular evolution or structural bioinformatics, but would like to take the advantage of published predictions to plan new experiments. We have therefore decided to survey the published literature and databases for experimental data and predictions concerning the structure of all Type II REases with sequences available in REBASE, and to make new predictions for the great majority that had no such information available. We carried out a search for additional homologs of Type II REases, not yet available in REBASE, and clustered all sequences to identify groups of close homologs that are likely to share very similar structures as well as substrate specificities (isoschizomers or nearly-isoschizomers). As a result, we provide the very first classification of all Type II REase sequences into families and superfamilies, and a comprehensive structural census. We also provide a list of prospective candidates for crystallographic analyses, with two priorities in mind: (i) maximization of structural coverage (availability of structural templates for confident modeling of a possibly largest number of sequences significantly related to these templates), and (ii) high-resolution structural characterization of folds that are either completely new or at least have not been reported among Type II REases.
METHODS
Sequence analyses
Sequence searches of the nonredundant (nr) and environmental samples (env_nr) database were carried out using a locally installed version of PSI-BLAST ( 50 ). Gapped blast algorithm (blastpgp) was used with default parameters [BLOSUM62 substitution matrix, gap open penalty 11, gap extension penalty 1, without iterating and with expectation (E) value threshold of 0.02].
To identify (sub)families of closely related sequences and visualize similarities within and between all genuine REases and their homologs we used CLANS (CLuster ANalysis of Sequences), a Java utility based on the Fruchterman-Reingold graph layout algorithm ( 51 ). CLANS uses the P -values of high-scoring segment pairs (HSPs) obtained from an N × N BLAST search, to compute attractive and repulsive forces between each sequence pair in a user-defined dataset. A 3D or 2D representation is achieved by randomly seeding sequences in the arbitrary distance space. The sequences are then moved within this environment according to the force vectors resulting from all pairwise interactions and the process is repeated to convergence.
Groups of two or more sequences that formed clusters were extracted from the CLANS output and aligned using MUSCLE ( 52 ). In cases of low sequence similarity, alignments were also constructed with other programs, MUMMALS ( 53 ), MAFFT ( 54 ) and PROBCONS ( 55 ), and checked for consistency. Those sequences of REase homologs, which could be aligned to true REases, but exhibited deletions (>30% of the alignment missing) were discarded. Manual adjustments were introduced into the alignments to preserve the continuity of secondary structure elements, either observed in crystal structures of representative family members, or predicted computationally (see below).
Domain assignment for proteins was performed mainly by Conserved Domain Database search service ( 56 ) with default parameters. Additional searches were made using HHPRED ( 57 ) against the database of all available sequence profiles. If a reliable multiple sequence alignment for a given sequence was available (see above), it was used as a query instead of a single sequence.
Structure prediction
Protein structure prediction was carried out using a new version ( http://genesilico.pl/meta2/ ) of the GeneSilico MetaServer ( 58 ), which is a gateway for a variety of methods for making predictions and analyzing their results. For each REase subfamily, at least one representative sequence was submitted, and often additional predictions were made for individual domains, other members and whole alignments. Secondary structure was predicted using a consensus of PSIPRED ( 59 ), PROFsec ( 60 ), PROF ( 61 ), SABLE ( 62 ), JNET ( 63 ), JUFO ( 64 ), PORTER ( 65 ), SSPRO2 ( 66 ) and SAM-T02 ( 67 ). Solvent accessibility for individual residues was predicted with SABLE ( 62 ), ACCPRO2 ( 66 ) and JNET ( 63 ). The fold-recognition (FR) analysis (attempt to match the query sequence to known protein structures) was carried out using a series of methods: PDB-BLAST [local implementation of a PSI-BLAST ( 50 ) search against sequences of proteins from PDB], HHSEARCH ( 68 ), FORTE ( 69 ), SAM-T02 ( 67 ), 3DPSSM ( 70 ), INBGU ( 71 ), FUGUE ( 72 ), mGENTHREADER ( 73 ) and SPARKS ( 74 ). Target-template alignments reported by these methods were compared, evaluated and ranked by the PCONS server ( 75 ) to identify the preferred template.
We have not attempted to build three-dimensional models for all REases, as currently this analysis is too demanding; it usually requires iterative comparative modeling of the core and model evaluation often accompanied by de novo folding of variable parts, with a lot of manual intervention and time-consuming calculations, which can take weeks or even months per protein [see previously published examples, e.g. ( 76 )]. The alignments published in this work, will however serve as a convenient starting point for building complete models in the future, when experimental data to directly test the models become available, and it will be worthwhile to invest time and computing power.
RESULTS
Identification of known and putative REases
We retrieved 1637 sequences of all Type II REases (genuine and putative enzymes, including sequences from metagenomics projects) from REBASE (edition 25 January 2008). For these sequences, we carried out preliminary clustering with CLANS ( 51 ), to detect groups of proteins exhibiting BLAST P -value <0.001 in pairwise comparisons (see Methods section for details). The results (data not shown) revealed four large clusters of 471, 221, 125 and 42 sequences, comprising all experimentally characterized Type IIC enzymes (including Type IIG and Type IIB) and their closest homologs, and a large number of very small clusters and ORFans. By definition, all known type IIC enzymes possess in the same polypeptide a nuclease domain and a DNA:m 6 A MTase domain. While the nuclease domains exhibit relatively low similarity (characteristic for REases of all types), the MTase domains exhibit very high sequence conservation (typical for MTases), leading all Type IIC enzymes to cluster together—regardless of the presence or absence of similarity between their non-MTase parts of the sequence. Preliminary clustering revealed also several other smaller clusters of proteins that shared sequence similarity in various kind of non-nuclease domains (such as the GHKL domain common to the ATPase/kinase superfamily ( 77 ) or the DEXDc helicase domain), but no similarity in known or predicted nuclease domains.
In order to cluster Type II REase sequences only with respect to similarity of their nuclease domains, we decided to identify all domains in sequences from REBASE and create a set of sequences from which all conserved non-nuclease domains have been deleted. This was made by retrieving sequences from sequence clusters, making multiple sequence alignments, assigning domains by CDD and HHPRED (see Methods section), followed by deletion of assigned non-nuclease domains. If necessary, additional subclustering and domain assignment was done for each cluster. We omitted very short sequences (<50 aa, e.g. from peptide sequencing), identical sequences and those lacking nuclease domains (e.g. due to truncation); this included partial sequences of some experimentally characterized enzymes, e.g. Aor13HI or PvuI.
To identify additional homologs not present in REBASE, we carried out BLAST searches of the nr database and environmental samples database (env_nr) using all Type II REase sequences (without conserved non-nuclease domains). For all BLAST hits, we performed domain assignment with the same procedure as for sequences from REBASE. Likewise, non-nuclease domains were identified and removed. As a result, we obtained a set of 3132 sequences in two categories: one comprising full-length sequences, and the other with promiscuous domains removed (i.e. REases comprising either exclusively nuclease domains, or nuclease domains with extensions that did not exhibit high similarity to domains in non-REase proteins). The latter set will be referred to the ‘nuclease domain’ set for simplicity.
Classification of REases
The nuclease domain dataset was clustered using CLANS ( Figure 1 ), which allowed us to classify all Type II REases into 190 subfamilies that contain mutually related proteins and ORFans that exhibit no easily detectable similarity of nuclease domain to proteins from other subfamilies. The distribution of size of these 190 subfamilies is shown in Figure 2 .
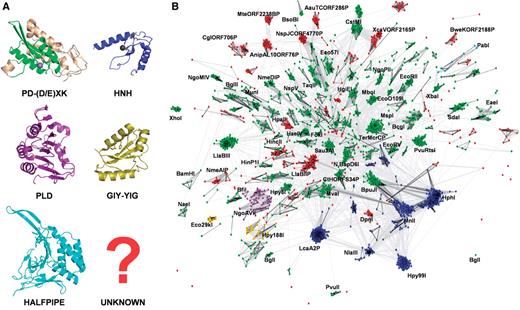
Clustering of Type II REase sequences and their assignment to three-dimensional folds. ( A ) Representative structures of nuclease domains of Type II REases or proteins sharing the same fold: PD-(D/E)XK: BamHI (3bam); the universally conserved core is indicated in green, nonconserved structures in gray, HNH: catalytic domain of T4 endonuclease VII (1en7), PLD: catalytic domain of R.BfiI (2c1l), GIY-YIG: catalytic domain of homing endonuclease I-TevI (1mk0), HALFPIPE: R.PabI (2dvy). ( B ) Results of clustering of Type II REases from REBASE and their homologs in the nr and env_nr database with CLANS (with promiscuous domains, such as MTase or GHKL domains, excluded from analysis). Structures in (A) and sequences in (B) are colored according to the their assignment to fold families (see below): PD-D(E)XK: green, HNH: blue, GIY-YIG: yellow, PLD: magenta, HALFPIPE: cyan, unclassified: red. Connections between dots represent the degree of pairwise sequence similarity, as quantified by BLAST P -value (the darker the line, the higher similarity). The whole ‘galaxy’ of REases is held together by a certain level of ‘background’ similarity between different (often unrelated) sequences that is due to pure chance. Thus, while connections within dense clusters practically always reflect high similarity and evolutionary relationship, connections between clusters do not have to reflect their phylogenetic relationships (although they often do, especially in the case of close connections with multiple dark lines). All subfamilies with >20 members or with representatives with solved X-ray structures have been labeled by the name of their representative sequence.
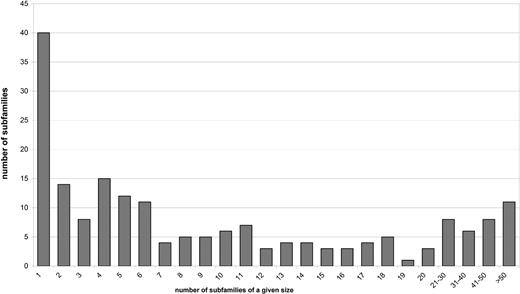
The distribution of size (number of members) among REase subfamilies. Seventy-seven subfamilies (41% of all subfamilies) contain < 5 sequences, which makes it very difficult to analyze the patterns of sequence conservation and e.g. identify invariant residues that could form active sites.
For all confirmed and putative Type II REases in our dataset, we carried out an extensive survey of the published literature and databases to identify experimental data, structural predictions, sequence analyses and phylogenetic studies. Our aim was to collect all experimental data and reasonable predictions that could provide hints to the structural and evolutionary classification of Type II REases, i.e. assignment of sequences to structural folds, grouping of subfamilies into families and families into superfamilies. We were able to identify published crystallographic evidence for members of 23 subfamilies, published structural prediction supported by experiment (e.g. mutagenesis) for members of additional 20 subfamilies and published predictions that have so far not been tested for additional 21 subfamilies. For 126 subfamilies we could find neither experimental data nor reliable predictions, which made them priority targets for our structure prediction methods. Based on analysis of all types of data available as well as the results of our preliminary sequence analyses, we named each subfamily after one representative enzyme, which in our subjective opinion was best studied from the structural or functional point of view or which exhibited features that were most typical for a given subfamily.
For 126 subfamilies that comprised structurally uncharacterized proteins and for any of the previously mentioned subfamilies where we had any doubts about the correctness of the published structural assignments, we carried out structure prediction via the GeneSilico MetaServer ( 58 ) using the protein Fold Recognition (FR) approach (see Methods section). The interpretation of FR results and selection of the best template was aided by analyzing the patterns of residue conservation in the light of predicted secondary structure, both in the target subfamily and in the putative templates. In a few particularly difficult cases the fold prediction was aided by building three-dimensional models using the FRankenstein's Monster approach ( 78 , 79 ) and analysis of sequence–structure compatibility in 3D using a series of Model Quality Assessment Programs ( 80 ) (see Methods section for details). The FR analysis allowed us to predict 3D folds and identify putative homology between Type II REase subfamilies and proteins of known structure, including all previously solved structures of Type II REases and their homologs. We have also used HHSEARCH ( 68 ) to perform a series of pairwise profile-to-profile comparisons for all alignments of subfamilies represented as profile hidden Markov models (HMMs) that include information both about sequence conservation (if more than one sequence is available) and secondary structure predicted by PSIPRED ( 59 ). This type of analysis allowed us to identify putative homology between different Type II REase subfamilies, including those for which no experimental structural information is available. Combination of structure and sequence-oriented searches allowed us to make fold predictions based on the principle of transitivity of homology. For example if subfamily A was found to be homologous to subfamily B, and the same sequence region in subfamily B that was matched with subfamily A was also found to match a structure of a known fold characteristic for subfamily C, then subfamily A was predicted to be homologous to subfamily C regardless of the absence of a direct match.
As a result of the aforementioned analyses, we confirmed all previously reported 3D fold predictions, and made new predictions for 52 subfamilies. Thus, as a result of our survey, we assigned three-dimensional folds to 1528 Type II REase sequences and their homologs based on previously published analyses and our alignments, and we made new predictions about the fold and active site for 1027 Type II REase sequences and their homologs. For 577 Type II REase sequences and their homologs (i.e. 18.4% of all sequences; 73 subfamilies among 190 subfamilies total), we could not make any structural assignment, based either on literature and database searches or on our new bioinformatic analyses. The results of our survey are summarized in Table 1 . Sequence alignments of core residues for representatives of all ‘assignable’ subfamilies are shown in Figure 3 [PD-(D/E)XK superfamily, 98 or 51.6% subfamilies], Figure 4 (HNH superfamily, 14 or 7.4% subfamilies), Figure 5 (PLD superfamily, 2 or 1.1% subfamilies) and Figure 6 (GIY-YIG superfamily, 2 or 1.1% subfamiles). We found no new subfamilies from the HALFPIPE superfamily compared to the previously published study, therefore readers are referred to the original publication for comparative analysis ( 40 , 81 ).
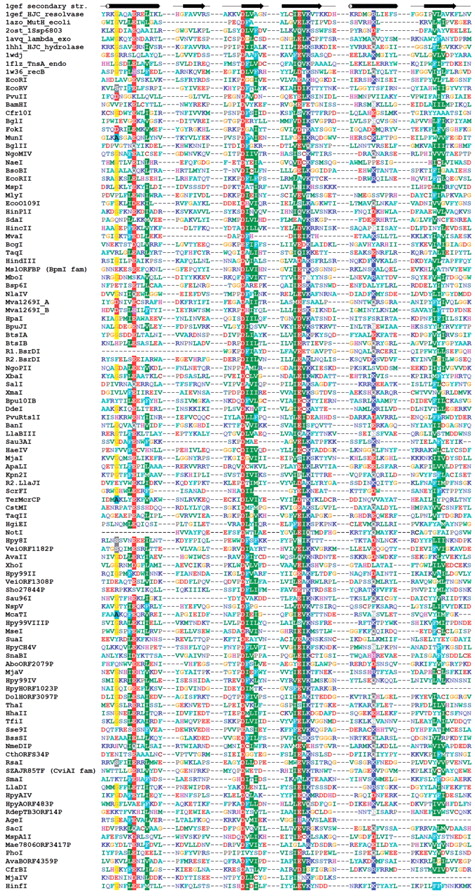
Sequence alignment of representative Type II REases from all subfamilies of the PD-(D/E)XK superfamily. Sequences of REases are preceded with sequences of several proteins from this superfamily with solved crystal structures and with typical secondary structure representation (of 1gef Holiday junction resolvase). Amino acids are colored according to physico-chemical properties of their side chains (negatively charged: red; positively charged: blue, violet; hydrophilic: gray; hydrophobic: green, magenta, yellow). Residues with more than 50% sequence conservation are shaded. Nonconserved sequence linkers between conserved blocks have been omitted for clarity.

Sequence alignment of representative Type II REases from all subfamilies of the HNH superfamily. Sequences of REases are preceded with sequences of several proteins from this superfamily with solved crystal structures and with typical secondary structure representation (of 1en7 T4 endonuclease VII). Amino acids are colored according to physico-chemical properties of their side chains (negatively charged: red; positively charged: blue, violet; hydrophilic: gray; hydrophobic: green, magenta, yellow). Residues with more than 50% sequence conservation are shaded.

Sequence alignment of representative Type II REases from the PLD superfamily. Sequences of REases are preceded with a sequence of Nuc nuclease (1BYR) from the PLD superfamily and with the secondary structure of R.BfiI (2c1l). Amino acids are colored according to physico-chemical properties of their side chains (negatively charged: red; positively charged: blue, violet; hydrophilic: gray; hydrophobic: green, magenta, yellow). Residues with more than 70% sequence conservation are shaded.
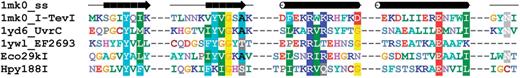
Sequence alignment of representative Type II REases from the GIY-YIG superfamily. Sequences of two REases are preceded by sequences of GIY-YIG members with solved crystal structures and with the secondary structure of I-TevI homing endonuclease (1mk0). Amino acids are colored according to physico-chemical properties of their side chains (negatively charged: red; positively charged: blue, violet; hydrophilic: gray; hydrophobic: green, magenta, yellow). Residues with more than 70% sequence conservation are shaded. Nonconserved sequence linkers between conserved blocks have been omitted for clarity.
3D-fold classification for Type II REase subfamilies
| Family name | Number of members | Type of evidence | Reference | Subtype | Reliability |
|---|---|---|---|---|---|
| A) PD-(D/E)XK superfamily | |||||
| EcoRI | 14 | X-ray EcoRI | ( 14 ) | P | 4 |
| EcoRV | 14 | X-ray EcoRV | ( 15 ) | P | 4 |
| PvuII | 5 | X-ray PvuII | ( 90 ) | P | 4 |
| BamHI | 5 | X-ray BamHI | ( 91 ) | P | 4 |
| Cfr10I | 6 | X-ray Cfr10I | ( 26 ) | P,F | 4 |
| BglI | 17 | X-ray BglI | ( 31 ) | P,F | 4 |
| FokI | 9 | X-ray FokI | ( 92 ) | S | 4 |
| MunI | 6 | X-ray MunI | ( 93 ) | P | 4 |
| BglII | 17 | X-ray BglII | ( 94 ) | P | 4 |
| NgoMIV | 11 | X-ray NgoMIV | ( 95 ) | F,P | 4 |
| NaeI | 7 | X-ray NaeI | ( 96 ) | E | 4 |
| BsoBI | 8 | X-ray BsoBI | ( 25 ) | P | 4 |
| EcoRII | 45 | X-ray EcoRII | ( 97 ) | P,E | 4 |
| MspI | 4 | X-ray MspI | ( 98 ) | P | 4 |
| MlyI | 45 | X-ray N.BspD6I | ( 99 ) | S,P | 4 |
| EcoO109I | 10 | X-ray EcoO109I | ( 100 ) | P | 4 |
| HinP1I | 6 | X-ray HinP1I | ( 88 ) | P | 4 |
| SdaI | 43 | X-ray SdaI | ( 28 ) | P | 4 |
| HincII | 5 | X-ray HincII | ( 101 ) | P | 4 |
| MvaI | 36 | X-ray MvaI | ( 76 , 102 ) | P | 4 |
| NotI | 23 | X-ray NotI | Lambert et al . to be published (PDB 3brv) | P | 4 |
| BcgI | 31 | Mutagenesis BcgI | ( 103 ) | C | 3 |
| TaqI | 6 | Mutagenesis TaqI | ( 104 ) | P | 3 |
| HindIII | 11 | Mutagenesis HindIII | ( 105 ) | P | 3 |
| Eco57I | 37 | Mutagenesis Eco57I | ( 106 ) | C | 3 |
| MboI | 40 | Mutagenesis MboI | ( 43 ) | P | 3 |
| Bsp6I | 15 | Mutagenesis Bsp6I | ( 47 ) | P | 3 |
| NlaIV | 10 | Mutagenesis, CD NlaIV | ( 46 ) | P | 3 |
| Mva1269I | 3 | Mutagenesis Mva1269I | ( 45 ) | S | 3 |
| HpaI | 1 | Mutagenesis HpaI | ( 48 ) | P | 3 |
| BpuJI | 73 | Mutagenesis BpuJI | ( 107 ) | S | 3 |
| BtsIA | 1 | Mutagenesis BtsIA | ( 108 ) | S | 3 |
| BtsIB | 1 | Mutagenesis BtsIB | ( 108 ) | S | 3 |
| R2.BsrDI | 3 | Mutagenesis R2.BsrDI | ( 108 ) | S | 3 |
| R1.BsrDI | 2 | Mutagenesis R1.BsrDI | ( 108 ) | S | 3 |
| NgoPII | 21 | Mutagenesis NgoPII | J.M.B. and coworkers, unpublished data | P | 3 |
| XbaI | 15 | Sequence analysis | ( 109 ) | P | 2 |
| SalI | 15 | Sequence analysis | ( 109 ) | P | 2 |
| XmaI | 9 | Sequence analysis | ( 110 ) | E,P | 2 |
| Bpu10IB | 18 | Sequence analysis | ( 111 ) | S,P | 2 |
| DdeI | 5 | Sequence analysis | ( 111 ) | P | 2 |
| PvuRts1 | 46 | Sequence analysis | ( 22 , 34 ) | ? | 2 |
| BanI | 8 | Sequence analysis | ( 22 , 112 ) | P | 2 |
| LlaBIII | 152 | Sequence analysis | ( 113 ) | C | 2 |
| Sau3AI | 36 | Sequence analysis | ( 22 , 114 ) | E,P | 2 |
| HaeIV | 20 | Sequence analysis | ( 115 ) | C | 2 |
| MjaI | 6 | Sequence analysis | ( 22 ) | P | 2 |
| ApaLI | 1 | Sequence analysis | ( 22 ) | P | 2 |
| Kpn2I | 7 | Sequence analysis | ( 27 ) | P | 2 |
| R2.LlaJI | 16 | Sequence analysis | ( 116 ) | P | 2 |
| ScrFI | 13 | Sequence analysis | ( 76 ) | P | 2 |
| TerMcrCP | 112 | Sequence analysis | This work | ? | 2 |
| CstMI | 76 | Sequence analysis | This work | C | 2 |
| TaqII | 35 | Sequence analysis | This work | C,S | 2 |
| HgiEI | 25 | Sequence analysis | This work | P | 2 |
| Hpy8I | 20 | Sequence analysis | This work | P | 2 |
| VeiORF1182P | 19 | Sequence analysis | This work | P | 2 |
| AvaII | 18 | Sequence analysis | This work | P | 2 |
| XhoI | 16 | Sequence analysis | This work | P | 2 |
| Hpy99II | 16 | Sequence analysis | This work | P | 2 |
| VeiORF1308P | 14 | Sequence analysis | This work | C | 2 |
| Sho27844P | 14 | Sequence analysis | This work | S | 2 |
| Sau96I | 13 | Sequence analysis | This work | P | 2 |
| NspV | 12 | Sequence analysis | This work | P | 2 |
| McaTI | 11 | Sequence analysis | This work | P | 2 |
| Hpy99VIIIP | 10 | Sequence analysis | This work | P | 2 |
| MseI | 9 | Sequence analysis | This work | P | 2 |
| SuaI | 6 | Sequence analysis | This work | P | 2 |
| HpyCH4V | 5 | Sequence analysis | This work | P | 2 |
| SnaBI | 5 | Sequence analysis | This work | P | 2 |
| AboORF2079P | 4 | Sequence analysis | This work | P | 2 |
| MjaV | 4 | Sequence analysis | This work | P | 2 |
| Hpy99IV | 4 | Sequence analysis | This work | P | 2 |
| HpyHORF1023P | 4 | Sequence analysis | This work | P | 2 |
| DolHORF3097P | 4 | Sequence analysis | This work | ? | 2 |
| ThaI | 3 | Sequence analysis | This work | P | 2 |
| HhaII | 2 | Sequence analysis | This work | P | 2 |
| TfiI | 1 | Sequence analysis | This work | P | 2 |
| Sse9I | 1 | Sequence analysis | This work | P | 2 |
| BssSI | 1 | Sequence analysis | This work | S | 1 |
| NmeDIP | 46 | Sequence analysis | This work | ? | 1 |
| CthORFS34P | 30 | Sequence analysis | This work | P | 1 |
| RsaI | 11 | Sequence analysis | This work | P | 1 |
| CviAI | 9 | Sequence analysis | This work | P | 1 |
| SmaI | 8 | Sequence analysis | This work | P | 1 |
| LlaDI | 7 | Sequence analysis | This work | P | 1 |
| HpyAIV | 6 | Sequence analysis | This work | P | 1 |
| HpyAORF483P | 4 | Sequence analysis | This work | P | 1 |
| RdepTB3ORF14P | 4 | Sequence analysis | This work | P | 1 |
| AgeI | 4 | Sequence analysis | This work | P | 1 |
| SacI | 3 | Sequence analysis | This work | P | 1 |
| MspA1I | 3 | Sequence analysis | This work | P | 1 |
| Mae7806ORF3417P | 3 | Sequence analysis | This work | P | 1 |
| PhoI | 2 | Sequence analysis | This work | P | 1 |
| AvaBORF4359P | 2 | Sequence analysis | This work | P | 1 |
| CfrBI | 2 | Sequence analysis | This work | P | 1 |
| MjaIV | 1 | Sequence analysis | This work | P | 1 |
| HinfI | 1 | Sequence analysis | This work | P | 1 |
| B) HNH superfamily | |||||
| KpnI | 2 | Mutagenesis | ( 37 ) | P | 3 |
| MnlI | 117 | Mutagenesis | ( 44 ) | S | 3 |
| HphI | 282 | Mutagenesis | ( 49 ) | S | 3 |
| Eco31I | 11 | Mutagenesis | ( 117 ) | S | 3 |
| NlaIII | 82 | Sequence analysis | ( 34 , 35 ) | P | 2 |
| MboII | 7 | Sequence analysis | ( 34 , 35 ) | S | 2 |
| SapI | 4 | Sequence analysis | ( 34 , 35 ) | S | 2 |
| SphI | 4 | Sequence analysis | ( 34 , 35 ) | P | 2 |
| NspI | 5 | Sequence analysis | ( 35 ) | P | 2 |
| Hin4II | 12 | Sequence analysis | ( 41 ) | S | 2 |
| LcaA2P | 229 | Sequence analysis | This work | ? | 2 |
| Hpy99I | 121 | Sequence analysis | This work | P | 2 |
| Mae7806ORF5066P | 13 | Sequence analysis | This work | P | 2 |
| PacI | 4 | Sequence analysis | This work | P | 1 |
| C) PLD superfamily | |||||
| BfiI | 3 | X-ray BfiI | ( 36 ) | S | 4 |
| NgoAVII | 50 | Sequence analysis | This work | P | 2 |
| D) GIY-YIG superfamily | |||||
| Eco29kI | 10 | Mutagenesis Eco29kI | ( 38 ) | P | 3 |
| Hpy188I | 23 | Sequence analysis | Mikihiko Kawai (University of Tokyo), personal communication | P | 2 |
| E) HALFPIPE superfamily | |||||
| PabI | 8 | X-ray PabI | ( 40 ) | P | 4 |
| Family name | Number of members | Type of evidence | Reference | Subtype | Reliability |
|---|---|---|---|---|---|
| A) PD-(D/E)XK superfamily | |||||
| EcoRI | 14 | X-ray EcoRI | ( 14 ) | P | 4 |
| EcoRV | 14 | X-ray EcoRV | ( 15 ) | P | 4 |
| PvuII | 5 | X-ray PvuII | ( 90 ) | P | 4 |
| BamHI | 5 | X-ray BamHI | ( 91 ) | P | 4 |
| Cfr10I | 6 | X-ray Cfr10I | ( 26 ) | P,F | 4 |
| BglI | 17 | X-ray BglI | ( 31 ) | P,F | 4 |
| FokI | 9 | X-ray FokI | ( 92 ) | S | 4 |
| MunI | 6 | X-ray MunI | ( 93 ) | P | 4 |
| BglII | 17 | X-ray BglII | ( 94 ) | P | 4 |
| NgoMIV | 11 | X-ray NgoMIV | ( 95 ) | F,P | 4 |
| NaeI | 7 | X-ray NaeI | ( 96 ) | E | 4 |
| BsoBI | 8 | X-ray BsoBI | ( 25 ) | P | 4 |
| EcoRII | 45 | X-ray EcoRII | ( 97 ) | P,E | 4 |
| MspI | 4 | X-ray MspI | ( 98 ) | P | 4 |
| MlyI | 45 | X-ray N.BspD6I | ( 99 ) | S,P | 4 |
| EcoO109I | 10 | X-ray EcoO109I | ( 100 ) | P | 4 |
| HinP1I | 6 | X-ray HinP1I | ( 88 ) | P | 4 |
| SdaI | 43 | X-ray SdaI | ( 28 ) | P | 4 |
| HincII | 5 | X-ray HincII | ( 101 ) | P | 4 |
| MvaI | 36 | X-ray MvaI | ( 76 , 102 ) | P | 4 |
| NotI | 23 | X-ray NotI | Lambert et al . to be published (PDB 3brv) | P | 4 |
| BcgI | 31 | Mutagenesis BcgI | ( 103 ) | C | 3 |
| TaqI | 6 | Mutagenesis TaqI | ( 104 ) | P | 3 |
| HindIII | 11 | Mutagenesis HindIII | ( 105 ) | P | 3 |
| Eco57I | 37 | Mutagenesis Eco57I | ( 106 ) | C | 3 |
| MboI | 40 | Mutagenesis MboI | ( 43 ) | P | 3 |
| Bsp6I | 15 | Mutagenesis Bsp6I | ( 47 ) | P | 3 |
| NlaIV | 10 | Mutagenesis, CD NlaIV | ( 46 ) | P | 3 |
| Mva1269I | 3 | Mutagenesis Mva1269I | ( 45 ) | S | 3 |
| HpaI | 1 | Mutagenesis HpaI | ( 48 ) | P | 3 |
| BpuJI | 73 | Mutagenesis BpuJI | ( 107 ) | S | 3 |
| BtsIA | 1 | Mutagenesis BtsIA | ( 108 ) | S | 3 |
| BtsIB | 1 | Mutagenesis BtsIB | ( 108 ) | S | 3 |
| R2.BsrDI | 3 | Mutagenesis R2.BsrDI | ( 108 ) | S | 3 |
| R1.BsrDI | 2 | Mutagenesis R1.BsrDI | ( 108 ) | S | 3 |
| NgoPII | 21 | Mutagenesis NgoPII | J.M.B. and coworkers, unpublished data | P | 3 |
| XbaI | 15 | Sequence analysis | ( 109 ) | P | 2 |
| SalI | 15 | Sequence analysis | ( 109 ) | P | 2 |
| XmaI | 9 | Sequence analysis | ( 110 ) | E,P | 2 |
| Bpu10IB | 18 | Sequence analysis | ( 111 ) | S,P | 2 |
| DdeI | 5 | Sequence analysis | ( 111 ) | P | 2 |
| PvuRts1 | 46 | Sequence analysis | ( 22 , 34 ) | ? | 2 |
| BanI | 8 | Sequence analysis | ( 22 , 112 ) | P | 2 |
| LlaBIII | 152 | Sequence analysis | ( 113 ) | C | 2 |
| Sau3AI | 36 | Sequence analysis | ( 22 , 114 ) | E,P | 2 |
| HaeIV | 20 | Sequence analysis | ( 115 ) | C | 2 |
| MjaI | 6 | Sequence analysis | ( 22 ) | P | 2 |
| ApaLI | 1 | Sequence analysis | ( 22 ) | P | 2 |
| Kpn2I | 7 | Sequence analysis | ( 27 ) | P | 2 |
| R2.LlaJI | 16 | Sequence analysis | ( 116 ) | P | 2 |
| ScrFI | 13 | Sequence analysis | ( 76 ) | P | 2 |
| TerMcrCP | 112 | Sequence analysis | This work | ? | 2 |
| CstMI | 76 | Sequence analysis | This work | C | 2 |
| TaqII | 35 | Sequence analysis | This work | C,S | 2 |
| HgiEI | 25 | Sequence analysis | This work | P | 2 |
| Hpy8I | 20 | Sequence analysis | This work | P | 2 |
| VeiORF1182P | 19 | Sequence analysis | This work | P | 2 |
| AvaII | 18 | Sequence analysis | This work | P | 2 |
| XhoI | 16 | Sequence analysis | This work | P | 2 |
| Hpy99II | 16 | Sequence analysis | This work | P | 2 |
| VeiORF1308P | 14 | Sequence analysis | This work | C | 2 |
| Sho27844P | 14 | Sequence analysis | This work | S | 2 |
| Sau96I | 13 | Sequence analysis | This work | P | 2 |
| NspV | 12 | Sequence analysis | This work | P | 2 |
| McaTI | 11 | Sequence analysis | This work | P | 2 |
| Hpy99VIIIP | 10 | Sequence analysis | This work | P | 2 |
| MseI | 9 | Sequence analysis | This work | P | 2 |
| SuaI | 6 | Sequence analysis | This work | P | 2 |
| HpyCH4V | 5 | Sequence analysis | This work | P | 2 |
| SnaBI | 5 | Sequence analysis | This work | P | 2 |
| AboORF2079P | 4 | Sequence analysis | This work | P | 2 |
| MjaV | 4 | Sequence analysis | This work | P | 2 |
| Hpy99IV | 4 | Sequence analysis | This work | P | 2 |
| HpyHORF1023P | 4 | Sequence analysis | This work | P | 2 |
| DolHORF3097P | 4 | Sequence analysis | This work | ? | 2 |
| ThaI | 3 | Sequence analysis | This work | P | 2 |
| HhaII | 2 | Sequence analysis | This work | P | 2 |
| TfiI | 1 | Sequence analysis | This work | P | 2 |
| Sse9I | 1 | Sequence analysis | This work | P | 2 |
| BssSI | 1 | Sequence analysis | This work | S | 1 |
| NmeDIP | 46 | Sequence analysis | This work | ? | 1 |
| CthORFS34P | 30 | Sequence analysis | This work | P | 1 |
| RsaI | 11 | Sequence analysis | This work | P | 1 |
| CviAI | 9 | Sequence analysis | This work | P | 1 |
| SmaI | 8 | Sequence analysis | This work | P | 1 |
| LlaDI | 7 | Sequence analysis | This work | P | 1 |
| HpyAIV | 6 | Sequence analysis | This work | P | 1 |
| HpyAORF483P | 4 | Sequence analysis | This work | P | 1 |
| RdepTB3ORF14P | 4 | Sequence analysis | This work | P | 1 |
| AgeI | 4 | Sequence analysis | This work | P | 1 |
| SacI | 3 | Sequence analysis | This work | P | 1 |
| MspA1I | 3 | Sequence analysis | This work | P | 1 |
| Mae7806ORF3417P | 3 | Sequence analysis | This work | P | 1 |
| PhoI | 2 | Sequence analysis | This work | P | 1 |
| AvaBORF4359P | 2 | Sequence analysis | This work | P | 1 |
| CfrBI | 2 | Sequence analysis | This work | P | 1 |
| MjaIV | 1 | Sequence analysis | This work | P | 1 |
| HinfI | 1 | Sequence analysis | This work | P | 1 |
| B) HNH superfamily | |||||
| KpnI | 2 | Mutagenesis | ( 37 ) | P | 3 |
| MnlI | 117 | Mutagenesis | ( 44 ) | S | 3 |
| HphI | 282 | Mutagenesis | ( 49 ) | S | 3 |
| Eco31I | 11 | Mutagenesis | ( 117 ) | S | 3 |
| NlaIII | 82 | Sequence analysis | ( 34 , 35 ) | P | 2 |
| MboII | 7 | Sequence analysis | ( 34 , 35 ) | S | 2 |
| SapI | 4 | Sequence analysis | ( 34 , 35 ) | S | 2 |
| SphI | 4 | Sequence analysis | ( 34 , 35 ) | P | 2 |
| NspI | 5 | Sequence analysis | ( 35 ) | P | 2 |
| Hin4II | 12 | Sequence analysis | ( 41 ) | S | 2 |
| LcaA2P | 229 | Sequence analysis | This work | ? | 2 |
| Hpy99I | 121 | Sequence analysis | This work | P | 2 |
| Mae7806ORF5066P | 13 | Sequence analysis | This work | P | 2 |
| PacI | 4 | Sequence analysis | This work | P | 1 |
| C) PLD superfamily | |||||
| BfiI | 3 | X-ray BfiI | ( 36 ) | S | 4 |
| NgoAVII | 50 | Sequence analysis | This work | P | 2 |
| D) GIY-YIG superfamily | |||||
| Eco29kI | 10 | Mutagenesis Eco29kI | ( 38 ) | P | 3 |
| Hpy188I | 23 | Sequence analysis | Mikihiko Kawai (University of Tokyo), personal communication | P | 2 |
| E) HALFPIPE superfamily | |||||
| PabI | 8 | X-ray PabI | ( 40 ) | P | 4 |
| Family name | Number of members | Subtype | Reliability |
|---|---|---|---|
| F) Unknown superfamily | |||
| LlaBIIP | 66 | C | – |
| XcaVORF2165P | 52 | C | – |
| AnipAL1ORF76P | 46 | C | – |
| NspJCORF4770P | 42 | C | – |
| AauTCORF286P | 23 | C | – |
| HpaII | 24 | P,E | – |
| CglORF706P | 21 | C | – |
| DpnI | 20 | M | – |
| MteORF2238BP | 18 | C | – |
| Fsp4HI | 18 | P | – |
| NmeAIP | 18 | P | – |
| BseRI | 17 | C,S | – |
| BtlORF114P | 17 | C | – |
| BweKORF2188P | 13 | P | – |
| EcoUTORF4938P | 12 | P | – |
| HaeII | 11 | P | – |
| MgiORF5513P | 11 | C | – |
| CviJI | 10 | P | – |
| HgiDI | 10 | P | – |
| AvaIII | 9 | P | – |
| TerORF950P | 8 | ? | – |
| RshI | 6 | P | – |
| ApeKI | 6 | P | – |
| LlaBI | 6 | P | – |
| XmnI | 6 | P | – |
| HaeIII | 5 | P | – |
| BstXI | 5 | P | – |
| SuaMcrB2P | 5 | ? | – |
| AccI | 5 | P | – |
| LxxORF2510P | 5 | ? | – |
| LweSORF291P | 4 | P | – |
| HgaI | 4 | S | – |
| BhaI | 4 | S | – |
| CglP6P | 3 | P | – |
| NheI | 2 | P | – |
| GviORF2740P | 2 | C | – |
| NcoI | 2 | P | – |
| CviAII | 2 | P | – |
| AluI | 2 | P | – |
| Rca13841ORF3082P | 2 | C | – |
| AatII | 2 | P | – |
| TspMI | 2 | P | – |
| LlaIA | 1 | ? | – |
| Lmo19115ORF1P | 1 | ? | – |
| RspRSORF4066P | 1 | P | – |
| BsuMIA | 1 | P | – |
| EsaSS1430P | 1 | C | – |
| BssHII | 1 | P | – |
| EsaNPORF9P | 1 | S | – |
| TspRI | 1 | P | – |
| BalI | 1 | P | – |
| AhdI | 1 | P | – |
| BsuRI | 1 | P | – |
| EsaSS157P | 1 | ? | – |
| CviQI | 1 | P | – |
| BlopNAC1P | 1 | P | – |
| BslIA | 1 | P | – |
| SspI | 1 | P | – |
| SonORF4P | 1 | P | – |
| BsrGI | 1 | P | – |
| BslIB | 1 | P | – |
| GurRORF3275P | 1 | P | – |
| FpsJIPORF858P | 1 | P | – |
| HgiDII | 1 | P | – |
| BseMII | 1 | S | – |
| BspLU11III | 1 | C,S | – |
| CwaWHORF3980P | 1 | ? | – |
| HauORF1126P | 1 | P | – |
| Mae7806ORF1639AP | 1 | C | – |
| PcaJCMORF748P | 1 | C | – |
| PmoSJORF1273P | 1 | C | – |
| UmeRCIORF389P | 1 | C | – |
| TmaI | 1 | P | – |
| Family name | Number of members | Subtype | Reliability |
|---|---|---|---|
| F) Unknown superfamily | |||
| LlaBIIP | 66 | C | – |
| XcaVORF2165P | 52 | C | – |
| AnipAL1ORF76P | 46 | C | – |
| NspJCORF4770P | 42 | C | – |
| AauTCORF286P | 23 | C | – |
| HpaII | 24 | P,E | – |
| CglORF706P | 21 | C | – |
| DpnI | 20 | M | – |
| MteORF2238BP | 18 | C | – |
| Fsp4HI | 18 | P | – |
| NmeAIP | 18 | P | – |
| BseRI | 17 | C,S | – |
| BtlORF114P | 17 | C | – |
| BweKORF2188P | 13 | P | – |
| EcoUTORF4938P | 12 | P | – |
| HaeII | 11 | P | – |
| MgiORF5513P | 11 | C | – |
| CviJI | 10 | P | – |
| HgiDI | 10 | P | – |
| AvaIII | 9 | P | – |
| TerORF950P | 8 | ? | – |
| RshI | 6 | P | – |
| ApeKI | 6 | P | – |
| LlaBI | 6 | P | – |
| XmnI | 6 | P | – |
| HaeIII | 5 | P | – |
| BstXI | 5 | P | – |
| SuaMcrB2P | 5 | ? | – |
| AccI | 5 | P | – |
| LxxORF2510P | 5 | ? | – |
| LweSORF291P | 4 | P | – |
| HgaI | 4 | S | – |
| BhaI | 4 | S | – |
| CglP6P | 3 | P | – |
| NheI | 2 | P | – |
| GviORF2740P | 2 | C | – |
| NcoI | 2 | P | – |
| CviAII | 2 | P | – |
| AluI | 2 | P | – |
| Rca13841ORF3082P | 2 | C | – |
| AatII | 2 | P | – |
| TspMI | 2 | P | – |
| LlaIA | 1 | ? | – |
| Lmo19115ORF1P | 1 | ? | – |
| RspRSORF4066P | 1 | P | – |
| BsuMIA | 1 | P | – |
| EsaSS1430P | 1 | C | – |
| BssHII | 1 | P | – |
| EsaNPORF9P | 1 | S | – |
| TspRI | 1 | P | – |
| BalI | 1 | P | – |
| AhdI | 1 | P | – |
| BsuRI | 1 | P | – |
| EsaSS157P | 1 | ? | – |
| CviQI | 1 | P | – |
| BlopNAC1P | 1 | P | – |
| BslIA | 1 | P | – |
| SspI | 1 | P | – |
| SonORF4P | 1 | P | – |
| BsrGI | 1 | P | – |
| BslIB | 1 | P | – |
| GurRORF3275P | 1 | P | – |
| FpsJIPORF858P | 1 | P | – |
| HgiDII | 1 | P | – |
| BseMII | 1 | S | – |
| BspLU11III | 1 | C,S | – |
| CwaWHORF3980P | 1 | ? | – |
| HauORF1126P | 1 | P | – |
| Mae7806ORF1639AP | 1 | C | – |
| PcaJCMORF748P | 1 | C | – |
| PmoSJORF1273P | 1 | C | – |
| UmeRCIORF389P | 1 | C | – |
| TmaI | 1 | P | – |
Families are named after the subjectively chosen most representative and/or best studied candidate. The number of members and subtypes of its members (according to REBASE, ‘?’ means no subtype information present) are indicated. The description of subtypes can be found in ref. ( 1 ). Very briefly, P indicates orthodox dimeric enzymes that recognize a single palindromic site, S indicates enzymes that cut at a fixed distance from an asymmetric site, E indicates enzymes that require an additional effector site, F indicates tetrameric enzymes that cut two sites, C indicates enzymes comprising REase and MTase activities in the same polypeptide and M indicates enzymes that cleave modified DNA. The type of evidence supporting the assignment is described, including the type of analysis and references to the key publication(s). Our subjective assessment of the confidence level for different 3D-fold assignments is indicated: 4 indicates certain, high-resolution experimental information (e.g. from crystallography), 3 indicates prediction supported by low-resolution experimental data (e.g. mutagenesis), 2 indicates confident, but purely theoretical prediction that remains to be tested experimentally, 1 indicates purely theoretical prediction with some level of uncertainty (e.g. poor scores, problems with identification of a full set of catalytic residues based on the model etc.).
3D-fold classification for Type II REase subfamilies
| Family name | Number of members | Type of evidence | Reference | Subtype | Reliability |
|---|---|---|---|---|---|
| A) PD-(D/E)XK superfamily | |||||
| EcoRI | 14 | X-ray EcoRI | ( 14 ) | P | 4 |
| EcoRV | 14 | X-ray EcoRV | ( 15 ) | P | 4 |
| PvuII | 5 | X-ray PvuII | ( 90 ) | P | 4 |
| BamHI | 5 | X-ray BamHI | ( 91 ) | P | 4 |
| Cfr10I | 6 | X-ray Cfr10I | ( 26 ) | P,F | 4 |
| BglI | 17 | X-ray BglI | ( 31 ) | P,F | 4 |
| FokI | 9 | X-ray FokI | ( 92 ) | S | 4 |
| MunI | 6 | X-ray MunI | ( 93 ) | P | 4 |
| BglII | 17 | X-ray BglII | ( 94 ) | P | 4 |
| NgoMIV | 11 | X-ray NgoMIV | ( 95 ) | F,P | 4 |
| NaeI | 7 | X-ray NaeI | ( 96 ) | E | 4 |
| BsoBI | 8 | X-ray BsoBI | ( 25 ) | P | 4 |
| EcoRII | 45 | X-ray EcoRII | ( 97 ) | P,E | 4 |
| MspI | 4 | X-ray MspI | ( 98 ) | P | 4 |
| MlyI | 45 | X-ray N.BspD6I | ( 99 ) | S,P | 4 |
| EcoO109I | 10 | X-ray EcoO109I | ( 100 ) | P | 4 |
| HinP1I | 6 | X-ray HinP1I | ( 88 ) | P | 4 |
| SdaI | 43 | X-ray SdaI | ( 28 ) | P | 4 |
| HincII | 5 | X-ray HincII | ( 101 ) | P | 4 |
| MvaI | 36 | X-ray MvaI | ( 76 , 102 ) | P | 4 |
| NotI | 23 | X-ray NotI | Lambert et al . to be published (PDB 3brv) | P | 4 |
| BcgI | 31 | Mutagenesis BcgI | ( 103 ) | C | 3 |
| TaqI | 6 | Mutagenesis TaqI | ( 104 ) | P | 3 |
| HindIII | 11 | Mutagenesis HindIII | ( 105 ) | P | 3 |
| Eco57I | 37 | Mutagenesis Eco57I | ( 106 ) | C | 3 |
| MboI | 40 | Mutagenesis MboI | ( 43 ) | P | 3 |
| Bsp6I | 15 | Mutagenesis Bsp6I | ( 47 ) | P | 3 |
| NlaIV | 10 | Mutagenesis, CD NlaIV | ( 46 ) | P | 3 |
| Mva1269I | 3 | Mutagenesis Mva1269I | ( 45 ) | S | 3 |
| HpaI | 1 | Mutagenesis HpaI | ( 48 ) | P | 3 |
| BpuJI | 73 | Mutagenesis BpuJI | ( 107 ) | S | 3 |
| BtsIA | 1 | Mutagenesis BtsIA | ( 108 ) | S | 3 |
| BtsIB | 1 | Mutagenesis BtsIB | ( 108 ) | S | 3 |
| R2.BsrDI | 3 | Mutagenesis R2.BsrDI | ( 108 ) | S | 3 |
| R1.BsrDI | 2 | Mutagenesis R1.BsrDI | ( 108 ) | S | 3 |
| NgoPII | 21 | Mutagenesis NgoPII | J.M.B. and coworkers, unpublished data | P | 3 |
| XbaI | 15 | Sequence analysis | ( 109 ) | P | 2 |
| SalI | 15 | Sequence analysis | ( 109 ) | P | 2 |
| XmaI | 9 | Sequence analysis | ( 110 ) | E,P | 2 |
| Bpu10IB | 18 | Sequence analysis | ( 111 ) | S,P | 2 |
| DdeI | 5 | Sequence analysis | ( 111 ) | P | 2 |
| PvuRts1 | 46 | Sequence analysis | ( 22 , 34 ) | ? | 2 |
| BanI | 8 | Sequence analysis | ( 22 , 112 ) | P | 2 |
| LlaBIII | 152 | Sequence analysis | ( 113 ) | C | 2 |
| Sau3AI | 36 | Sequence analysis | ( 22 , 114 ) | E,P | 2 |
| HaeIV | 20 | Sequence analysis | ( 115 ) | C | 2 |
| MjaI | 6 | Sequence analysis | ( 22 ) | P | 2 |
| ApaLI | 1 | Sequence analysis | ( 22 ) | P | 2 |
| Kpn2I | 7 | Sequence analysis | ( 27 ) | P | 2 |
| R2.LlaJI | 16 | Sequence analysis | ( 116 ) | P | 2 |
| ScrFI | 13 | Sequence analysis | ( 76 ) | P | 2 |
| TerMcrCP | 112 | Sequence analysis | This work | ? | 2 |
| CstMI | 76 | Sequence analysis | This work | C | 2 |
| TaqII | 35 | Sequence analysis | This work | C,S | 2 |
| HgiEI | 25 | Sequence analysis | This work | P | 2 |
| Hpy8I | 20 | Sequence analysis | This work | P | 2 |
| VeiORF1182P | 19 | Sequence analysis | This work | P | 2 |
| AvaII | 18 | Sequence analysis | This work | P | 2 |
| XhoI | 16 | Sequence analysis | This work | P | 2 |
| Hpy99II | 16 | Sequence analysis | This work | P | 2 |
| VeiORF1308P | 14 | Sequence analysis | This work | C | 2 |
| Sho27844P | 14 | Sequence analysis | This work | S | 2 |
| Sau96I | 13 | Sequence analysis | This work | P | 2 |
| NspV | 12 | Sequence analysis | This work | P | 2 |
| McaTI | 11 | Sequence analysis | This work | P | 2 |
| Hpy99VIIIP | 10 | Sequence analysis | This work | P | 2 |
| MseI | 9 | Sequence analysis | This work | P | 2 |
| SuaI | 6 | Sequence analysis | This work | P | 2 |
| HpyCH4V | 5 | Sequence analysis | This work | P | 2 |
| SnaBI | 5 | Sequence analysis | This work | P | 2 |
| AboORF2079P | 4 | Sequence analysis | This work | P | 2 |
| MjaV | 4 | Sequence analysis | This work | P | 2 |
| Hpy99IV | 4 | Sequence analysis | This work | P | 2 |
| HpyHORF1023P | 4 | Sequence analysis | This work | P | 2 |
| DolHORF3097P | 4 | Sequence analysis | This work | ? | 2 |
| ThaI | 3 | Sequence analysis | This work | P | 2 |
| HhaII | 2 | Sequence analysis | This work | P | 2 |
| TfiI | 1 | Sequence analysis | This work | P | 2 |
| Sse9I | 1 | Sequence analysis | This work | P | 2 |
| BssSI | 1 | Sequence analysis | This work | S | 1 |
| NmeDIP | 46 | Sequence analysis | This work | ? | 1 |
| CthORFS34P | 30 | Sequence analysis | This work | P | 1 |
| RsaI | 11 | Sequence analysis | This work | P | 1 |
| CviAI | 9 | Sequence analysis | This work | P | 1 |
| SmaI | 8 | Sequence analysis | This work | P | 1 |
| LlaDI | 7 | Sequence analysis | This work | P | 1 |
| HpyAIV | 6 | Sequence analysis | This work | P | 1 |
| HpyAORF483P | 4 | Sequence analysis | This work | P | 1 |
| RdepTB3ORF14P | 4 | Sequence analysis | This work | P | 1 |
| AgeI | 4 | Sequence analysis | This work | P | 1 |
| SacI | 3 | Sequence analysis | This work | P | 1 |
| MspA1I | 3 | Sequence analysis | This work | P | 1 |
| Mae7806ORF3417P | 3 | Sequence analysis | This work | P | 1 |
| PhoI | 2 | Sequence analysis | This work | P | 1 |
| AvaBORF4359P | 2 | Sequence analysis | This work | P | 1 |
| CfrBI | 2 | Sequence analysis | This work | P | 1 |
| MjaIV | 1 | Sequence analysis | This work | P | 1 |
| HinfI | 1 | Sequence analysis | This work | P | 1 |
| B) HNH superfamily | |||||
| KpnI | 2 | Mutagenesis | ( 37 ) | P | 3 |
| MnlI | 117 | Mutagenesis | ( 44 ) | S | 3 |
| HphI | 282 | Mutagenesis | ( 49 ) | S | 3 |
| Eco31I | 11 | Mutagenesis | ( 117 ) | S | 3 |
| NlaIII | 82 | Sequence analysis | ( 34 , 35 ) | P | 2 |
| MboII | 7 | Sequence analysis | ( 34 , 35 ) | S | 2 |
| SapI | 4 | Sequence analysis | ( 34 , 35 ) | S | 2 |
| SphI | 4 | Sequence analysis | ( 34 , 35 ) | P | 2 |
| NspI | 5 | Sequence analysis | ( 35 ) | P | 2 |
| Hin4II | 12 | Sequence analysis | ( 41 ) | S | 2 |
| LcaA2P | 229 | Sequence analysis | This work | ? | 2 |
| Hpy99I | 121 | Sequence analysis | This work | P | 2 |
| Mae7806ORF5066P | 13 | Sequence analysis | This work | P | 2 |
| PacI | 4 | Sequence analysis | This work | P | 1 |
| C) PLD superfamily | |||||
| BfiI | 3 | X-ray BfiI | ( 36 ) | S | 4 |
| NgoAVII | 50 | Sequence analysis | This work | P | 2 |
| D) GIY-YIG superfamily | |||||
| Eco29kI | 10 | Mutagenesis Eco29kI | ( 38 ) | P | 3 |
| Hpy188I | 23 | Sequence analysis | Mikihiko Kawai (University of Tokyo), personal communication | P | 2 |
| E) HALFPIPE superfamily | |||||
| PabI | 8 | X-ray PabI | ( 40 ) | P | 4 |
| Family name | Number of members | Type of evidence | Reference | Subtype | Reliability |
|---|---|---|---|---|---|
| A) PD-(D/E)XK superfamily | |||||
| EcoRI | 14 | X-ray EcoRI | ( 14 ) | P | 4 |
| EcoRV | 14 | X-ray EcoRV | ( 15 ) | P | 4 |
| PvuII | 5 | X-ray PvuII | ( 90 ) | P | 4 |
| BamHI | 5 | X-ray BamHI | ( 91 ) | P | 4 |
| Cfr10I | 6 | X-ray Cfr10I | ( 26 ) | P,F | 4 |
| BglI | 17 | X-ray BglI | ( 31 ) | P,F | 4 |
| FokI | 9 | X-ray FokI | ( 92 ) | S | 4 |
| MunI | 6 | X-ray MunI | ( 93 ) | P | 4 |
| BglII | 17 | X-ray BglII | ( 94 ) | P | 4 |
| NgoMIV | 11 | X-ray NgoMIV | ( 95 ) | F,P | 4 |
| NaeI | 7 | X-ray NaeI | ( 96 ) | E | 4 |
| BsoBI | 8 | X-ray BsoBI | ( 25 ) | P | 4 |
| EcoRII | 45 | X-ray EcoRII | ( 97 ) | P,E | 4 |
| MspI | 4 | X-ray MspI | ( 98 ) | P | 4 |
| MlyI | 45 | X-ray N.BspD6I | ( 99 ) | S,P | 4 |
| EcoO109I | 10 | X-ray EcoO109I | ( 100 ) | P | 4 |
| HinP1I | 6 | X-ray HinP1I | ( 88 ) | P | 4 |
| SdaI | 43 | X-ray SdaI | ( 28 ) | P | 4 |
| HincII | 5 | X-ray HincII | ( 101 ) | P | 4 |
| MvaI | 36 | X-ray MvaI | ( 76 , 102 ) | P | 4 |
| NotI | 23 | X-ray NotI | Lambert et al . to be published (PDB 3brv) | P | 4 |
| BcgI | 31 | Mutagenesis BcgI | ( 103 ) | C | 3 |
| TaqI | 6 | Mutagenesis TaqI | ( 104 ) | P | 3 |
| HindIII | 11 | Mutagenesis HindIII | ( 105 ) | P | 3 |
| Eco57I | 37 | Mutagenesis Eco57I | ( 106 ) | C | 3 |
| MboI | 40 | Mutagenesis MboI | ( 43 ) | P | 3 |
| Bsp6I | 15 | Mutagenesis Bsp6I | ( 47 ) | P | 3 |
| NlaIV | 10 | Mutagenesis, CD NlaIV | ( 46 ) | P | 3 |
| Mva1269I | 3 | Mutagenesis Mva1269I | ( 45 ) | S | 3 |
| HpaI | 1 | Mutagenesis HpaI | ( 48 ) | P | 3 |
| BpuJI | 73 | Mutagenesis BpuJI | ( 107 ) | S | 3 |
| BtsIA | 1 | Mutagenesis BtsIA | ( 108 ) | S | 3 |
| BtsIB | 1 | Mutagenesis BtsIB | ( 108 ) | S | 3 |
| R2.BsrDI | 3 | Mutagenesis R2.BsrDI | ( 108 ) | S | 3 |
| R1.BsrDI | 2 | Mutagenesis R1.BsrDI | ( 108 ) | S | 3 |
| NgoPII | 21 | Mutagenesis NgoPII | J.M.B. and coworkers, unpublished data | P | 3 |
| XbaI | 15 | Sequence analysis | ( 109 ) | P | 2 |
| SalI | 15 | Sequence analysis | ( 109 ) | P | 2 |
| XmaI | 9 | Sequence analysis | ( 110 ) | E,P | 2 |
| Bpu10IB | 18 | Sequence analysis | ( 111 ) | S,P | 2 |
| DdeI | 5 | Sequence analysis | ( 111 ) | P | 2 |
| PvuRts1 | 46 | Sequence analysis | ( 22 , 34 ) | ? | 2 |
| BanI | 8 | Sequence analysis | ( 22 , 112 ) | P | 2 |
| LlaBIII | 152 | Sequence analysis | ( 113 ) | C | 2 |
| Sau3AI | 36 | Sequence analysis | ( 22 , 114 ) | E,P | 2 |
| HaeIV | 20 | Sequence analysis | ( 115 ) | C | 2 |
| MjaI | 6 | Sequence analysis | ( 22 ) | P | 2 |
| ApaLI | 1 | Sequence analysis | ( 22 ) | P | 2 |
| Kpn2I | 7 | Sequence analysis | ( 27 ) | P | 2 |
| R2.LlaJI | 16 | Sequence analysis | ( 116 ) | P | 2 |
| ScrFI | 13 | Sequence analysis | ( 76 ) | P | 2 |
| TerMcrCP | 112 | Sequence analysis | This work | ? | 2 |
| CstMI | 76 | Sequence analysis | This work | C | 2 |
| TaqII | 35 | Sequence analysis | This work | C,S | 2 |
| HgiEI | 25 | Sequence analysis | This work | P | 2 |
| Hpy8I | 20 | Sequence analysis | This work | P | 2 |
| VeiORF1182P | 19 | Sequence analysis | This work | P | 2 |
| AvaII | 18 | Sequence analysis | This work | P | 2 |
| XhoI | 16 | Sequence analysis | This work | P | 2 |
| Hpy99II | 16 | Sequence analysis | This work | P | 2 |
| VeiORF1308P | 14 | Sequence analysis | This work | C | 2 |
| Sho27844P | 14 | Sequence analysis | This work | S | 2 |
| Sau96I | 13 | Sequence analysis | This work | P | 2 |
| NspV | 12 | Sequence analysis | This work | P | 2 |
| McaTI | 11 | Sequence analysis | This work | P | 2 |
| Hpy99VIIIP | 10 | Sequence analysis | This work | P | 2 |
| MseI | 9 | Sequence analysis | This work | P | 2 |
| SuaI | 6 | Sequence analysis | This work | P | 2 |
| HpyCH4V | 5 | Sequence analysis | This work | P | 2 |
| SnaBI | 5 | Sequence analysis | This work | P | 2 |
| AboORF2079P | 4 | Sequence analysis | This work | P | 2 |
| MjaV | 4 | Sequence analysis | This work | P | 2 |
| Hpy99IV | 4 | Sequence analysis | This work | P | 2 |
| HpyHORF1023P | 4 | Sequence analysis | This work | P | 2 |
| DolHORF3097P | 4 | Sequence analysis | This work | ? | 2 |
| ThaI | 3 | Sequence analysis | This work | P | 2 |
| HhaII | 2 | Sequence analysis | This work | P | 2 |
| TfiI | 1 | Sequence analysis | This work | P | 2 |
| Sse9I | 1 | Sequence analysis | This work | P | 2 |
| BssSI | 1 | Sequence analysis | This work | S | 1 |
| NmeDIP | 46 | Sequence analysis | This work | ? | 1 |
| CthORFS34P | 30 | Sequence analysis | This work | P | 1 |
| RsaI | 11 | Sequence analysis | This work | P | 1 |
| CviAI | 9 | Sequence analysis | This work | P | 1 |
| SmaI | 8 | Sequence analysis | This work | P | 1 |
| LlaDI | 7 | Sequence analysis | This work | P | 1 |
| HpyAIV | 6 | Sequence analysis | This work | P | 1 |
| HpyAORF483P | 4 | Sequence analysis | This work | P | 1 |
| RdepTB3ORF14P | 4 | Sequence analysis | This work | P | 1 |
| AgeI | 4 | Sequence analysis | This work | P | 1 |
| SacI | 3 | Sequence analysis | This work | P | 1 |
| MspA1I | 3 | Sequence analysis | This work | P | 1 |
| Mae7806ORF3417P | 3 | Sequence analysis | This work | P | 1 |
| PhoI | 2 | Sequence analysis | This work | P | 1 |
| AvaBORF4359P | 2 | Sequence analysis | This work | P | 1 |
| CfrBI | 2 | Sequence analysis | This work | P | 1 |
| MjaIV | 1 | Sequence analysis | This work | P | 1 |
| HinfI | 1 | Sequence analysis | This work | P | 1 |
| B) HNH superfamily | |||||
| KpnI | 2 | Mutagenesis | ( 37 ) | P | 3 |
| MnlI | 117 | Mutagenesis | ( 44 ) | S | 3 |
| HphI | 282 | Mutagenesis | ( 49 ) | S | 3 |
| Eco31I | 11 | Mutagenesis | ( 117 ) | S | 3 |
| NlaIII | 82 | Sequence analysis | ( 34 , 35 ) | P | 2 |
| MboII | 7 | Sequence analysis | ( 34 , 35 ) | S | 2 |
| SapI | 4 | Sequence analysis | ( 34 , 35 ) | S | 2 |
| SphI | 4 | Sequence analysis | ( 34 , 35 ) | P | 2 |
| NspI | 5 | Sequence analysis | ( 35 ) | P | 2 |
| Hin4II | 12 | Sequence analysis | ( 41 ) | S | 2 |
| LcaA2P | 229 | Sequence analysis | This work | ? | 2 |
| Hpy99I | 121 | Sequence analysis | This work | P | 2 |
| Mae7806ORF5066P | 13 | Sequence analysis | This work | P | 2 |
| PacI | 4 | Sequence analysis | This work | P | 1 |
| C) PLD superfamily | |||||
| BfiI | 3 | X-ray BfiI | ( 36 ) | S | 4 |
| NgoAVII | 50 | Sequence analysis | This work | P | 2 |
| D) GIY-YIG superfamily | |||||
| Eco29kI | 10 | Mutagenesis Eco29kI | ( 38 ) | P | 3 |
| Hpy188I | 23 | Sequence analysis | Mikihiko Kawai (University of Tokyo), personal communication | P | 2 |
| E) HALFPIPE superfamily | |||||
| PabI | 8 | X-ray PabI | ( 40 ) | P | 4 |
| Family name | Number of members | Subtype | Reliability |
|---|---|---|---|
| F) Unknown superfamily | |||
| LlaBIIP | 66 | C | – |
| XcaVORF2165P | 52 | C | – |
| AnipAL1ORF76P | 46 | C | – |
| NspJCORF4770P | 42 | C | – |
| AauTCORF286P | 23 | C | – |
| HpaII | 24 | P,E | – |
| CglORF706P | 21 | C | – |
| DpnI | 20 | M | – |
| MteORF2238BP | 18 | C | – |
| Fsp4HI | 18 | P | – |
| NmeAIP | 18 | P | – |
| BseRI | 17 | C,S | – |
| BtlORF114P | 17 | C | – |
| BweKORF2188P | 13 | P | – |
| EcoUTORF4938P | 12 | P | – |
| HaeII | 11 | P | – |
| MgiORF5513P | 11 | C | – |
| CviJI | 10 | P | – |
| HgiDI | 10 | P | – |
| AvaIII | 9 | P | – |
| TerORF950P | 8 | ? | – |
| RshI | 6 | P | – |
| ApeKI | 6 | P | – |
| LlaBI | 6 | P | – |
| XmnI | 6 | P | – |
| HaeIII | 5 | P | – |
| BstXI | 5 | P | – |
| SuaMcrB2P | 5 | ? | – |
| AccI | 5 | P | – |
| LxxORF2510P | 5 | ? | – |
| LweSORF291P | 4 | P | – |
| HgaI | 4 | S | – |
| BhaI | 4 | S | – |
| CglP6P | 3 | P | – |
| NheI | 2 | P | – |
| GviORF2740P | 2 | C | – |
| NcoI | 2 | P | – |
| CviAII | 2 | P | – |
| AluI | 2 | P | – |
| Rca13841ORF3082P | 2 | C | – |
| AatII | 2 | P | – |
| TspMI | 2 | P | – |
| LlaIA | 1 | ? | – |
| Lmo19115ORF1P | 1 | ? | – |
| RspRSORF4066P | 1 | P | – |
| BsuMIA | 1 | P | – |
| EsaSS1430P | 1 | C | – |
| BssHII | 1 | P | – |
| EsaNPORF9P | 1 | S | – |
| TspRI | 1 | P | – |
| BalI | 1 | P | – |
| AhdI | 1 | P | – |
| BsuRI | 1 | P | – |
| EsaSS157P | 1 | ? | – |
| CviQI | 1 | P | – |
| BlopNAC1P | 1 | P | – |
| BslIA | 1 | P | – |
| SspI | 1 | P | – |
| SonORF4P | 1 | P | – |
| BsrGI | 1 | P | – |
| BslIB | 1 | P | – |
| GurRORF3275P | 1 | P | – |
| FpsJIPORF858P | 1 | P | – |
| HgiDII | 1 | P | – |
| BseMII | 1 | S | – |
| BspLU11III | 1 | C,S | – |
| CwaWHORF3980P | 1 | ? | – |
| HauORF1126P | 1 | P | – |
| Mae7806ORF1639AP | 1 | C | – |
| PcaJCMORF748P | 1 | C | – |
| PmoSJORF1273P | 1 | C | – |
| UmeRCIORF389P | 1 | C | – |
| TmaI | 1 | P | – |
| Family name | Number of members | Subtype | Reliability |
|---|---|---|---|
| F) Unknown superfamily | |||
| LlaBIIP | 66 | C | – |
| XcaVORF2165P | 52 | C | – |
| AnipAL1ORF76P | 46 | C | – |
| NspJCORF4770P | 42 | C | – |
| AauTCORF286P | 23 | C | – |
| HpaII | 24 | P,E | – |
| CglORF706P | 21 | C | – |
| DpnI | 20 | M | – |
| MteORF2238BP | 18 | C | – |
| Fsp4HI | 18 | P | – |
| NmeAIP | 18 | P | – |
| BseRI | 17 | C,S | – |
| BtlORF114P | 17 | C | – |
| BweKORF2188P | 13 | P | – |
| EcoUTORF4938P | 12 | P | – |
| HaeII | 11 | P | – |
| MgiORF5513P | 11 | C | – |
| CviJI | 10 | P | – |
| HgiDI | 10 | P | – |
| AvaIII | 9 | P | – |
| TerORF950P | 8 | ? | – |
| RshI | 6 | P | – |
| ApeKI | 6 | P | – |
| LlaBI | 6 | P | – |
| XmnI | 6 | P | – |
| HaeIII | 5 | P | – |
| BstXI | 5 | P | – |
| SuaMcrB2P | 5 | ? | – |
| AccI | 5 | P | – |
| LxxORF2510P | 5 | ? | – |
| LweSORF291P | 4 | P | – |
| HgaI | 4 | S | – |
| BhaI | 4 | S | – |
| CglP6P | 3 | P | – |
| NheI | 2 | P | – |
| GviORF2740P | 2 | C | – |
| NcoI | 2 | P | – |
| CviAII | 2 | P | – |
| AluI | 2 | P | – |
| Rca13841ORF3082P | 2 | C | – |
| AatII | 2 | P | – |
| TspMI | 2 | P | – |
| LlaIA | 1 | ? | – |
| Lmo19115ORF1P | 1 | ? | – |
| RspRSORF4066P | 1 | P | – |
| BsuMIA | 1 | P | – |
| EsaSS1430P | 1 | C | – |
| BssHII | 1 | P | – |
| EsaNPORF9P | 1 | S | – |
| TspRI | 1 | P | – |
| BalI | 1 | P | – |
| AhdI | 1 | P | – |
| BsuRI | 1 | P | – |
| EsaSS157P | 1 | ? | – |
| CviQI | 1 | P | – |
| BlopNAC1P | 1 | P | – |
| BslIA | 1 | P | – |
| SspI | 1 | P | – |
| SonORF4P | 1 | P | – |
| BsrGI | 1 | P | – |
| BslIB | 1 | P | – |
| GurRORF3275P | 1 | P | – |
| FpsJIPORF858P | 1 | P | – |
| HgiDII | 1 | P | – |
| BseMII | 1 | S | – |
| BspLU11III | 1 | C,S | – |
| CwaWHORF3980P | 1 | ? | – |
| HauORF1126P | 1 | P | – |
| Mae7806ORF1639AP | 1 | C | – |
| PcaJCMORF748P | 1 | C | – |
| PmoSJORF1273P | 1 | C | – |
| UmeRCIORF389P | 1 | C | – |
| TmaI | 1 | P | – |
Families are named after the subjectively chosen most representative and/or best studied candidate. The number of members and subtypes of its members (according to REBASE, ‘?’ means no subtype information present) are indicated. The description of subtypes can be found in ref. ( 1 ). Very briefly, P indicates orthodox dimeric enzymes that recognize a single palindromic site, S indicates enzymes that cut at a fixed distance from an asymmetric site, E indicates enzymes that require an additional effector site, F indicates tetrameric enzymes that cut two sites, C indicates enzymes comprising REase and MTase activities in the same polypeptide and M indicates enzymes that cleave modified DNA. The type of evidence supporting the assignment is described, including the type of analysis and references to the key publication(s). Our subjective assessment of the confidence level for different 3D-fold assignments is indicated: 4 indicates certain, high-resolution experimental information (e.g. from crystallography), 3 indicates prediction supported by low-resolution experimental data (e.g. mutagenesis), 2 indicates confident, but purely theoretical prediction that remains to be tested experimentally, 1 indicates purely theoretical prediction with some level of uncertainty (e.g. poor scores, problems with identification of a full set of catalytic residues based on the model etc.).
Analysis of domain architectures
3D fold assignment of nuclease domains together with assignment of non-nuclease domains enabled us to study the diversity of domain organization of confirmed and putative Type II REases. We found out that REases show great variety of possible compositions as we observed 50 different types of domain fusions and rearrangements ( Figure 7 ). The most frequently found domains in REases (apart from nuclease domains) are: MTase domains, variants of helix–turn–helix (HTH) DNA-binding domains (e.g. ‘winged helix’, wH) and different kinds of domains associated with helicase or ATPase functions (DEXD-box, GHKL). Interestingly, in seven subfamilies (e.g.: R.MboI, R.SdaI) MTase domains are present only in one or a few members. This observation suggests that translational fusions of REase and MTase domains occurred independently multiple times in the evolution, and has been facilitated by the frequent occurrence of REase and MTase domains in operons (i.e. transcriptional fusions).

A variety of primary structures (domain architectures on the sequence level) in confirmed and putative Type II REases. Sequences are aligned by their nuclease domains. Drawing in scale, length of PD-D(E)XK domain corresponds to 110 aa. Some very long sequences are broken for the clarity of presentation.
Characterization of selected subfamilies
Although a complete description of all new fold assignments and all domain organizations is beyond the limits of a single publication, we would like to describe in more detail the most interesting or most intriguing (in a few cases potentially controversial) new findings and predictions:
R.LlaBIIP: this long protein (1461 aa) appears to be a fusion of HsdR-like and HsdM-like subunits, comprising the putative ATP-dependent translocase and MTase modules. However, the N-terminal region appears to lack the PD-D(E)XK domain common to HsdR subunits. Instead, the N-terminus contains a putative helical domain HEPN found in nucleotidyltransferases (aa 1–130), and another putative domain (aa 130–250), which shows no sequence or secondary structure similarity to any known nuclease domains. It would be very interesting to test experimentally whether R.LlaBIIP (and in particular its unusual N-terminal region) exhibits a nuclease activity.
R.CviAI (GATC specific) ( 82 ) is predicted to be a PD-(D/E)XK superfamily member, yet it shows no obvious similarity to other GATC-specific enzymes (e.g. neither the R.MboI nor the R.Sau3AI subfamily). Thus, we predict that its substrate specificity represents a case of convergent evolution within the same structural scaffold, used multiple times to independently develop recognition of the same DNA sequence.
R.HgiDII contains two domains. As mentioned earlier, the N-terminal domain belongs to the GHKL superfamily, which includes e.g. the MutL enzyme involved in DNA mismatch repair [where MutH is the associated nuclease from the PD-(D/E)XK superfamily]. The C-terminal domain of R.HgiDII remains unassigned to any of the known REase folds, or in fact to any known fold or protein family. Interestingly, among four other subfamilies of REases that exhibit the GHKL domain in the N-terminus, one (R.VeiORF1182P) contains the C-terminal domain of the PD-(D/E)XK fold, and in three others (R.NmeAIP, R.EcoUTORF4938P and R.LweSORF291P) the C-terminal extension is apparently different from that in either R.HgiDII or R.VeiORF1182P. The C-terminal domain of R.NmeAIP shows significant similarity to an uncharacterized protein family dubbed ‘Hypoth_Ymh’ in PFAM (CDD search e-value 3e-22). On the other hand, the C-terminus of R.EcoUTORF4938P exhibits similarity to a signal transduction histidine kinase domain from the GHKL superfamily (CDD search e-value 3e-8) with conserved N, D, F and G motifs required for the catalytic activity ( 83 ). However, middle parts of both R.NmeAIP and R.EcoUTORF4938P remain unassigned to any known protein family and may contain additional domains. It will be very interesting to determine experimentally the role of the unassigned domains in GHKL-containing REases, and if they turn out to be responsible for the REase activity, they would constitute interesting candidates for new folds (and thereby, for structure determination by X-ray crystallography).
R.DpnI is a representative of a large family of REases that cleave GATC sequence only if the adenosine is methylated to m 6 A. We identified a putative Zn-binding region in the N-terminal part of their sequences (a conserved tetrad of Cys residues), but thus far we failed to determine its relationship to any known protein family or any known protein structure. Thus, we propose R.DpnI as an attractive target for structure determination by X-ray crystallography.
R.HphI: the analysis of this subfamily has been published ( 49 ), but we believe it is worth re-emphasizing that many members of this subfamily are most likely not Type II REases, as they lack MTase neighbors. Thus, it has been predicted that they might belong to another category of selfish nucleases, perhaps similar to homing endonucleases (HEases).
R.LcaA2P is a very close relative of HEases I-HmuI, I-HmuII and I-BsoI that act as nicking enzymes (BLAST e-value: 6e-11 with I-HmuI). Many other members of the LcaA2P family are therefore most likely HEases rather than Type II REases. On the other hand, it will be very interesting to determine whether R.LcaA2P is functional, and if it is—whether it acts as a nicking enzyme or as a ‘normal’ dsDNA nuclease and whether its activity can be inhibited by DNA methylation by the putative MTase encoded by the neighboring gene (M.LcaA2P). Should cleavage by LcaA2P be prevented by methylation, this enzyme may be considered an evolutionary intermediate between REases and HEases.
R.NgoAVIIP: sequences from this subfamily are confidently predicted to belong to the PLD superfamily, based on results of both FR and HHSEARCH analyses (e.g. FFAS score—23.9 to R.BfiI REase, HHSEARCH e-value 1.9e-14 to the profile of the R.BfiI subfamily). Moreover, analysis of the multiple sequence alignment reveals that putative catalytic residues are present. However, thus far efforts to detect the nuclease activity of R.NgoAVIIP have remained unsuccessful (V. Siksnys, IBT Vilnius, Lithuania, personal communication). Interestingly, the C-terminal domain of R.NgoAVIIP shows significant similarity (HHPRED e-value 7e-19) to the C-terminal domain of proteins from another nuclease subfamily (R.Fsp4HI), but they do not seem to share any detectable similarity in the catalytic domain. Thus far, we were unable to identify a known nuclease domain in R.Fsp4HI therefore we propose it as an interesting candidate for further experimental analysis. It would be worthwhile to identify catalytic residues in this nuclease and to check whether its mode of action resembles other REases or other enzymes from the PLD family.
Hypothetical protein SAV_2336 (gi:29828878): this very long protein (1667 aa) from Streptomyces avermitilis shows clear similarity to R.NaeI enzyme from the PD-D(E)XK superfamily in its C-terminus (aa 1359-1657 BLAST e-value 2e-28, alignment spanning the catalytic domain and the wH DNA-binding domain, suggesting that SAV_2336 binds two copies of the target DNA sequence, like R.NaeI). The N-terminal part of SAV_2336 sequence shows significant similarity to the VWA-type domain of unknown function from CO-oxidizing operons in bacteria (HHPRED e-value 1e-06). The central part of SAV_2336 is related to ATPase domains from the MalT family of transcription regulators (e-value <1e-06). This combination of multiple domains that may be involved not only in restriction, but also other aspects of nucleic acid metabolism, makes SAV_2336 an attractive target for experimental analyses.
R.PhoI shows remote similarity to archaeal Holliday junction resolvases from the PD-(D/E)XK superfamily (HHSEARCH hit to PFAM profile for archaeal Holliday junction resolvases Hjc with probability 58.4%) and an expected pattern of secondary structures associated with the catalytic core. However, its catalytic residues appear to be missing, as the PD-(D/E)XK motif is replaced by a PI-ERL variant. One possible explanation is that this protein exhibits an extreme case of catalytic residue migration to alternative locations in protein structure, as described earlier individually for the (D/E) residue in R.Cfr10I ( 84 ) and for the K residue in putative cyanobacterial nucleases ( 29 ) and in the R.SdaI subfamily ( 28 ). However, we found only one close homolog of R.PhoI, which provided insufficient information to predict catalytic residues based on residue conservation, and the preliminary model (data not shown) revealed no good candidates for a spatially reorganized active site. Thus, if R.PhoI is indeed active as a REase and if our 3D fold prediction is correct, it will be very interesting to determine its exact mode of action in vitro , especially its ability to catalyze the phosphodiester bond hydrolysis.
Some catalytically inactive mutants of Type IV REase McrA have been shown to restrict phage growth in vivo , presumably due to unproductive site-specific binding of the protein to a phage DNA, which could disrupt the phage development program at an early stage ( 85 ). It will be interesting to determine if REases, such as R.NgoAVIIP and perhaps also R.PhoI, that may be inactive as nucleases, can nevertheless function as REases in vivo , and if this activity can be inhibited by site-specific methylation by the cognate MTase.
Putative REases from the MjaORF1200P subfamily (four sequences in the REBASE set) are most likely RNA MTases rather than REases. According to the fold-recognition analysis [recently published as a separate article ( 86 )], these proteins show clear similarity to the SPOUT superfamily of RNA MTases, and they exhibit no additional domains or residues that would suggest them to act as REases. We suspect that they were (most likely incorrectly) assigned as Type II REases due to the genomic association of MjaORF1200P (ORF MJ1199) with a putative DNA:m 5 C MTase (M.MjaORF1200P).
Putative REases from the BceAUORF42P subfamily (three sequences in the REBASE set) are most likely Type III rather than Type II REases. In database searches they show clear similarity to Type III Res subunits and they are genetically associated with homologs of Type III MTase subunits.
R.SauN315ORF189P: members of this family show significant sequence similarity and similar domain organization to Type I REase HsdR proteins [e.g. e-value 3.5e-47 for a HHSEARCH alignment with the N terminus of R subunit of Type I restriction enzyme (HSDR_N) profile from the PFAM database].
R.EcoCH14P: sequence of this short protein (95 aa) is similar (HHPRED E-value 4e-05) to a C-terminal helical domain found in Type I REase HsdR proteins and implicated in binding to the Type I MTase complex rather than in the nuclease activity.
Distribution of 3D folds among confirmed and putative REases
From the aforementioned examples it is quite clear that the correctness of our estimated 3D fold distribution among REases is influenced not only by the quality of bioinformatic methods and the confidence in individual predictions or the availability of experimental data to support structural predictions, but also by the confidence in assignment of a given protein as a REase candidate. In particular, our analysis revealed a number of protein families comprising REases, in which some (or even most) members are most likely not REases, but fulfill some other function. Therefore, it is interesting to compare the distribution of 3D fold assignments in sets of experimentally validated Type II REases versus the expanded dataset comprising also putative enzymes.
To this end, we divided all sequences of Type II REases and their homologs into classes on the basis of their source:
CONFIRMED set: all sequences from REBASE with nuclease activity confirmed experimentally;
PREDICTED set: sequences from REBASE without direct experimental confirmation, excluding the data from environmental DNA sequencing projects;
NR set: homologs of sequences from sets 1–2 that are not present in REBASE, but were identified by us in the nr database at the NCBI; and
ENV set: putative REases in REBASE predicted from environmental DNA sequencing projects and identified by us in the environmental samples database (env_nr).
For each of these classes, we additionally created a ‘purged’ variant, from which we removed sequences above the level of 90% sequence identity. We used the following hierarchy of importance (from the most important to the least important): CONFIRMED set > PREDICTED set > NR set > ENV set. Thus, we removed all sequences from environmental samples not present in REBASE that exhibited ≥90% sequence identity to any of the sequences from ‘higher classes’, then the same was applied to all putative REases from nr and so on. Finally, if several genuine REases exhibited ≥90% sequence identity to each other, only one of them was retained. We have also considered an additional PUTATIVE set, which is a sum of PREDICTED, NR and ENV sets, thus contains all sequences that have NOT been experimentally confirmed to function as REases. Table 2 shows the number of sequences present in each of the original and purged datasets and in each fold. The fractions of enzymes assigned to different folds for the CONFIRMED set, PREDICTED set and for the PUTATIVE set, purged at maximum 90% identity, are shown in Figure 8 .
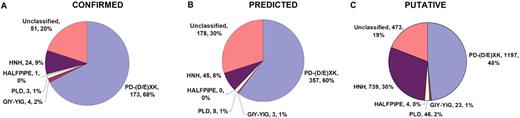
Fraction of enzymes assigned to different folds, purged at maximum 90% identity. ( A ) Confirmed REases from REBASE; ( B ) putative REASES from REBASE; ( C ) putative REASES from REBASE and all homologs found nonredundant (nr) and environmental samples (env_nr) NCBI database.
Number of endonucleases exhibiting different folds and different sources
| SET\Family | PD-(D/E)XK | GIY-YIG | PLD | HALFPIPE | HNH | Unclassified | Sum |
|---|---|---|---|---|---|---|---|
| CONFIRMED | 199 (173) | 6 (4) | 3 (3) | 1 (1) | 24 (24) | 56 (51) | 289 (256) |
| PREDICTED | 460 (357) | 7 (3) | 9 (8) | 0 (0) | 112 (45) | 203 (178) | 791 (591) |
| NR | 401 (358) | 3 (3) | 17 (15) | 7 (4) | 359 (322) | 137 (121) | 924 (823) |
| ENV | 508 (482) | 17 (17) | 24 (23) | 0 (0) | 398 (372) | 181 (174) | 1128 (1068) |
| PUTATIVE (PREDICTED + NR + ENV) | 1369 (1197) | 27 (23) | 50 (46) | 7 (4) | 869 (739) | 521 (473) | 2843 (2482) |
| ALL (CONFIRMED + PUTATIVE) | 1568 (1370) | 33 (27) | 53 (49) | 8 (5) | 893 (763) | 577 (524) | 3132 (2738) |
| SET\Family | PD-(D/E)XK | GIY-YIG | PLD | HALFPIPE | HNH | Unclassified | Sum |
|---|---|---|---|---|---|---|---|
| CONFIRMED | 199 (173) | 6 (4) | 3 (3) | 1 (1) | 24 (24) | 56 (51) | 289 (256) |
| PREDICTED | 460 (357) | 7 (3) | 9 (8) | 0 (0) | 112 (45) | 203 (178) | 791 (591) |
| NR | 401 (358) | 3 (3) | 17 (15) | 7 (4) | 359 (322) | 137 (121) | 924 (823) |
| ENV | 508 (482) | 17 (17) | 24 (23) | 0 (0) | 398 (372) | 181 (174) | 1128 (1068) |
| PUTATIVE (PREDICTED + NR + ENV) | 1369 (1197) | 27 (23) | 50 (46) | 7 (4) | 869 (739) | 521 (473) | 2843 (2482) |
| ALL (CONFIRMED + PUTATIVE) | 1568 (1370) | 33 (27) | 53 (49) | 8 (5) | 893 (763) | 577 (524) | 3132 (2738) |
CONFIRMED, all sequences from REBASE with nuclease activity confirmed experimentally; PREDICTED, sequences from REBASE without direct experimental confirmation, excluding the data from environmental DNA sequencing projects; NR, homologs of sequences from sets CONFIRMED and PREDICTED that are not present in REBASE, but were identified by us in the non-redundant (nr) database at the NCBI; ENV, Putative REases from environmental DNA sequencing projects, predicted by REBASE or by us in env_nr database. PUTATIVE = PREDICTED + NR + ENV. Values in parentheses correspond to the number of sequences with no more than 90% sequence identity.
Number of endonucleases exhibiting different folds and different sources
| SET\Family | PD-(D/E)XK | GIY-YIG | PLD | HALFPIPE | HNH | Unclassified | Sum |
|---|---|---|---|---|---|---|---|
| CONFIRMED | 199 (173) | 6 (4) | 3 (3) | 1 (1) | 24 (24) | 56 (51) | 289 (256) |
| PREDICTED | 460 (357) | 7 (3) | 9 (8) | 0 (0) | 112 (45) | 203 (178) | 791 (591) |
| NR | 401 (358) | 3 (3) | 17 (15) | 7 (4) | 359 (322) | 137 (121) | 924 (823) |
| ENV | 508 (482) | 17 (17) | 24 (23) | 0 (0) | 398 (372) | 181 (174) | 1128 (1068) |
| PUTATIVE (PREDICTED + NR + ENV) | 1369 (1197) | 27 (23) | 50 (46) | 7 (4) | 869 (739) | 521 (473) | 2843 (2482) |
| ALL (CONFIRMED + PUTATIVE) | 1568 (1370) | 33 (27) | 53 (49) | 8 (5) | 893 (763) | 577 (524) | 3132 (2738) |
| SET\Family | PD-(D/E)XK | GIY-YIG | PLD | HALFPIPE | HNH | Unclassified | Sum |
|---|---|---|---|---|---|---|---|
| CONFIRMED | 199 (173) | 6 (4) | 3 (3) | 1 (1) | 24 (24) | 56 (51) | 289 (256) |
| PREDICTED | 460 (357) | 7 (3) | 9 (8) | 0 (0) | 112 (45) | 203 (178) | 791 (591) |
| NR | 401 (358) | 3 (3) | 17 (15) | 7 (4) | 359 (322) | 137 (121) | 924 (823) |
| ENV | 508 (482) | 17 (17) | 24 (23) | 0 (0) | 398 (372) | 181 (174) | 1128 (1068) |
| PUTATIVE (PREDICTED + NR + ENV) | 1369 (1197) | 27 (23) | 50 (46) | 7 (4) | 869 (739) | 521 (473) | 2843 (2482) |
| ALL (CONFIRMED + PUTATIVE) | 1568 (1370) | 33 (27) | 53 (49) | 8 (5) | 893 (763) | 577 (524) | 3132 (2738) |
CONFIRMED, all sequences from REBASE with nuclease activity confirmed experimentally; PREDICTED, sequences from REBASE without direct experimental confirmation, excluding the data from environmental DNA sequencing projects; NR, homologs of sequences from sets CONFIRMED and PREDICTED that are not present in REBASE, but were identified by us in the non-redundant (nr) database at the NCBI; ENV, Putative REases from environmental DNA sequencing projects, predicted by REBASE or by us in env_nr database. PUTATIVE = PREDICTED + NR + ENV. Values in parentheses correspond to the number of sequences with no more than 90% sequence identity.
In all datasets analyzed in this work, the largest number of structurally classifiable enzymes always belong to PD-(D/E)XK superfamily. PD-D(E)XK family is overrepresented in the CONFIRMED set (68%) compared to PREDICTED and PUTATIVE sets (60 and 48%, respectively). This is caused by the fact that this family is the most intensively studied [e.g. almost all enzymes with structures solved by X-ray crystallography belong to the PD-(D/E)XK fold]. On the contrary, HNH superfamily, the second largest in all datasets, is overrepresented in the PUTATIVE set (30%) compared to the CONFIRMED and PREDICTED sets (9 and 8%, respectively). As mentioned earlier, this might be due to the fact that some of the genuine REases from the HNH superfamily (e.g. R.HphI) exhibit similarity to putative nucleases that are in fact unlikely to function as REases, thus distorting the PUTATIVE set by inclusion of potential false positives. In the case of R.HphI family, only 20% of R.HphI homologs had detectable MTase neighbors within 5000 bp ( 49 ). On the other hand, virtually all experimentally characterized, ‘orthodox’ Type II REases encoded in completely sequenced genomes, whose sequences are available in REBASE (including all experimentally characterized members of the R.HphI family) do possess a MTase neighbor ( 8 ).
Distribution of DNA cleavage preferences among folds of REases
An interesting question to be asked is whether REases from particular folds exhibit preferences for certain DNA sequences and/or cleavage patterns (length of 3′ or 5′ overhangs). Should that be the case, the experimental characterization of products of cleavage could aid the prediction of folds (structure) or vice versa . To answer this question, we have manually aligned DNA recognition sequences for all type II REases from the ‘CONFIRMED’ set (see Supplementary Table 1 ). The features of DNA sequences taken into account were, in order of importance: the cleavage pattern (in some cases with a tolerance of up to 1 bp), the distance between recognition site and cleavage site, the site of methylation by a cognate MTase (if known) and the DNA sequence. We also made a histogram of cleavage patterns for REases from different folds ( Figure 9 ). It shows that the preferred cleavage patterns are indeed different for REases from different folds. REases from the PD-(D/E)XK family show high preference for 5′ overhangs or blunt ends, while REases from the HNH superfamily prefer to generate 1-nt or 4-nt 3′ ends or 4-nt 5′ ends. Interestingly, in our dataset there is not a single case of REases with the same recognition sequence and cleavage pattern that would have different folds, while the probability of such situation in case of random distribution of known cleavage patterns to Type II REases from different families is <10 −6 (data not shown). This finding suggests that the knowledge of the target sequence and cleavage pattern could be used as a predictor for the 3D fold assignment. Interestingly, one of the enzymes for which we failed to predict the structure using bioinformatic methods, i.e. R.HpaII ( 87 ), cleaves the same DNA sequence (C′CG,G) as another enzyme of known structure, namely R.HinP1I from the PD-(D/E)XK superfamily ( 88 ). Neither secondary structure prediction nor ‘sequence gazing’ allowed us to propose any reliable candidate of the PD-(D/E)XK motif in R.HpaII, therefore we propose it as a valuable target for experimental structure determination by X-ray crystallography.
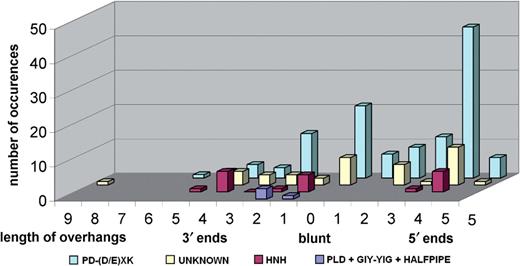
Number of Type II REases from different folds leaving 5′ or 3′ overhangs of different length or blunt ends.
CONCLUSIONS
The results of our bioinformatics analysis provide the very first classification of all Type II REase sequences into families and superfamilies, and a comprehensive structural census. We believe that our results will be very useful for experimental researchers. First, a number of particularly interesting candidates for crystallographic analyses are proposed, with two priorities in mind: (i) high-resolution structural characterization of folds that are either completely new or at least have not been reported among Type II REases, and (ii) maximization of structural coverage (availability of structural templates for confident modeling of a possibly largest number of sequences significantly related to these templates). Second, our delineation of sequence-related groups of REases that exhibit differences in substrate specificity suggests that detailed comparative analyses (that are beyond the scope of this article) could provide insight into the molecular basis of different specificity. Such groups of nucleases appear to require a smaller number of mutations to change the substrate preference and therefore they may be particularly useful targets for experimental protein engineering aiming at development of enzymes with new specificities. Finally, the observed correlation between the structural folds and the patterns of cleavage (length of ends) provides evidence to support the earlier prediction that the phenotypes of REases may correlate with their evolutionary relationships ( 89 ). Thus, structural predictions for putative REases (e.g. those identified by genome sequencing) may aid in prediction of their cleavage patterns and thereby simplify the planning of experiments to characterize them functionally. Conversely, functional characterization of enzymes with unknown structure may provide hints as to their 3D folds. Indeed, the recently characterized REase R.PabI with unusual DNA recognition sequence and cleavage pattern ( 39 ) turned out to exhibit a completely new type of structure. Although these correlations should by no means be taken as a rule, they may help experimentalists in prioritization of experiments, aiming at identification and characterization of proteins with particular features of interest.
ACKNOWLEDGEMENTS
We thank Rich Roberts, Alfred Pingoud, Virgis Siksnys, Matthias Bochtler, Mikihiko Kawai, Ichizo Kobayashi and members of the Bujnicki laboratory (in particular Jan Kosinski) for stimulating discussions on structural, functional and evolutionary classification of Type II REases and contributing various unpublished materials during the work on this article. We also thank Jan Kosinski for critical reading of the manuscript. This analysis was funded by the NIH (Fogarty International Center grant R03 TW007163-01). Funding to pay the Open Access publication charges for this paper has been waived by Oxford University Press—NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement . None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments