





Abstract
We describe a new method for relative quantification of 40 different DNA sequences in an easy to perform reaction requiring only 20 ng of human DNA. Applications shown of this multiplex ligation-dependent probe amplification (MLPA) technique include the detection of exon deletions and duplications in the human BRCA1, MSH2 and MLH1 genes, detection of trisomies such as Down’s syndrome, characterisation of chromosomal aberrations in cell lines and tumour samples and SNP/mutation detection. Relative quantification of mRNAs by MLPA will be described elsewhere. In MLPA, not sample nucleic acids but probes added to the samples are amplified and quantified. Amplification of probes by PCR depends on the presence of probe target sequences in the sample. Each probe consists of two oligonucleotides, one synthetic and one M13 derived, that hybridise to adjacent sites of the target sequence. Such hybridised probe oligonucleotides are ligated, permitting subsequent amplification. All ligated probes have identical end sequences, permitting simultaneous PCR amplification using only one primer pair. Each probe gives rise to an amplification product of unique size between 130 and 480 bp. Probe target sequences are small (50–70 nt). The prerequisite of a ligation reaction provides the opportunity to discriminate single nucleotide differences.
Received February 22, 2002; Revised and Accepted April 20, 2002.
INTRODUCTION
Changes in copy number of certain specific chromosomal sequences are frequently implicated in the cause of, or disposition to, human diseases and syndromes. Such changes include the presence of an extra copy of a complete chromosome as in Down’s syndrome, deletions of up to several million base pairs as in DiGeorge syndrome and deletions or duplications of smaller chromosomal fragments such as a single exon. For instance, >60% of Duchenne and Becker muscular dystrophy cases are due to deletions or duplications of one or more exons of the DMD gene ( 1 ). Deletion or duplication of one or more exons of the BRCA1 or the MLH1/MSH2 genes predispose the carrier to breast and colon cancer ( 2 , 3 ), respectively. In the same way, copy number changes of specific chromosomal regions are very common in tumours and are an important factor influencing gene expression ( 4 ). Analysis of these copy number changes is important for treatment as exemplified by the introduction of ERBB2-specific antibodies for the treatment of breast cancer patients with ERBB2 gene amplification ( 5 ).
At present, many different techniques are used for the detection of copy number changes of chromosomal sequences including standard chromosome analysis, comparative genomic hybridisation (CGH) ( 6 ), fluorescent in situ hybridisation (FISH) ( 7 ), BAC arrays ( 8 ), Southern blots and loss of heterozygosity (LOH) ( 9 ) assays. Most of these techniques are not able to detect deletions or duplications of single exons. Besides they are time consuming, difficult to implement as multiplex assays (FISH, LOH) or require large amounts of sample DNA (Southern blots). The conventional methods for mutation detection that are based on PCR amplification of single exons from genomic DNA will not detect most exon deletions and duplications as a normal allele is also present. Whilst oligonucleotide and cDNA microarrays are still insufficiently sensitive and reproducible to detect a deletion or duplication of one copy of a small chromosomal sequence like a single exon, real-time PCR provides the possibility to detect several fold amplification of chromosomal sequences; however, its use in a multiplex assay is severely limited by spectral overlap of the fluorescent dyes used. In addition, the presence of multiple primer pairs in a multiplex reaction reduces the robustness of PCRs and the reliability of the quantification. Yet the use of multiplex techniques is essential for the routine detection of exon deletions and duplications in genes like BRCA1, with 24 exons, or DMD, having 79 exons. Analysis of the large variety of aberrations found in tumours also requires multiplex nucleic acid analysis methods that need to be very sensitive in view of the often restrictive amounts of sample available.
Nucleic acid fingerprinting techniques, such as amplified fragment length polymorphism (AFLP) ( 10 ) and differential mRNA display ( 11 ), have clearly demonstrated that PCR can be used for simultaneous reproducible amplification of many DNA fragments in one reaction, provided that only a single primer set is used for amplification of all these fragments. AFLP can be used to determine the relative copy number of more than 50 random sequences in a single reaction. Multiplex amplifiable probe hybridisation (MAPH) ( 12 ) is a similar method in which not random fragments but 40 different specific target sequences are detected and quantified. MAPH uses oligonucleotide probes that hybridise to specific nucleic acid sequences. Each hybridised probe can be simultaneously amplified with the use of a single primer pair and yields an amplification product of unique size. The copy number of target sequences is reflected in the relative intensities of the MAPH probe amplification products ( 12 ). However, like Southern blotting, MAPH requires immobilisation of sample nucleic acids and tedious washing of unbound (amplifiable) probes making it difficult to implement in a routine diagnostic setting. Here, we introduce a related technique named multiplex ligation-dependent probe amplification (MLPA) that is more sensitive and easier to use. As in MAPH, added oligonucleotide probes rather than sample nucleic acids are amplified. However, immobilisation of sample nucleic acids and removal of excess probes is not necessary.
MATERIALS AND METHODS
Probe preparation
Each MLPA probe consists of one short synthetic oligonucleotide and one, phage M13-derived, long probe oligonucleotide. The short synthetic oligonucleotide of each probe contains a target-specific sequence (21–30 nt) at the 3′ end and a common 19 nt sequence, identical to the labelled PCR primer, at the 5′ end. Preparation of the long MLPA probe oligonucleotide is outlined in Figure 1 . For the preparation of each long MLPA probe, a target sequence-specific oligonucleotide of 25–43 nt is cloned in one of the M13-derived SALSA vectors. Each clone obtained is used to infect a 1-l culture of Escherichia coli strain TG1. Single stranded DNA is purified by polyethyleneglycol precipitation of phage particles, heat disruption of the virus and cetyl-trimethyl-ammonium bromide precipitation of the DNA. This single stranded DNA is made partially double stranded at the Eco RV and Bsm I sites by the annealing of two short oligonucleotides. Digestion by Eco RV and Bsm I results in the formation of a large 7200 nt M13 fragment and the MLPA probe oligonucleotide (80–420 nt). This probe oligonucleotide contains the 25–43 nt target-specific sequence at the 5′ phosphorylated end, a 36 nt sequence that contains the complement of the unlabelled PCR primer and is common to all probes at the 3′ end, and a stuffer sequence of variable length in between. It is not necessary to remove the large 7000 nt M13 fragment before use. Melting temperatures of all probe target-specific sequences are >65°C at 100 mM NaCl. For the design of the hybridising probe parts, sequence information available from the public databases was used. Except for the labelled PCR primers, synthetic oligonucleotides were not purified.
MLPA analysis
DNA samples were diluted with TE to 5 µl and were heated at 98°C for 5 min in 200 µl tubes in a thermocycler with a heated lid (Biometra Uno II). After addition of 1.5 µl salt solution (1.5 M KCl, 300 mM Tris–HCl pH 8.5, 1 mM EDTA) mixed with 1.5 µl probe mix (1–4 fmol of each synthetic probe oligonucleotide and each M13-derived oligonucleotide in TE), samples were heated for 1 min at 95°C and then incubated for 16 h at 60°C. Ligation of annealed oligonucleotides was performed by diluting the samples to 40 µl with dilution buffer (2.6 mM MgCl 2 , 5 mM Tris–HCl pH 8.5, 0.013% non-ionic detergents, 0.2 mM NAD) containing 1 U Ligase-65 enzyme, and incubation for 15 min at 54°C. The ligase enzyme was inactivated by heating at 98°C for 5 min and ligation products were amplified by PCR. For most experiments, 10 µl of the ligation reaction was added to 30 µl PCR buffer. While at 60°C, 10 µl of a buffered solution containing the PCR primers (10 pmol), dNTPs (2.5 nmol) and 2.5 U Taq polymerase (Promega) or SALSA polymerase (MRC-Holland) were added. Alternatively, the 10 µl solution containing PCR primers, dNTPs and polymerase was added to the complete MLPA reactions while at 60°C. PCR was for 33 cycles (30 s at 95°C, 30 s at 60°C and 1 min at 72°C). Samples amplified with one unlabelled and one IRD-800 labelled primer (LICOR) were analysed on a 6.5% acrylamide slab gel in a LICOR IR2 system. Samples amplified with one unlabelled and one D4‐labelled primer (Research Genetics) were analysed on a Beckman CEQ2000 capillary electrophoresis system. MLPA reactions resulting in only 10–12 amplification products were analysed by electrophoresis on ethidium bromide stained agarose gels. Two-fold differences in relative amounts of target sequences could be scored by visual examination.
The sequence of the labelled primer is 5′-GGGTTCCCTAAGGGTTGGA-3′ and that of the unlabelled primer is 5′-GTGCCAGCAAGATCCAATCTAGA-3′. The exact gene and sequence recognised by each probe used in this article can be found at www.mrc-holland.com .
RESULTS
An outline of the MLPA reaction is shown in Figure 2 . DNA (20–500 ng) is denatured and fragmented by a 5 min heat treatment at 98°C. MLPA probes are added and allowed to hybridise for 16 h at 60°C in a thermocycler with a heated lid. Dilution buffer including the ligase enzyme is added and ligation is allowed to proceed for 15 min at 54°C. After heat inactivation of the ligase and addition of PCR primers, dNTPs and polymerase, PCR amplification of the ligation products is started. One PCR primer is fluorescently or isotopically labelled. Amplification products are detected and quantified by capillary electrophoresis (see Figs 4 – 8 ) or traditional gel electrophoresis (Fig. 3 ).
Preparation and design of MLPA probes
Ligation-dependent PCR has been described previously ( 13 – 15 ). Several modifications were made to make the technique simple to perform, more reproducible and sensitive, and suitable for true multiplex analysis.
Each MLPA probe consists of two oligonucleotides that can be ligated to each other when hybridised to a target sequence. All ligated probes have identical sequences at their 5′ and 3′ ends, permitting simultaneous amplification in a PCR containing only one primer pair. Each probe gives rise to an amplification product of unique size between 130 and 480 bp. One of the two oligonucleotides of each MLPA probe is chemically synthesised and has a common sequence used for PCR amplification at the 5′ end and a target-specific sequence at the 3′ end. Each second probe oligonucleotide contains a sequence of 25–43 nt at the 5′ end that is able to hybridise to the target sequence immediately adjacent to the first probe oligonucleotide, a common sequence used for PCR amplification at the 3′ end, and a stuffer sequence of 19–370 nt in between. Chemically synthesised oligonucleotides of this size (80–440 nt) are not commercially available in the quality needed for MLPA. We therefore use single stranded DNA from M13 clones containing small target-specific sequences for the preparation of these oligonucleotides. The M13 DNA is made partially double stranded by the annealing of complementary oligonucleotides and is digested by two restriction endonucleases (Fig. 1 ). One of these enzymes cuts the DNA outside its recognition sequence, resulting in a 5′ phosphorylated end that is perfectly complementary to the target sequence. Most probe mixes made contain 35–42 probes with length differences between consecutive amplification products of 6 or 9 bp. All probes used in a specific MLPA probe mix are made in a different M13-derived vector and have different stuffer and hybridising sequences. Amplification products of different probes have common sequences only at their ends in order to prevent heteroduplex formation during later stages of the amplification reaction when competition between duplex formation and PCR primer annealing takes place. We prepared a set of 118 different M13-derived MLPA vectors, each containing a stuffer sequence of different length and sequence. Target sequence-specific synthetic oligonucleotides can easily be inserted in these vectors, allowing flexibility to create all required fragment lengths. Probes consisting of two synthetic oligonucleotides and resulting in amplification products of 94–124 bp were also successfully used.
The non-hybridising stuffer sequences of our M13-derived probe oligonucleotides provide the advantage that the amplification characteristics of a large part of each amplicon are known. Short hybridising sequences also have an advantage in mutation detection and SNP analysis as sequences close to each other can be analysed without competition between probes with overlapping target sequences.
Reaction conditions
All M13-derived MLPA probe oligonucleotides contain the complement sequence of one of the two PCR primers at their 3′ end, and will thus be linearly amplified during the PCR. This PCR primer is unlabelled in order to prevent background signals. To prevent most of this PCR primer being consumed by linear amplification, low amounts of probe oligonucleotides have to be used. If 10 fmol of each of the 40 probes were to be linearly amplified, this would consume all 10 pmol unlabelled primer in only 25 PCR cycles. Despite the use of low amounts of probes, hybridisation of probe oligonucleotides to their target sequences has to be complete in order to obtain reproducible results. We use a 16 h hybridisation period in an 8 µl reaction volume containing 1–4 fmol of most probe oligonucleotides. Hybridisation kinetics differed slightly for each oligonucleotide. Some probes required the presence of up to 8 fmol. Once hybridisation of all probes is complete, prolonged incubation did not influence relative probe signal strength.
Commercially available ligases, as well as several ligases cloned at MRC-Holland, were tested for use in MLPA reactions. The NAD requiring Ligase-65 enzyme chosen is active at 50–65°C, but can easily be heat inactivated before the start of the amplification reaction. Ligase-65 is very sensitive to probe-target mismatches next to the ligation site (Fig. 3 ). Within the range of 20–500 ng human sample DNA, MLPA results were not influenced by the amount of DNA used. Some non-specific amplification products appeared when samples containing <20 ng human chromosomal DNA were analysed. Most experiments were performed using 50–100 ng human sample DNA.
Probe signal strengths
The hybridising parts of our probes were designed to detect human sequences that are present in a single copy/haploid genome. However, the relative signal strength of different probes (relative peak area of each probe amplification product) is not equal. Apart from the copy number of the probe target sequence, the major factors influencing the relative signal strength of MLPA probes proved to be the amount of polymerase used in the PCR and the nature of the first nucleotide following the labelled PCR primer. The MgCl 2 concentration in the PCR did not have substantial effects. The KCl concentration, however, influenced the relative peak sizes of some probe amplification products.
The polymerase activity during the PCR influenced the relative signal strength of a minority of the probes. Relative peak areas of 5–10% of the probes made decreased >25% when 2.5-fold lower amounts of polymerase were used, whilst the relative peak areas of 2% of the probes strongly decreased when 2–4 times higher amounts of polymerase were used. These probes were replaced.
The influence of the nature of the first nucleotide following the PCR primer sequence on the relative peak size was unexpected. In our short synthetic probe oligonucleotides the PCR primer sequence is immediately followed by the variable target-specific sequence. Probes in which the PCR primer sequence was followed by an adenine had a >2-fold lower average signal strength. In all 25 cases examined, replacement of this A nucleotide resulted in increased signal strength. Signal strength increased in the order A < T < G < C. Average signal strength of probes in which the unlabelled primer was first elongated with an adenine residue was also significantly lower as compared with other probes. This finding may be of use for the design of primers for traditional multiplex PCR. The vast majority of recently made MLPA probes, designed to detect human single copy DNA sequences on chromosomes 1–22, have signal strengths between 50 and 150% of the average signal strength. This indicates a maximum difference in amplification efficiency during each PCR cycle of <2% from average for each probe amplification product.
Probe specificity
As shown in Figure 3 , omitting one of the short probe oligonucleotides, or replacing it by an identical oligonucleotide having a mismatch at the 3′ end, prevented the appearance of probe amplification products. Sensitivity of probe signals to mismatches close to the ligation site depended on the amount of ligase used and the duration of the ligation reaction (not shown). Under our experimental conditions a mismatch at the 3′ end of the short probe oligonucleotide completely prevented the appearance of probe amplification products. Mismatches at 4–6 nt from the ligation site had no or only small effects (Fig. 3 ). This sensitivity to a mismatch at the ligation site was, in some cases, used to distinguish target sequences from pseudogenes or related genes.
Detection of trisomies
In order to test the linearity of probe signal with small changes in target sequence copy number, we prepared a mix of 40 probes, including four probes each for chromosomes X and Y and eight probes each for chromosomes 13, 18 and 21 sequences. Part of the capillary electrophoresis patterns obtained on four DNA samples are shown in Figure 4 . For relative quantification purposes we divided each peak area by the sum of all probe peak areas of that sample. The ratio of each individual probe relative area was then normalised to that obtained on a control sample. In Table 1 , this ratio is shown for each probe using female control, triple X, triple 13, triple 18 and Down’s syndrome (trisomy 21) DNA. Results show that the relative probe signals obtained for each probe reflected the relative amount of the probe target sequences in the sample. The excellent reproducibility of relative signals obtained enabled the detection of a single extra copy of a probe target sequence per diploid genome. Highest deviations of the expected values were obtained for the triple 13 DNA sample that was derived from a cell line. An increase in relative peak area of the chromosome 8-specific MYC probe (1.30), and a lower than expected relative peak area of the chromosome 13-specific RB1 (1.30) and DLEU 1 probes (1.25) might be due to chromosomal aberrations in some cells of our cell line. The DLEU1 gene and the tumour suppressor RB1 are located close (<1 Mb) to each other. A probe specific for a different sequence of the MYC gene (relative signal 1.35) also indicated an increase in copy number of this oncogene in our triple 13 cell line.
Detection of exon deletions in the human BRCA1, MLH1 and MSH2 genes
The human BRCA1 gene is involved in hereditary disposition for breast cancer. In The Netherlands >30% of hereditary disposition for BRCA1-related breast cancer is due to deletions of one or more of the 24 BRCA1 exons ( 2 ). Deletion of one or more exons can be proved by Southern blotting provided that the deletion does not extend beyond the probe boundaries. Known deletions can be tested by PCR. We prepared MLPA probes for each BRCA1 exon. Samples known from specific PCRs ( 2 ) to be heterozygote for a deletion of either exon 13 or exon 22 were easily identified by MLPA as they resulted in an ∼2-fold reduction of relative probe signal for these probes (Fig. 5 ). DNA samples from 850 individuals suspected of hereditary disposition for breast cancer have been tested by MLPA. Several new as well as several previously described aberrations of the human BRCA1 gene were detected (F.B.L.Hogervorst, P.M.Nederlof, J.J.P.Gille, C.J.McElgunn, M.Grippeling, R.Pruntel, R.Regnerus, T.van Welsem, F.H.Menko, I.Kluijt, C.Dommering, S.Verhoef, J.Schouten, L.J.van ’t Veer and G.Pals, manuscript in preparation).
Genomic deletions of parts of the human MLH1 and MSH2 genes are a frequent cause of hereditary non-polyposis colon cancer (HNPCC) ( 3 ). An MLPA probe mix was prepared containing probes for each of the 19 MLH1 exons and each of the 16 MSH2 exons, as well as seven probes for genes on other chromosomes. Using this probe mix, all six different known exon deletions tested could easily be identified by MLPA. Screening of a large number of DNA samples from HNPCC patients is in progress. Results obtained on a sample known to contain in one chromosome a deletion of exons 1–6 of the MSH2 gene are shown in Figure 6 .
Detection of gains and losses of chromosomal regions
For analysis of tumour DNA, three MLPA probe mixes have been made, each containing 41 different probes specific for human single copy DNA sequences. Most target sequences were chosen in chromosomal regions often deleted or amplified in various types of tumours. These probe mixes were used to analyse DNA from diffuse large B-cell lymphomas (DLBCL) as well as DNA from the SkBr3 cell line. Results obtained with one of the probe mixes on a DLBCL DNA sample and the SkBr3 DNA are shown in Figure 7 .
SkBr3 is known to contain amplified ERBB2 (HER2/neu) and MYC loci ( 16 , 17 ). Relative peak areas for two different MYC-specific MLPA probes were increased 5.3 and 6.1 times compared with control human DNA. Relative peak areas of three different ERBB2 probes were increased 5.3, 6.5 and 6.0 times, respectively. This compares well with the 6.6 times amplification of ERBB2 in SkBr3 as measured by FISH ( 4 ). Exact amplification levels are difficult to determine as the number of chromosomal aberrations is large and the relative copy numbers are calculated by comparison with the total peak area of a sample. The SkBr3 cell line is known to be substantially tetraploid and to contain several abnormal chromosomes ( 17 ). Apart from many other aberrations, our SkBr3 DNA sample completely lacked the target sequence for a CDH1 probe on chromosome 16q22. Major parts of chromosomes 7 and 20 appeared to have a 1.5–2-fold increased relative copy number whereas BRCA1-specific probes had a 2-fold lower relative peak area.
The DNA from the (male) DLBCL lymphoma showed a 2.4–3.0-fold increase in relative peak area for each of three different BCL2-specific probes. Probes for DCC and PMAIP1, which are located on chromosome 18 within 11 Mb of BCL2, were also increased 2–3-fold whereas CDH2, which is located 37 Mb from the BCL2 locus, was not increased. Probes for CDKN2A and CDKN2B were both reduced by 82%, whereas two chromosome Y probes were reduced by 73 and 82%, respectively, indicating homozygous deletion of 9p21 and chromosome Y in the tumour and a high percentage of tumour cells in the sample. All seven probes surrounding the HLA region on chromosome 6p had a 25–56% reduction in relative peak area.
The same probe mixes were used to analyse 15 DNA samples extracted from formalin-treated paraffin-embedded primary breast cancer tissues. Reversal of formaldehyde cross-linking was performed by prolonged heating of the DNA ( 18 ). Most of the results obtained by MLPA corresponded with results obtained on these samples by CGH (not shown).
SNP and mutation detection
MLPA probe signal is completely absent if the short probe oligonucleotide has a mismatch at the 3′ nucleotide when annealed to the target sequence (Fig. 3 ). This sensitivity of the ligase for a mismatch next to the ligation site can be used to distinguish two sequences differing in only one or a few nucleotides, such as SNPs and various mutations. In order to obtain a signal from both alleles, which can be distinguished by the length of the probe amplification products, we used probes for the two alleles that have the M13-derived oligonucleotide in common, as outlined in Figure 8 . Two different short synthetic probe oligonucleotides are used that differ at both the site of the mutation/SNP and also by 3 nt in length. These two oligonucleotides will compete with each other for binding to both target sequences as a single mismatch at the end of the probe oligonucleotide will usually not be sufficient to destabilise the hybrids. However, ligation of hybrids with mismatches next to the ligation site will occur with very low efficiency. In Figure 8 , MLPA results are shown in which a DNA sample of a cystic fibrosis patient homozygote for the ΔF508 mutation is compared with samples from a heterozygote and a control DNA. The probe designed resulted in a 3 bp longer amplification product for the ΔF508 mutation as compared with the probe for the wild-type DNA sequence.
DISCUSSION
Ligation-dependent PCR has been described previously ( 13 – 15 ). The major modifications made in order to make this technique suitable for multiplex analysis are the use of much lower amounts of probes combined with longer hybridisation periods, the use of probe oligonucleotides made by digestion of M13-derived single stranded DNA with a restriction endonuclease that cuts outside its recognition sequence and the introduction of the heat-inactivatable (95°C), thermo-stable (60°C) Ligase-65 enzyme. The MLPA technique presented here permits relative quantification of 40 different target sequences in an extremely easy to perform reaction. Only a thermal cycler and electrophoresis equipment are needed. Design and preparation of an MLPA probe mix is time consuming, but once clones have been made, a one litre bacterial culture of each clone provides sufficient M13 DNA for more than 500 000 MLPA reactions.
Sensitivity
In order to obtain reproducible results, we currently need the presence of at least 20 ng human DNA, corresponding to approximately 3000 cells or 6000 single copy target sequences. Experiments to increase sensitivity are in progress.
Probe specificity
When 40 probes are present in one probe mix, not 40 but 1600 different sample sequences are able to align one short and one long oligonucleotide and make them a substrate for ligation. However, there are several indications that specificity of our probe mixes is very high. (i) As shown in Figure 3 , omitting one of the short probe oligonucleotides or replacing it by an identical oligonucleotide that has a mismatch at the 3′ end prevented the appearance of the probe amplification product. (ii) All seven probes specific for sequences on human chromosome Y did not give any signal on female DNA. The 28 probes specific for sequences on chromosomes X, 21, 18 and 13 gave on average a 1.44-fold higher signal on the respective trisomy DNA samples as compared with control DNA (Table 1 ). This is close to the theoretical 1.50 increase in relative signal, and compares well with the 1.31-fold higher relative signal obtained with cDNA microarrays upon analysis of human trisomy DNA ( 19 ). (iii) Three probe mixes containing a total of 100 probes were made in which each probe was designed to be cDNA specific as the two probe oligonucleotides hybridise to sequences separated by introns in human chromosomal DNA. No significant signal was obtained with these probe mixes when human chromosomal DNA was used as a template. However, as will be shown elsewhere, most probes did give strong, reproducible signals on cDNA samples. (iv) The short synthetic probe oligonucleotides range in size from 40 to 49 nt. If more than one short probe oligonucleotide could be ligated to a specific M13-derived probe oligonucleotide this would, in most cases, have been apparent by the appearance of extra bands.
The major cause of the specificity of the MLPA probes will be the need for two probe oligonucleotides to anneal at immediately adjacent sites of a target nucleic acid. In order to increase specificity, MLPA hybridisation and ligation reactions are performed at 54–60°C. Thermophilic NAD requiring ligases are not able to ligate oligonucleotides that leave a one base gap or have an overlap when bound to their target sequence ( 20 , 21 ). Furthermore, the Ligase-65 enzyme used is very sensitive to mismatches between probes and target sequences next to the ligation site. This can be used to distinguish target sequences from pseudogenes or related genes. NAD requiring ligases are, in general, most sensitive to mismatches at the last nucleotide (3′ OH site) of the short probe oligonucleotide ( 20 , 21 ). Under our experimental conditions a mismatch at the 3′ end of the short probe oligonucleotide completely prevented the appearance of probe amplification products.
Reproducibility
Reproducibility of our results depended on the apparatus used, capillary electrophoresis being superior to fluorescent detection on slab gels, on the software used for peak detection, the method used for normalisation of data and the purity of DNA samples. We found that DNA degradation to small fragments did not influence results, whereas the presence of PCR inhibitors, such as small remnants of phenol, did. In general, the standard deviation of results obtained was between 4 and 10% for each probe.
Probe signal strength
We have shown that relative probe signal strengths depend on the relative amounts of the target sequences present (Table 1 and Figs 4 – 7 ). An almost linear, up to 5-fold increase in relative probe signal could be obtained by addition of increasing amounts of synthetic probe target sequences to human DNA samples (not shown). Addition of higher amounts of synthetic probe target sequences resulted in a non-linear further increase in relative probe signal.
We observed that the nature of the first nucleotide following the primer has a large effect on the relative peak size of the amplification products. When the first nucleotide to be incorporated was an adenine, a small peak size was usually obtained. The 2–3-fold difference in relative signal strength between fragments that are first elongated with an adenine as compared with a cytosine reflects an ∼3% difference in amplification efficiency during each PCR cycle. This effect might be due to increased stabilisation of annealed primers by elongation with a C or G as compared with the incorporation of an A. Primers that are not, or not sufficiently, elongated will be melted during the transition from annealing temperature to the elongation temperature, but might also be more easily displaced by the complementary strand during the last PCR cycles.
Applications
Human trisomies could easily be identified by MLPA (Table 1 ). Aberrant copy numbers of human chromosomes 13, 18, 21, X and Y are not always lethal. Many techniques currently used for detection of trisomies are based on variable number of tandem repeats and rely on the presence of three different alleles. Only first meiotic disjunctions can be demonstrated. As an MLPA test can include up to eight different probes for each chromosome, aberrant copy numbers can be detected in small fetal DNA samples with high accuracy.
As the sequence recognised by an MLPA probe is only 50–70 nt long, MLPA is very useful for the detection of deletions and amplifications of single exons. We used MLPA for the detection of deletions and amplifications in the human BRCA1 gene and the MLH1/MSH2 genes. Screening of blood-derived DNA samples from 850 patients suspected to have a hereditary cause of their breast cancer revealed several new BRCA1 genomic aberrations (F.B.L.Hogervorst, P.M.Nederlof, J.J.P.Gille, C.J.McElgunn, M.Grippeling, R.Pruntel, R.Regnerus, T.van Welsem, F.H.Menko, I.Kluijt, C.Dommering, S.Verhoef, J.Schouten, L.J.van ’t Veer and G.Pals, manuscript in preparation). Most exon deletions and duplications in human DNA will not be detected by conventional methods for mutation detection that are based on PCR amplification of single exons from genomic DNA as a normal allele is also present. Southern blots should reveal most deletions and duplications, but are time consuming, require large amounts of DNA and will not reveal deletions extending beyond the probes used. One of the samples tested contained an exon 13 duplication. This previously described duplication ( 22 ) was confirmed by PCR. Detection during routine screening of DNA samples of an increase in copy number from two to three of a single exon shows the sensitivity of MLPA.
MLPA is a sensitive method for the detection of ERBB2 (Her2/neu) amplification in DNA samples extracted from frozen breast cancer tissues as well as from DNA extracted from paraffin-embedded formaldehyde-fixed breast cancer tissues (P.M.Nederlof and M.J. van de Vijver, manuscript in preparation). MLPA analysis of primary breast tumour and lymphoma DNA samples, using probes located in chromosomal regions in which aberrations often occur in tumours, showed several different gains and losses of loci in many samples. Many of these aberrations could be confirmed by results of other MLPA probes recognising sequences of the same gene or a gene located nearby. Other aberrations corresponded to the CGH data for these samples. As the amount of sample DNA required is very low, MLPA may be useful for the analysis of small preneoplastic lesions. Besides, these probe mixes may provide DNA fingerprints that prove clonality of tumours and should be useful for identification and comparison of cell lines. Chromosomal rearrangements and numerical changes result in large differences in behaviour between ‘identical’ cell lines obtained from different laboratories ( 23 ).
In agriculture, MLPA can be used for high-throughput multiplex homozygote/heterozygote discrimination. MLPA analysis of mRNAs will be described elsewhere.
MLPA probes binding at target sequences containing a restriction endonuclease site close to the probe ligation site do not give a signal when the target DNA is digested by that enzyme as the two probe oligonucleotides bind to different DNA fragments (not shown). MLPA reactions on DNA samples digested by methylation-sensitive restriction enzymes may therefore be used for quantification of DNA methylation at those sites.
Conclusion
We have presented here the MLPA technique that has proved to be sufficiently sensitive, reproducible and sequence specific, to allow detection of the gain or loss of one copy of a single exon in human DNA samples as small as 20 ng. In comparison to DNA arrays, MLPA is cheap, more sensitive and requires much less sample preparation time. The equipment needed is present in many molecular biology laboratories. Although limited to 40 target sequences/reaction, this is sufficient for many routine tests in which only a specific question needs to be solved.
ACKNOWLEDGEMENTS
The authors thank Maria Worsham (Henry Ford Hospital, Detroit) for providing DNA from the SkBr3 cell line, M. Spaargaren and S. T. Pals (AMC, Amsterdam) for providing DLBCL DNA samples and Tibor van Welsem and Petra Nederlof (Netherlands Cancer Institute, Amsterdam) for providing DNA samples from primary breast tumours. We thank Rahanna Nasrullah, Peter den Harder and Yordi Coffa for excellent technical assistance. C.J.M. is a recipient of a Marie Curie Industrial Host Fellowship. J.P.S. thanks Jesus Calmero for many years of excellent technical assistance and the Instrumentmakerij Biologie and the Biology faculty of the Free University of Amsterdam for 17 years of support.
To whom correspondence should be addressed. Tel: +31 20 4447218; Fax: +31 20 6891149; Email: schouten@mrc-holland.com
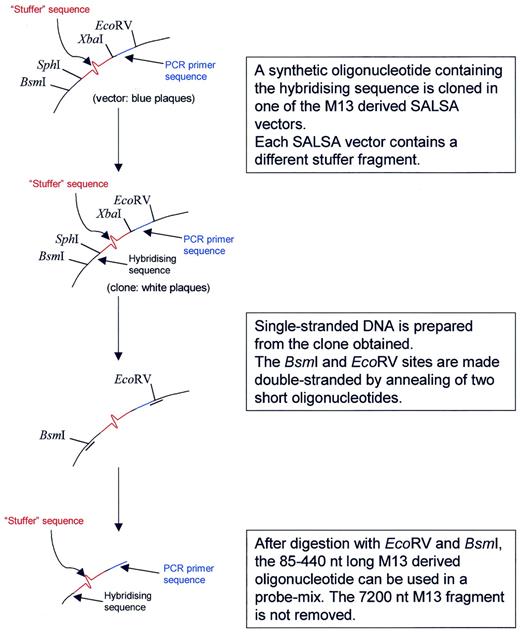
Figure 1. Preparation of the long M13-derived MLPA probe oligonucleotides. The basic MLPA vector M214 was derived from M13mp18 ( 24 ) by destroying the single Bsm I site and replacement of the polylinker site by a synthetic oligonucleotide containing from 5′ to 3′, a new Bsm I site, an Sph I and Xba I site, a sequence complementary to one of the two SALSA PCR primer sequences and an Eco RV site. A collection of 118 different SALSA vectors was prepared by insertion of stuffer fragments in the Sph I and Xba I sites. Each stuffer fragment has a different sequence and length (19–370 bp, 3 bp increments). Stuffer fragments up to 55 nt were synthetic; longer fragments were made by PCR amplification of T7 phage sequences. For each MLPA probe, a synthetic 30–43 nt long oligonucleotide containing the hybridising sequence is cloned in the Sph I and Bsm I sites of one of the SALSA vectors. Single stranded DNA is prepared from the clone obtained and is digested by Bsm I and Eco RV as indicated in the figure.
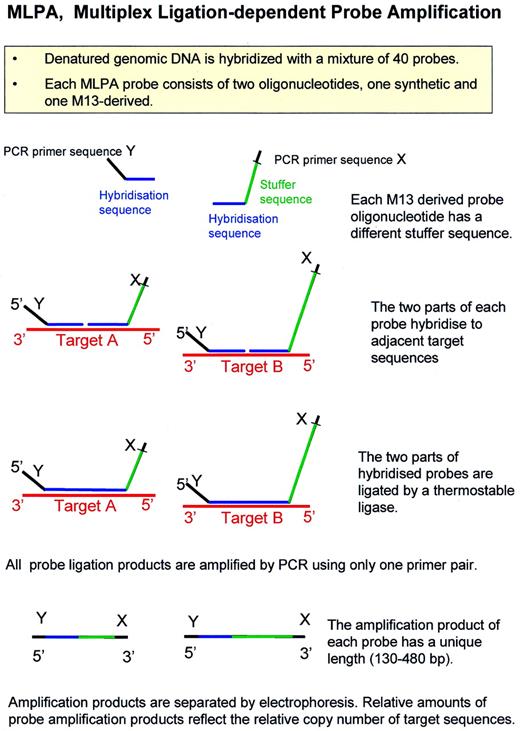
Figure 2. Outline of the MLPA reaction.
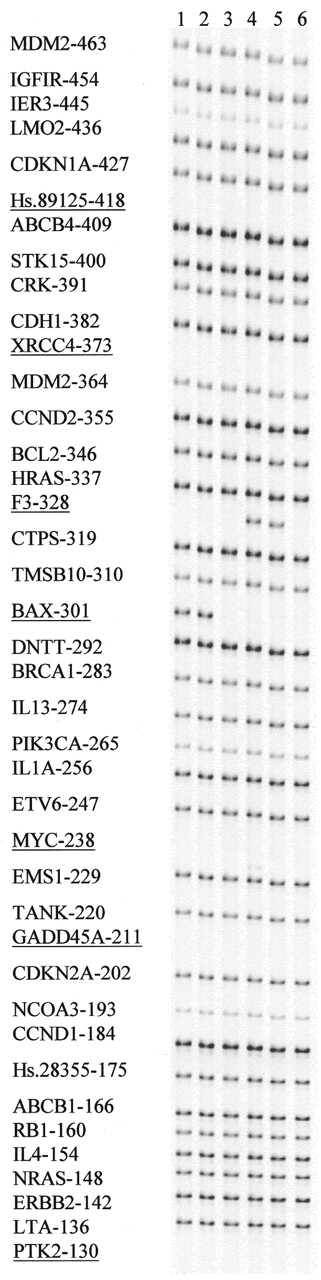
Figure 3. Sensitivity of MLPA analysis to mismatches in the short probe oligonucleotide. MLPA reactions were performed on 100 ng samples of human DNA. Amplification products were separated on a denaturing acrylamide gel (LICOR). Length (bp) as well as gene HUGO name is indicated for each probe amplification product. The probe mix contained 33 complete probes. For seven other (underlined) probes, only the long M13-derived oligonucleotide was included in each test. The part of the gel shown shows the result of addition of different short probe oligonucleotides for two of these probes to the probe mix: lane 1, Bax-specific short probe oligonucleotide, resulting in a 301 bp extra amplification product. Lane 2, as lane 1 but with a mismatch (T/T) at the fourth nucleotide from the ligation site. Lane 3, as lane 1 but with a mismatch (A/C) at the 3′ nucleotide of the BAX short probe oligonucleotide. Lane 4, F3-specific short probe oligonucleotide, resulting in a 328 bp extra amplification product. Lane 5, as lane 4 but with a mismatch (G/G) at the fourth nucleotide from the ligation site. Lane 6, as lane 4 but with a mismatch (G/G) at the 3′ nucleotide of the F3 short probe oligonucleotide.
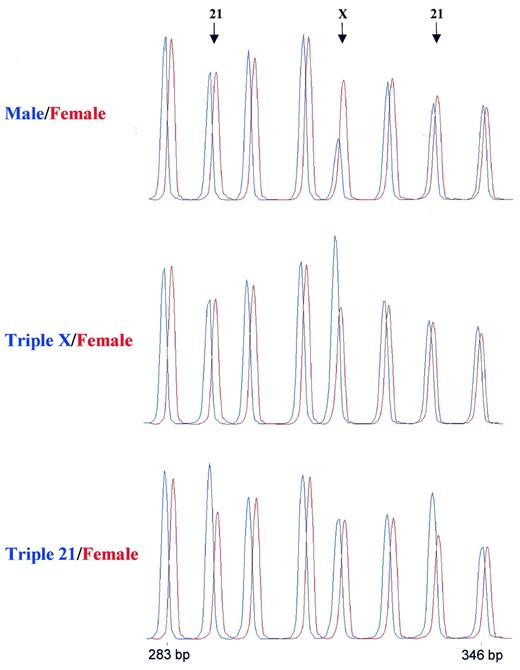
Figure 4. Detection of trisomies by MLPA. Samples containing 100 ng DNA were analysed by MLPA using probe mix P001. Male and female control DNA was obtained from Promega. Blood-derived DNA from a triple X and a female triple 21 individual were provided by the Department of Clinical Genetics, Free University of Amsterdam. Reactions were analysed by capillary electrophoresis (Beckman CEQ2000). In each case the female control DNA is shown in red. Probe mix P001 contains 40 probes. Only part of the resulting electropherogram is shown. Arrows indicate the positions of a 292 bp amplification product of a probe specific for the TFF1 gene on chromosome 21, a 319 bp amplification product of a probe specific for the L1CAM gene on the X chromosome and a 337 bp amplification product of a probe specific for the APP gene on chromosome 21. The other probes were specific for chromosome 3 (283 bp), chromosome 18 (301 and 346 bp), chromosome 13 (310 bp) and chromosome 1 (328 bp). A complete list of genes in this probe mix can be found in Table 1.
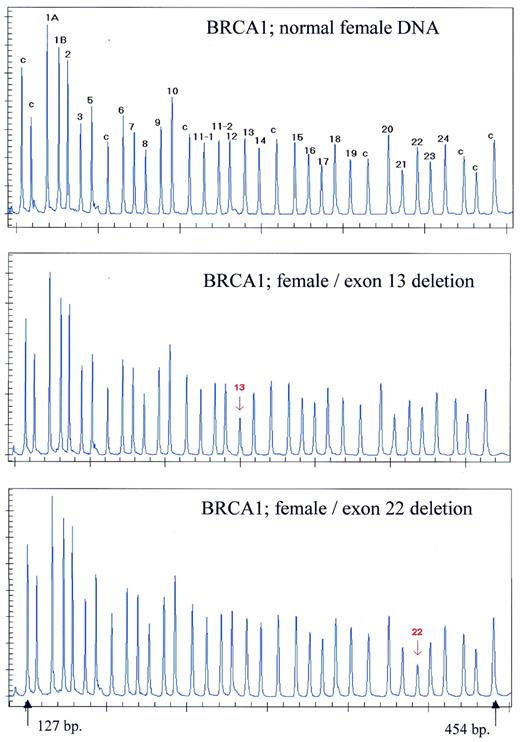
Figure 5. Detection of BRCA1 exon deletions by MLPA. Samples containing ∼100 ng DNA were analysed by MLPA using probe mix P002. Female control DNA was obtained from Promega. Blood-derived DNA from individuals known to contain an exon 13 or an exon 22 deletion were provided by the Department of Clinical Genetics, Free University of Amsterdam. Reactions were analysed by capillary electrophoresis. Probe mix P002 contains 34 probes. Nine probes recognising non-BRCA1 sequences on various chromosomes are indicated by a ‘c’. The exon recognised by the BRCA1-specific probes is indicated by a number. Probes for both alternative exons 1 are indicated as 1A and 1B. Exon 4 is not present in the normal BRCA1 gene transcript. Two probes specific for the first and last parts of exon 11 are included as this exon is very large (3.4 kb). Exon deletions are apparent by an ∼50% reduction in peak area of a specific probe. The exact gene and sequence recognised by each probe of the P002 probe mix can be found on the www.mrc-holland.com website.
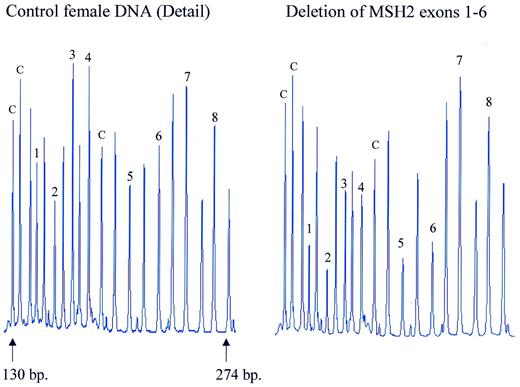
Figure 6. Detection of MSH2 exon deletions by MLPA. Samples containing ∼100 ng DNA were analysed by MLPA using probe mix P003. Female control DNA was obtained from Promega. Blood-derived DNA from an individual known to contain a deletion of exons 1–6 of the MSH2 gene was provided by the Department of Clinical Genetics, Free University of Amsterdam. Reactions were analysed by capillary electrophoresis (Beckman CEQ2000). Probe mix P003 contains 42 probes. Probes are present for each of the 19 MLH1 and each of the 16 MSH2 exons. Seven probes recognise other sequences on various chromosomes. Only part of the electropherogram is shown. The exon recognised by the MSH2-specific probes is indicated by a number. Control probes are indicated by a ‘c’. The remaining probes are specific for MLH1 exons 1–9. Exon deletions are apparent by an ∼50% reduction in peak area of a specific probe. The exact gene and sequence recognised by each probe of the P003 probe mix can be found at the www.mrc-holland.com website.
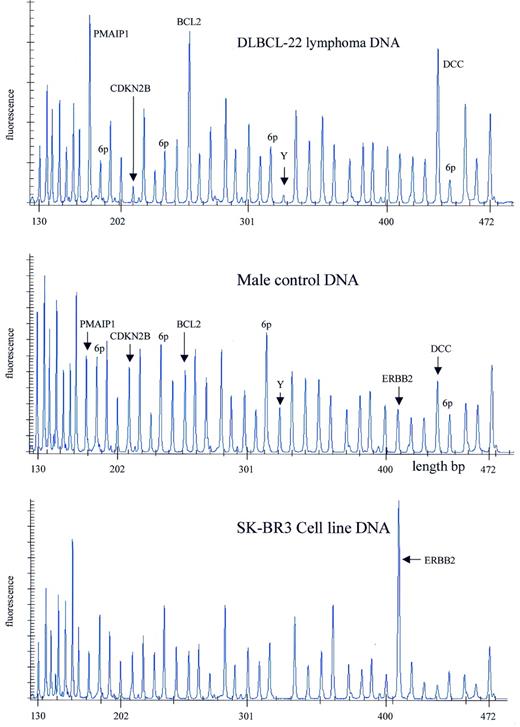
Figure 7. Detection of gains and losses of human chromosomal sequences by MLPA. Samples containing ∼100 ng DNA were analysed by MLPA using probe mix P006. Male control DNA was obtained from Promega. DNA isolated from a frozen DLBCL lymphoma was provided by the Department of Pathology, Amsterdam Medical Center. DNA isolated from the SkBr3 cell line was provided by the Henry Ford Hospital in Detroit. Reactions were analysed by capillary electrophoresis (Beckman CEQ2000). Probe mix P006 contains 41 probes. Probes specific for the chromosome 18 genes PMAIP1, BCL2 and DCC, as well as the CDKN2B gene on chromosome 9 and the ERBB2 gene on chromosome 17 are indicated. A probe recognising the SRY gene on the Y chromosome is indicated by Y. Four probes recognising the KIAA0170, BAK1, TNF and IER3 genes, all in the HLA region of chromosome 6, are indicated by 6p. The exact gene, sequence and chromosomal location recognised by each probe of the P006 probe mix can be found at the www.mrc-holland.com website.
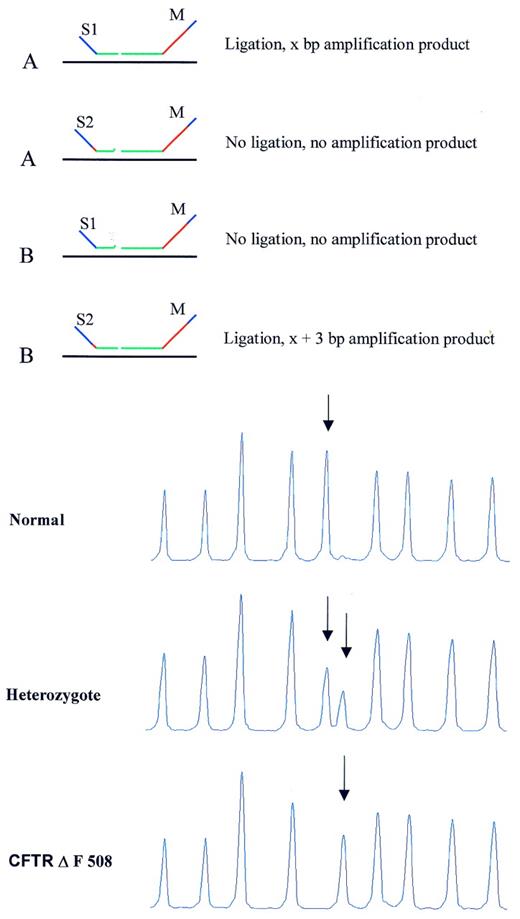
Figure 8. MLPA analysis of SNPs/mutations resulting in amplification products for both alleles with different lengths. (Top) Schematic drawing. Target sequences A and B differ in 1 nt. The short synthetic probe oligonucleotides S1 and S2 differ also in 1 nt, each being specific for one SNP sequence. In addition, S1 and S2 differ in length due to the presence of a 3 nt stuffer sequence between the hybridising sequence and the PCR primer sequence of oligonucleotide S2. Both oligonucleotides can anneal to target sequences A and B and will compete with each other for binding. A mismatch adjacent to the ligation site, prevents oligonucleotide ligation to the common M13-derived probe oligonucleotide M when S1 is bound to B, or when S2 is bound to target sequence A. (Bottom) Capillary electrophoresis profile. Detection of the CFTR ΔF508 mutation by MLPA. DNA samples (100 ng) of a control individual, a heterozygote and a homozygote carrier of the CFTR ΔF508 mutation (3 nt deletion) were tested by MLPA. The probe mix contained 40 probes including a probe for the wild type CFTR sequence and one specific for this mutation. These two probes have the M13-derived probe oligonucleotide in common. The short synthetic probe oligonucleotides differ at the site of the mutation, which is located at the 3′ end of the oligonucleotide as well as in length. Only part of the capillary electrophoresis profile is shown. Arrows indicate wild type (202 bp) and mutation specific probe (205 bp) amplification products.
Detection of trisomies by MLPA
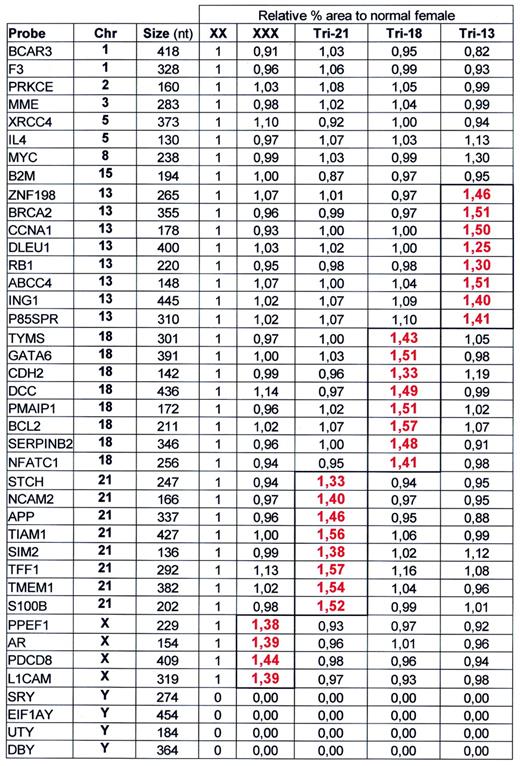
Samples containing ∼100 ng DNA were analysed by MLPA using probe mix P001. Male and female control DNA was obtained from Promega. Blood-derived DNA from triple 21 and triple X individuals as well as DNA from a triple 13 and a triple 18 cell line were provided by the Department of Clinical Genetics, Free University of Amsterdam. Reactions were analysed by capillary electrophoresis on a Beckman CEQ2000. Peak area of each probe amplification product was divided by the combined peak area of all 40 probes. The resulting relative peak area of each probe amplification product was divided by the relative peak area of that probe obtained on female control DNA. The presence of two copies of a probe target sequence/diploid genome should therefore result in a relative signal of 1.00. The presence of three copies of a probe target sequence/diploid genome should result in a 1.50 relative probe signal. The exact sequence recognised by each probe of the P001 probe mix can be found at the www.mrc-holland.com website.
Detection of trisomies by MLPA
Samples containing ∼100 ng DNA were analysed by MLPA using probe mix P001. Male and female control DNA was obtained from Promega. Blood-derived DNA from triple 21 and triple X individuals as well as DNA from a triple 13 and a triple 18 cell line were provided by the Department of Clinical Genetics, Free University of Amsterdam. Reactions were analysed by capillary electrophoresis on a Beckman CEQ2000. Peak area of each probe amplification product was divided by the combined peak area of all 40 probes. The resulting relative peak area of each probe amplification product was divided by the relative peak area of that probe obtained on female control DNA. The presence of two copies of a probe target sequence/diploid genome should therefore result in a relative signal of 1.00. The presence of three copies of a probe target sequence/diploid genome should result in a 1.50 relative probe signal. The exact sequence recognised by each probe of the P001 probe mix can be found at the www.mrc-holland.com website.
References
Norman,A.M., Thomas,N.S.T., Kingston,H.M. and Harper,P.S. (
Petrij-Bosch,A., Peelen,T., van Vliet,M., van Eijk,R., Olmer,R., Drusedau,M., Hogervorst,F.B., Hageman,S., Arts,P.J., Ligtenberg,M.J. et al . (
Wijnen,J., van der Klift,H., Vasen,H., Khan,P.M., Menko,F., Tops,C., Meijers Heijboer,H., Lindhout,D., Moller,P. and Fodde,R. (
Kauraniemi,P., Barlund,M., Monni,O. and Kallioniemi,A. (
Leyland-Jones,B. and Smith,I. (
Kallioniemi,A., Kallioniemi,O.P., Rutovitz,D., Gray,J.W., Waldman,F. and Pinkel,D. (
Klinger,K., Landes,G., Shook,D., Harvey,R., Lopez,L., Locke,P., Lerner,T., Osathanondh,R., Leverone,B., Houseal,T. et al . (
Snijders,A.M., Nowak,N., Segraves,R., Blackwood,S., Brown,N., Conroy,J., Hamilton,G., Hindle,A.K., Huey,B., Kimura,K. et al . (
Devilee,P., Cleton-Jansen,A.-M. and Cornelisse,C.J. (
Vos,P., Hogers,R., Bleeker,M., Reijans,M., van de Lee,T., Hornes,M., Frijters,A., Pot,J., Peleman,J. and Kuiper,M. (
Liang,P. and Pardee,A.B. (
Armour,J.A., Sismani,C., Patsalis,P.C. and Cross,G. (
Hsuih,T.C.H., Park,Y.N., Zaretski,C., Wu,F., Tyagi,S., Kramer,F.R., Sperling,R. and Zhang,D.Y. (
Carrino,J.J. (
Barany,F. and Lubin,M. (
Pauletti,G., Godolphin,W., Press,M.F. and Slamon,D.J. (
Feo,S., Di Liegro,C., Jones,T., Read,M. and Fried,M. (
Schouten,J.P. (
Pollack,J.R., Perou,C.M., Alizadeh,A.A., Eisen,M.B., Pergamenschikov,A., Williams,C.F., Jeffrey,S.S., Botstein,D. and Brown,P.O. (
Tong,J., Cao,W. and Barany,F. (
Housby,J.H. and Southern,E.M. (
BRCA1 Exon 13 Duplication Screening Group (
Jones,C., Payne,J., Wells,D., Delhanty,J.D., Lakhany,S.R. and Kortenkamp,A. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments