





Abstract
Background. The podocyte is bathed in an angiotensin II (AngII)-rich ultrafiltrate, but the impact of AngII on podocyte pathobiology is not well known. Because podocytes play a direct role in the glomerular basement membrane (GBM) thickening of diabetes, the α3(IV) collagen chain was examined. Podocyte expression of α3(IV) collagen may involve the transforming growth factor-β (TGF-β) and vascular endothelial growth factor (VEGF) systems.
Methods. Cultured mouse podocytes were treated with various doses of AngII for selected periods of time, with or without inhibitors of TGF-β and VEGF signalling, SB-431542 and SU5416, respectively. TGF-β1 and VEGF were assayed by enzyme-linked immunosorbent assay (ELISA); α3(IV) collagen, TGF-β type II receptor and phospho-Smad2 were assayed by immunoblotting.
Results. AngII ≥10−10 M was found to stimulate the production of α3(IV) collagen significantly in as short a time as 3 h. The expression of α3(IV) collagen was influenced by the TGF-β system, but AngII did not increase the podocyte's production of TGF-β1 ligand; rather, it increased the expression of the TGF-β type II receptor and activated the TGF-β signalling system through Smad2. Despite the TGF-β receptor upregulation, synergy between AngII and TGF-β1 to boost α3(IV) collagen production was not observed. However, blockade of TGF-β signalling with SB-431542 prevented AngII from stimulating α3(IV) collagen production. Podocyte expression of α3(IV) collagen was also increased by the autocrine activity of VEGF. Podocytes were stimulated to secrete VEGF by 10−10 M or higher AngII after 48 h. Blockade of the endogenous VEGF activity by SU5416 prevented AngII-stimulated α3(IV) collagen production.
Conclusions. AngII stimulates the podocyte to produce α3(IV) collagen protein via mechanisms involving TGF-β and VEGF signalling. Alterations in α3(IV) collagen production may contribute to GBM thickening and perhaps proteinuria in diabetes.
Introduction
Activation of the intrarenal renin–angiotensin system (RAS) is an important characteristic of diabetic kidney disease, and an expanding body of evidence supports a prime role for this system in the progressive development of proteinuria and the loss of kidney function. Under normal conditions, the glomerular level of angiotensin II (AngII) is elevated some 1000-fold above the plasma concentration of AngII (∼10−8 M vs ∼10−11 M, respectively) [1]. An intriguing concept has emerged that AngII not only mediates intraglomerular hypertension but also behaves as a growth factor that contributes to diabetic hypertrophy and sclerosis [2]. These so-called ‘non-haemodynamic’ effects of AngII have been investigated in many kidney cell lines. However, it remains unclear how elevated levels of AngII in diabetic nephropathy affect the podocyte.
Located at the periphery of each glomerular capillary loop, podocytes are bathed in an ultrafiltrate that is rich in AngII. In addition, these cells possess both the AT1 and AT2 receptors that mediate almost all of the physiological actions of AngII [3]. One of the non-haemodynamic effects of AngII is to stimulate the production of extracellular matrix [2], which in the case of the podocyte could be one of the novel chains of type IV collagen, a principal building block of the glomerular basement membrane (GBM). In particular, the α3 chain of collagen IV was selected for further investigation, because immunohistochemical studies on diabetic renal tissue have found that the subepithelial zone of the GBM is enriched in α3 and α4(IV) collagen [4]. The GBM thickening that results may be associated with the degree of proteinuria in diabetes [5].
The expression of α3(IV) collagen in podocytes is stimulated by at least two effector cytokines. Treatment with exogenous transforming growth factor-β1 (TGF-β1) markedly increases the production of this collagen IV chain in cultured podocytes [6,7]. In addition, the TGF-β signalling system mediates the stimulation of α3(IV) collagen production by high ambient glucose, perhaps due to upregulation of the TGF-β type II receptor (TβRII) rather than the TGF-β1 ligand [6]. When TGF-β is blocked by antibody therapy in the diabetic mouse, the thickening of the GBM is partially reversed [8], consistent with TGF-β playing an important role in the podocyte overexpression of GBM collagens such as α3(IV). Another cytokine that stimulates the production of α3(IV) collagen is vascular endothelial growth factor (VEGF). In cultured mouse podocytes, addition of exogenous VEGF164 increases α3(IV) protein, and inhibition of VEGF signalling by SU5416 (a pan-VEGF receptor inhibitor) prevents VEGF-stimulated α3(IV) collagen expression [7]. Furthermore, the endogenous VEGF that is constitutively secreted by the podocyte can act in an autocrine loop to sustain a basal level of α3(IV) production. This podocyte-derived VEGF is also responsible for part of TGF-β's stimulatory effect on α3(IV) collagen production [7]. Since TGF-β and VEGF activities can be stimulated by AngII [9,10], the intriguing possibility arises that these two cytokine systems may underlie certain effects of AngII on the podocyte.
The present study was undertaken to evaluate the impact of graded concentrations of AngII on the podocyte's production of a key GBM constituent, α3(IV) collagen. After evaluating the dose–response and time course relationships between AngII treatment and α3(IV) collagen production, we tested whether the TGF-β and VEGF systems might mediate AngII's effect on α3(IV) collagen. First, components of the TGF-β signalling system were analysed in terms of their response to AngII treatment, and then the TGF-β pathway was specifically inhibited to gauge its involvement in AngII-stimulated α3(IV) collagen production. Because TGF-β1 itself increases α3(IV) collagen expression, a synergy between TGF-β1 and AngII was investigated. Secondly, the effect of AngII on the podocyte secretion of VEGF was characterized. Specific blockade of the VEGF signalling pathway was used to determine the role of the VEGF autocrine loop in mediating the stimulatory action of AngII on the production of α3(IV) collagen.
Subjects and methods
Podocyte cell culture
Conditionally immortalized mouse podocytes were kindly provided by Dr Peter Mundel (Albert Einstein College of Medicine; Bronx, NY) and were handled as previously described [6,7]. The cells harbour a temperature-sensitive variant of the SV40 large T antigen (tsA58) that is inducible by γ-interferon and is stable at 33°C but is rapidly degraded at 37–39°C [11]. At 33°C, the large T antigen allows for cellular proliferation. The growing cells are maintained in collagen I-coated flasks in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin, 100 µg/ml streptomycin and 10 U/ml mouse recombinant γ-interferon. When the cells have reached confluence, they are passaged and allowed to differentiate at 37.5°C for 2 weeks without γ-interferon in serum-free Dulbecco's modified Eagle's medium (DMEM) containing 5.5 mM glucose, 100 U/ml penicillin and 100 µg/ml streptomycin. FCS was removed from the culture medium because it destroys AngII within minutes, verified by an AngII enzyme-linked immunosorbent assay (ELISA; SPI-bio; Massy Cedex, France). In serum-free medium, however, AngII is stable (data not shown). Podocyte differentiation was confirmed by the expression of α-synaptopodin [11].
Experimental treatment conditions
Differentiated podocytes were treated with exogenous AngII (Sigma, St Louis, MO) in concentrations ranging from 10−12 to 10−5 M. Treatment times ranged from 1 to 8 h if the experiment was conducted in a regular working day and up to 52 h if conducted over several days (see figure legends for details). Because AngII is degraded within 2 h when in contact with the cultured podocytes (by cell surface aminopeptidases [12]), the dose of AngII was replenished every 2 h during the working day; for experiments longer than a single working day, the AngII dose was replenished every 2 h during an 8 h working day, and then dosing was resumed in the morning of the subsequent working day.
In the AngII/TGF-β1 synergy experiment, recombinant human TGF-β1 (R&D Systems) was added at 2 ng/ml for 4 h. This dose and duration have been shown to significantly stimulate α3(IV) collagen production [6]. Concurrent with the AngII treatment in certain experiments, specific inhibitors of the TGF-β and VEGF signalling pathways were added: SB-431542, an inhibitor of the TGF-β type I receptor kinase (gift of Dr Nicholas Laping, GlaxoSmithKline; IC50 ∼100 nM) [6], and SU5416, an inhibitor of the VEGF receptor tyrosine kinases (gift of SUGEN/Pfizer; IC50 ∼1 μM for inhibition of receptor autophosphorylation) [7].
Western immunoblotting
Preparation of total lysate protein in RIPA buffer followed by SDS–PAGE (3–8% gradient, pre-cast gels from NuPAGE; Invitrogen, Carlsbad, CA) and wet transfer to a nitrocellulose membrane (Bio-Rad, Hercules, CA) were performed as described [6]. The membrane was blocked in 5% non-fat milk in Tris-buffered saline–0.1% Tween-20 (TBS-T) and probed overnight at 4°C with one of the following primary antibodies: human anti-α3(IV) collagen (University of Pennsylvania), rabbit anti-TβRII (Santa Cruz Biotechnology), rabbit anti-phospho-Smad2 (Cell Signaling Technology) or mouse anti-β-actin antibody (Sigma). After three washes in TBS-T, membranes were probed with the appropriate secondary antibody: goat anti-human (Jackson ImmunoResearch, West Grove, PA), donkey anti-rabbit (Amersham Biosciences, Piscataway, NJ) or sheep anti-mouse antibody (Amersham), each conjugated to horseradish peroxidase (HRP). The HRP-catalysed chemiluminescence reaction was developed with SuperSignal West Pico substrate (Pierce Biotechnology, Rockford, IL), allowing the detection of immunoreactive protein bands. The membrane was re-probed with β-actin to correct for small differences in loading. The resulting bands on film were quantitated by computer-assisted video densitometry. Using the ImageJ 1.29× software (National Institutes of Health, Bethesda, MD), the protein band density was measured. The amount of protein under control conditions was assigned a relative value of 100%.
TGF-β1 ELISA
Conditioned cell culture media were frozen at −20°C until assayed by a TGF-β1 ELISA kit, performed according to the manufacturer's instructions (R&D Systems). In brief, the supernatants of the podocyte cultures were acid activated with 1 M HCl and then neutralized with 1.2 M NaOH/0.5 M HEPES to measure total TGF-β1 (latent plus active fractions). Samples were applied to microtitre plates that had been pre-coated with TβRII. After a 3 h incubation, the wells were washed and a HRP-conjugated anti-TGF-β1 antibody was added to the wells for 1.5 h at room temperature. Following another wash, the substrate for HRP was added. The resulting chromogenic reaction was halted with a stop solution. The absorbance at 450 nm was measured in a microplate reader. TGF-β1 concentrations were determined from the standard curve and corrected for the amount of total cell protein (data expressed as pg of TGF-β1/mg of protein).
VEGF120,164 ELISA
Cell culture media supernatants were frozen at −20°C until assayed for VEGF using a commercial ELISA kit (R&D Systems). The supernatants were dispensed onto a microtitre plate coated with an anti-VEGF antibody that recognizes mouse VEGF120,164 and then sandwiched with a secondary anti-VEGF antibody conjugated to HRP. The resulting chromogenic reaction was stopped and read at 450 nm, and the concentration of VEGF was determined from the standard curve. VEGF concentrations were then corrected for the total protein concentrations, and the data were expressed as pg of VEGF/mg of protein.
Statistical analysis
Graphical data are displayed as the mean±SE for the number of independent experiments indicated in the figure legends. When comparing two independent sets of data, i.e. control vs an experimental group, the unpaired Student's t-test was used. When comparing control with multiple treatment conditions as in the dose–response and time course data, a two-way analysis of variance (ANOVA) with Bonferroni post-test was used. P<0.05 was considered statistically significant.
Results
Dose-response of the AngII effect on α3(IV) collagen production
The addition of AngII to the media of cultured mouse podocytes effectively stimulated the protein production of α3(IV) collagen over an 8 h period. A mild but noticeable increase in α3(IV) collagen was detected at 10−11 M AngII (Figure 1A). A 10-fold higher concentration, 10−10 M, of AngII stimulated α3(IV) production even more (Figure 1A). The dose-dependent effect of AngII on α3(IV) collagen production reached a plateau at 10−8 M, having increased the amount of α3(IV) collagen by some 50% above control (Figure 1A).
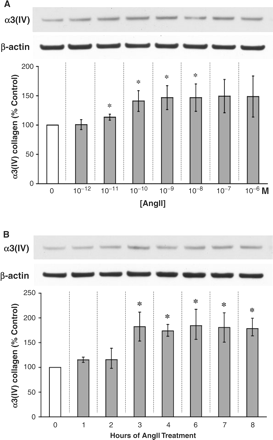
AngII affects α3(IV) collagen production by podocytes. (A) AngII dose–response: exogenous AngII was added to cultured mouse podocytes every 2 h for a total of 8 h at the concentrations indicated on the graph. From 10−11 to 10−8 M, AngII significantly stimulated the protein production of α3(IV) collagen by 13–49%. *P<0.05 vs control (0 M AngII) in nine independent experiments. (B) AngII time course: AngII at 10−8 M was added every 2 h (where feasible) for the time periods indicated on the graph. At 3 h up until 8 h at least, the stimulatory effect of AngII on α3(IV) collagen production was significant and sustained. *P<0.05 vs control (0 h) in six independent experiments.
Time course of the AngII effect on α3(IV) collagen
At a fixed dose of 10−8 M AngII, the stimulation of α3(IV) collagen production was evident in as short a time as 3 h (Figure 1B). Additional time of exposure to AngII did not appear to increase the quantity of α3(IV) collagen protein in the mouse podocytes further, but the effect of AngII persisted over the remainder of the 8 h incubation (Figure 1B).
AngII does not stimulate TGF-β1 secretion
Treatment with AngII doses, increasing in exponential orders of magnitude from 10−12 to 10−5 M, did not significantly raise the level of total TGF-β1 protein secreted into the cell culture media of mouse podocytes over an 8 h span (Figure 2A, left panel). Likewise, a fixed dose of 10−8 M AngII used for selected periods of time from 6 to 48 h failed to stimulate podocyte secretion of total TGF-β1 into the media supernatant (Figure 2A, right panel).
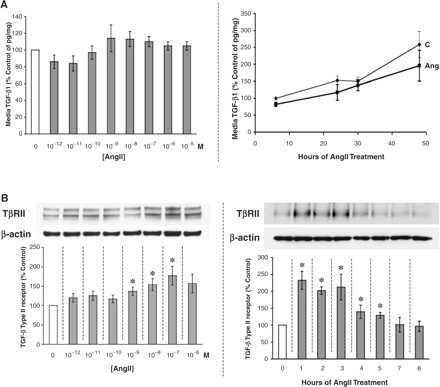
AngII stimulates the TGF-β system at the receptor level. (A) AngII does not significantly affect TGF-β1 secretion by the podocyte. AngII was added to cultured mouse podocytes every 2 h for 8 h at increasing doses (dose–response, left panel) or was added at 10−8 M for 6–48 h (time course, right panel). The concentration of TGF-β1 in the culture media was measured by a specific ELISA (R&D Systems) and corrected for the total cellular protein concentration (pg of TGF-β1/mg of protein); results are displayed as the percentage of the control in five independent experiments. (B) Increasing doses of exogenous AngII for 8 h upregulate the protein production of the signalling TGF-β type II receptor (TβRII) in podocytes, measured as a percentage of control by western immunoblotting. AngII-stimulated TβRII expression was significant and sustained after 10−9 M AngII. *P<0.05 vs control in four independent experiments (left panel). Increasing time of exposure to 10−8 M AngII (added every 2 h where feasible) had the greatest effect on the stimulation of TβRII production at 1–3 h, tapering by 7 h. *P<0.05 vs control (0 h) in six independent experiments (right panel). (C) AngII stimulates TGF-β signalling through the Smad2 pathway. AngII at 10−8 M was added to cultured podocytes for 8 h, with or without treatment with 1 μM SB-431542, a TGF-β signalling inhibitor. Quantities of phospho-Smad2 were significantly increased by AngII but were suppressed below baseline by SB-431542. *P<0.05 vs control and †P<0.05 vs AngII in eight independent experiments.
AngII increases TGF-β type II receptor abundance
Although physiological levels of AngII failed to increase TGF-β1 secretion, they stimulated the protein production of another component of the TGF-β system, the signalling TβRII. This increase in TβRII protein was dose dependent, becoming statistically significant at 10−9 M AngII (Figure 2B, left panel). In the time course experiments, a fixed dose of 10−8 M AngII had the greatest effect on the stimulation of TβRII production at 1–3 h; AngII's stimulatory effect was sustained until 6 h (Figure 2B, right panel).
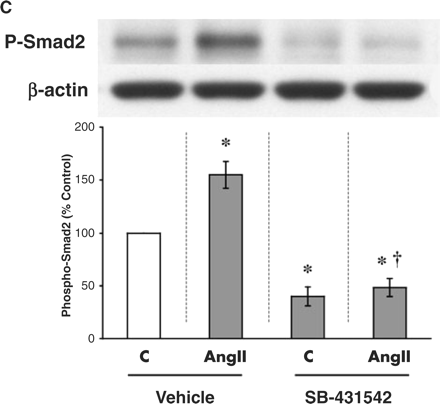
AngII activates TGF-β signalling
To demonstrate the functional significance of AngII-stimulated TβRII, evidence for the activation of the TGF-β signalling pathway was sought. After binding to TβRII, TGF-β signals primarily through the Smad2 and Smad3 pathways [13]. Activation of either pathway can be assessed by the phosphorylation of the Smad2 or Smad3 protein. Levels of phospho-Smad2 significantly increased by 55±13% with the addition of 10−8 M AngII for 8 h (Figure 2C). However, phosphorylation of Smad2 was blocked by an inhibitor of TGF-β signalling, SB-431542 (1 μM for 8 h), such that steady-state levels of phospho-Smad2 were suppressed below baseline (Figure 2C). In the presence of SB-431542, AngII could not stimulate Smad2 phosphorylation (Figure 2C).
TGF-β signalling mediates the effect of AngII on α3(IV) collagen
Consistent with the dose–response and time course data, AngII treatment at 10−8 M for 8 h increased α3(IV) collagen production by ∼30% (P<0.05 vs control, Figure 3). However, AngII-stimulated α3(IV) collagen production was prevented by pre-treatment with SB-431542 (1 μM for 8 h). This small molecule inhibitor blocks the kinase activity of TβRI, arresting the transmission of the TGF-β signal. Thus, the TGF-β pathway plays a role in the mechanism of AngII-stimulated production of α3(IV) collagen.
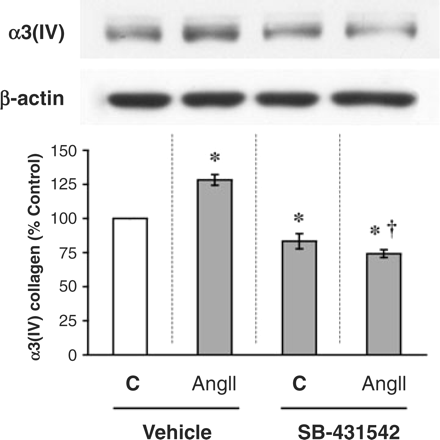
TGF-β signalling mediates the effect of AngII to stimulate α3(IV) collagen. AngII at 10−8 M was added to cultured podocytes for 8 h, with or without treatment with 1 μM SB-431542, a TGF-β signalling inhibitor. AngII stimulated α3(IV) collagen production by ∼30%, but this was prevented by concurrent treatment with SB-431542. In addition, SB-431542 lowered the baseline production of α3(IV) collagen in the absence of AngII. *P<0.05 vs control and †P<0.05 vs AngII in five independent experiments.
Combination of AngII and TGF-β1 exerts a greater effect than AngII alone
Cultured podocytes were treated with AngII at 10−8 M for 24 h; this increased the protein production of α3(IV) collagen by ∼30% (P<0.05 vs control, Figure 4). Treatment with TGF-β1 at 2 ng/ml for 4 h stimulated α3(IV) collagen production by ∼70% (P<0.05 vs control, Figure 4). However, the combination of AngII and TGF-β1, wherein AngII was added for the first 24 h and TGF-β1 was added for the last 4 h of the experiment, stimulated α3(IV) production the greatest, increasing collagen IV production by ∼80% (P<0.05 vs control or AngII, Figure 4).
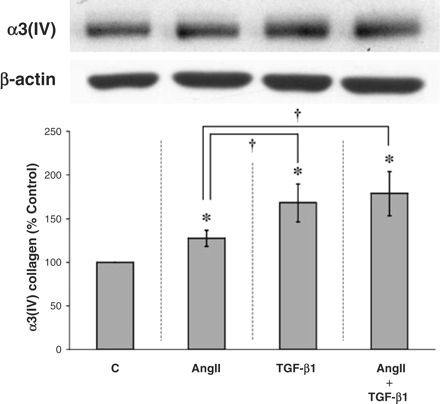
Combination treatment with AngII and TGF-β1 is more effective than AngII alone but not more effective than TGF-B1 alone. Cultured mouse podocytes were treated with 10−8 M AngII to upregulate the expression of the TGF-β type II receptor; then a submaximal dose (0.5 ng/ml) of TGF-β1 was added for the last 4 h of the experiment. Both AngII alone and TGF-β1 alone significantly increased the production of α3(IV) collagen, with TGF-β1 having the significantly greater effect. In comparison, the combination of AngII and TGF-β1 exerted a larger effect on α3(IV) collagen than AngII alone but not greater than TGF-β1 alone. *P<0.05 vs control and †P<0.05 vs AngII alone in five independent experiments.
AngII raises podocyte VEGF production
The podocyte is the primary source of VEGF in the glomerulus, and it can be stimulated to secrete more VEGF by AngII. Beginning at 10−10 M, AngII was found to increase VEGF protein production significantly, as measured in the culture media via ELISA (Figure 5A). Higher doses of AngII also significantly raised podocyte VEGF production. There appeared to be two peaks of VEGF stimulation, one at 10−10 M and the other at 10−6 M AngII (Figure 5A).
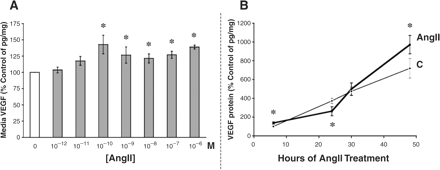
AngII increases VEGF secretion in a dose- and time-dependent manner. (A) Mouse podocytes that were treated with increasing doses of AngII over an 8 h period secreted significantly more VEGF120,164 into the culture media, as measured by a specific ELISA (R&D Systems). The concentration of VEGF was adjusted for the total cellular protein concentration (pg of VEGF/mg of protein), and the results are presented as the percentage of control. *P<0.05 vs control in four independent experiments. (B) Podocytes were treated with 10−8 M AngII from 6–48 h. Untreated control podocytes showed a steady rate of VEGF accumulation in the culture media over time (thin line). Compared with control, AngII treatment (thick line) significantly increased media VEGF levels at 6 h, but by 24 h VEGF quantities had significantly dropped. At 30 h, however, VEGF secretion in the AngII-treated podocytes had caught up to control levels, and at 48 h AngII-induced VEGF production significantly exceeded control. *P<0.05 vs corresponding time point control in seven independent experiments.
In a related set of experiments, the effect of a single AngII dose, 10−8 M, was observed over time. Starting at 6 h, the quantity of VEGF protein in the cell culture media of AngII-treated podocytes was already 30% greater than control (P<0.05, Figure 5B), consistent with the data of the dose–response curve (Figure 5A). With continued exposure to AngII, the media VEGF level dropped significantly below control at 24 h (Figure 5B), before catching up at 30 h and then surpassing control levels at 48 h (P<0.05, Figure 5B).
VEGF signalling system mediates AngII-stimulated α3(IV) collagen production
Because of the dose–response and time course characteristics of the AngII effect on VEGF production, cultured podocytes were treated with 10−6 M AngII for 52 h to ensure an increased ambient VEGF level. This podocyte-derived VEGF acts in an autocrine loop to stimulate the production of α3(IV) collagen [7]. Using this as an end-point, AngII stimulated the protein production of α3(IV) collagen by 37% (Figure 6). However, AngII's effect on α3(IV) collagen was completely prevented by an inhibitor of VEGF signalling, SU5416 (Figure 6) [7].
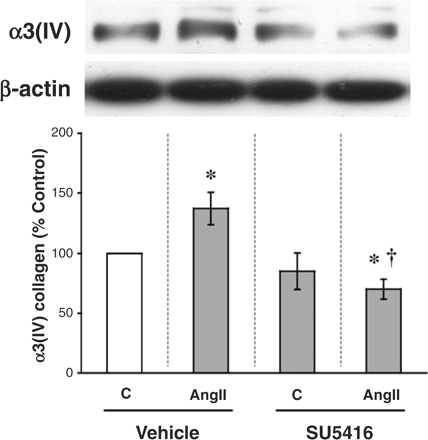
AngII-induced α3(IV) collagen production is also mediated by VEGF signalling. Cultured podocytes were exposed to 10−6 M AngII for 52 h, with or without treatment with 5 μM SU5416, a VEGF signalling inhibitor. AngII stimulated α3(IV) collagen production by 37%, but this was prevented by concurrent treatment with SU5416. *P<0.05 vs control and †P<0.05 vs AngII in five independent experiments.
Discussion
Although the pathophysiology of diabetic nephropathy has classically emphasized the haemodynamic mechanisms of AngII-induced renal injury, the non-haemodynamic actions of AngII, which can be investigated in cell culture, are equally important and are becoming more widely appreciated. The initial studies focused on the effects of AngII on mesangial, proximal tubule and glomerular endothelial cells and found that AngII could induce a number of pleiotropic effects on cell growth [14,15] and extracellular matrix expression [16], some of which are dependent upon the autocrine TGF-β system [9] and the small GTP-binding proteins [15]. With the advent of a reliable means to culture and study differentiated podocytes [11], the impact of an elevated AngII concentration on podocyte pathobiology can be examined.
Given the profibrotic nature of AngII in most of the renal cell types studied to date, it is reasonable to evaluate the effects of AngII on the podocyte's expression of extracellular matrix. Perhaps the most suitable set of matrix proteins to assay would be type IV collagen, because this branching, sheet-like collagen forms the meshwork of the GBM. Specifically, we assayed the novel α3 chain of type IV collagen because it is synthesized by the podocyte, and its relative abundance increases in the thickened GBM of diabetic nephropathy [17]. The expression of α3(IV) collagen previously was shown to be increased by AngII, but these experiments were performed in proximal tubule cells [18]. In our podocyte studies, AngII was found to stimulate α3(IV) collagen production significantly at 10−11 M, but the biologically significant effect seems to occurs at 10−10 M AngII and above (Figure 1).
The expression of α3(IV) collagen in mouse podocytes is potently stimulated by TGF-β1 treatment [6]. We investigated whether the TGF-β system might play a role in AngII-stimulated α3(IV) collagen production. AngII did not significantly stimulate TGF-β1 secretion into the culture media, but AngII did stimulate the expression of TβRII (Figure 2B). Perhaps because it upregulated TβRII, AngII was able to activate the TGF-β signalling system in podocytes, manifested by the increased phosphorylation of Smad2 that was preventable by a TGF-β signalling inhibitor, SB-431542 (Figure 2C). In this regard, the podocyte differs from other renal cell types that respond to AngII by increasing their expression of both TGF-β ligand and type II receptor [19].
One consequence of AngII stimulating the TGF-β system appears to be an increase in α3(IV) collagen production. The ability of AngII to augment α3(IV) collagen production was abrogated by specifically inhibiting the TGF-β type I receptor (Figure 3), through which TGF-β and TβRII ultimately signal. This indicates that the effect of AngII to stimulate α3(IV) collagen production is mediated by the TGF-β system. That treatment with SB-431542 alone significantly decreased α3(IV) collagen levels suggests that the basal rate of α3(IV) production is dependent upon endogenous TGF-β activity (Figure 3).
Another consequence of AngII-induced expression of TβRII could be a kind of synergy between AngII and TGF-β1. AngII stimulates α3(IV) collagen production in a TGF-β-dependent manner, but, by increasing the abundance of the TβRII, AngII may potentiate the effect of the ambient TGF-β. As expected, both AngII and TGF-β1 by themselves increased α3(IV) collagen levels, but the combination of AngII and TGF-β1 did not significantly raise α3(IV) collagen production above that found with TGF-β1 alone (Figure 4). Care was taken to allow the AngII-treated podocytes enough time to upregulate the production of TβRII, and afterwards to treat the podocytes with a low, submaximal dose of TGF-β1 (0.5 ng/ml) to allow the α3(IV) collagen level to increase if possible. Our results currently argue against a synergism between AngII and TGF-β1 treatments on the podocyte production of α3(IV) collagen.
Collagen IV expression in podocytes is also influenced by the endogenous VEGF system. This podocyte-derived VEGF recently has been discovered to act in an autocrine manner to exert profound physiological and pathophysiological effects in podocytes [7,20]. Lending credence to these observations, podocytes are known to possess certain receptors for VEGF, including VEGFR-1 (Flt-1), VEGFR-3 (Flt-4) and the accessory VEGF receptor—neuropilin-1 [7,20]. The VEGF autocrine loop in podocytes appears to function in intracellular calcium release and protection against cytotoxicity and apoptosis [20]. In addition, our laboratory has found that the VEGF autocrine loop plays an important role in the podocyte's expression of α3(IV) collagen [7]. Under standard conditions, the actions of endogenous VEGF maintain a basal level of α3(IV) collagen production, but supraphysiological doses of VEGF can augment α3(IV) expression [7]. Finally, podocyte-derived VEGF is stimulated by TGF-β1, and the ensuing increase in VEGF autocrine activity mediates part of TGF-β1's ability to stimulate α3(IV) collagen production [7].
The endogenous VEGF system may also mediate the effect of AngII to stimulate α3(IV) collagen expression. Podocyte VEGF secretion is increased by doses of AngII at 10−10 M or higher (Figure 5A) but, interestingly, VEGF production is actually decreased by AngII at 24 h (Figure 5B). Afterwards, continued treatment with AngII causes the VEGF production to catch up to control levels at 30 h and to surpass them at 48 h (Figure 5B). Given the time course relationship between AngII and VEGF, AngII may increase α3(IV) collagen expression at 48 h or later. We found that AngII stimulates α3(IV) protein production at 52 h, and this is prevented by an inhibitor of VEGF signalling, SU5416 (Figure 6). Together with the data from Figure 3, the findings suggest that the early effect of AngII on α3(IV) collagen is TGF-β dependent and that the late effect is VEGF dependent.
Ultimately, the impact of AngII on podocyte pathobiology with regard to diabetic nephropathy may lie in the alteration of α3(IV) collagen production. The absolute increase in this α collagen chain may help to explain the GBM thickening of diabetes [17], a kidney lesion that can be prevented by AngII antagonists. Despite the increase in the quantity of GBM, the ‘quality’ of GBM seems to suffer, because the thickened GBM loses its permselectivity and becomes more permeable to filtered proteins, most notably albumin. It is intriguing to postulate that the underlying basis may involve an alteration in the ratio of α3(IV) collagen to the other α chains that disrupts the podocyte's methodical assembly of the GBM. Consistent with this theory, the GBM in diabetes is enriched in α3 and α4(IV) collagen but relatively deficient in α5(IV) collagen [17]. According to our study, the disproportionate increase in the podocyte production of α3(IV) collagen may derive from the actions of AngII. Insofar as alterations in α3(IV) collagen contribute to GBM thickening and perhaps proteinuria in diabetes, a mechanism underlying the renoprotection by angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may be to shield the podocyte from the metabolic effect of AngII to activate the TGF-β and VEGF systems that transmit the signal to stimulate α3(IV) collagen production, highlighting the complex interplay between the three major cytokine systems paramount to podocyte biology in diabetes.
The authors wish to thank Ms Yuki Kasama (University of Pennsylvania) for technical assistance, Dr Peter Mundel (Albert Einstein College of Medicine) for donating the conditionally immortalized mouse podocyte cell line, and Dr Michael P. Madaio (University of Pennsylvania) for supplying the human polyclonal anti-α3(IV) collagen antibody. This work was supported by grants from the National Institutes of Health (DK-61537 to S.C.; DK-54608 and DK-44513 to F.N.Z.) and the American Diabetes Association (to F.N.Z.).
Conflict of interest statement. None declared.
References
Seikaly MG, Arant BS Jr, Seney FD Jr. Endogenous angiotensin concentrations in specific intrarenal fluid compartments of the rat.
Wolf G, Ziyadeh FN. The role of angiotensin II in diabetic nephropathy: emphasis on nonhemodynamic mechanisms.
Wang L, Flannery PJ, Spurney RF. Characterization of angiotensin II-receptor subtypes in podocytes.
Zhu D, Kim Y, Steffes MW, Groppoli TJ, Butkowski RJ, Mauer SM. Glomerular distribution of type IV collagen in diabetes by high resolution quantitative immunochemistry.
Caramori ML, Kim Y, Huang C et al. Cellular basis of diabetic nephropathy: 1. Study design and renal structural–functional relationships in patients with long-standing type 1 diabetes.
Iglesias-de la Cruz MC, Ziyadeh FN, Isono M et al. Effects of high glucose and TGF-β1 on the expression of collagen IV and vascular endothelial growth factor in mouse podocytes.
Chen S, Kasama Y, Lee JS, Jim B, Marin M, Ziyadeh FN. Podocyte-derived vascular endothelial growth factor mediates the stimulation of α3(IV) collagen production by transforming growth factor-β1 in mouse podocytes.
Chen S, Iglesias-de la Cruz MC, Jim B, Hong SW, Isono M, Ziyadeh FN. Reversibility of established diabetic glomerulopathy by anti-TGF-β antibodies in db/db mice.
Wolf G, Mueller E, Stahl RAK, Ziyadeh FN. Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor-β.
Pupilli C, Lasagni L, Romagnani P et al. Angiotensin II stimulates the synthesis and secretion of vascular permeability factor/vascular endothelial growth factor in human mesangial cells.
Mundel P, Reiser J, Zúñiga-Mejía-Borja A et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines.
Bakris GL, Re RN. Endothelin modulates angiotensin II-induced mitogenesis of human mesangial cells.
Massague J, Wotton D. Transcriptional control by the TGF-β/Smad signaling system.
Wolf G, Ziyadeh FN, Zahner G, Schroeder R, Stahl RAK. Angiotensin II is mitogenic for cultured rat glomerular endothelial cells.
Zeng L, Xu H, Chew TL et al. Simvastatin modulates angiotensin II signaling pathway by preventing Rac1-mediated upregulation of p27.
Wolf G, Haberstroh U, Neilson EG. Angiotensin II stimulates the proliferation and biosynthesis of type I collagen in cultured murine mesangial cells.
Funabiki K, Makita Y, Yamamoto M et al. Dissociated expression of collagen type IV subchains in diabetic kidneys of KKAy mice.
Wolf G, Kalluri R, Ziyadeh FN, Neilson EG, Stahl RA. Angiotensin II induces α3(IV) collagen expression in cultured murine proximal tubular cells.
Wolf G, Ziyadeh FN, Stahl RA. Angiotensin II stimulates expression of transforming growth factor β receptor type II in cultured mouse proximal tubular cells.
Author notes
1Renal-Electrolyte and Hypertension Division of the University of Pennsylvania, Philadelphia, PA, 3Division of Nephrology/Hypertension, Northwestern University Feinberg School of Medicine, Chicago, IL, USA, 2Cellular Biology Unit, Department of Biology, Universidad Autónoma de Madrid, Spain and 4Klinik für Innere Medizin III, University of Jena, Jena, Germany


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments