





Abstract
With the huge increase of protein data, an important problem is to estimate, within a large protein family, the number of sensible subsets for subsequent in-depth structural, functional, and evolutionary analyses. To tackle this problem, we developed a new program, Secator, which implements the principle of an ascending hierarchical method using a distance matrix based on a multiple alignment of protein sequences. Dissimilarity values assigned to the nodes of a deduced phylogenetic tree are partitioned by a new stopping rule introduced to automatically determine the significant dissimilarity values. The quality of the clusters obtained by Secator is verified by a separate Jackknife study. The method is demonstrated on 24 large protein families covering a wide spectrum of structural and sequence conservation and its usefulness and accuracy with real biological data is illustrated on two well-studied protein families (the Sm proteins and the nuclear receptors).
Introduction
With the rapid growth of the sequence databases, the number of sequences belonging to a particular functionally related protein family is increasing sharply. As a consequence, it is becoming more and more necessary for biologists to analyze the relationships existing between the numerous members of a protein family and categorize them into sensible subfamilies. Subfamilies are frequently representative of sets of proteins with related functions and/or distinct domain organizations resulting from different evolution histories. Clustering approaches have until now been focused on the discovery of groups of homologous proteins in entire protein databases (Wolf et al. 1999 ; Enright and Ouzounis 2000 ; Krause, Stoye, and Vingron 2000 ; Tatusov et al. 2000 ) based on single-sequence similarity search algorithms such as BLAST (Altschul et al. 1997 ) and FASTA (Pearson 1994 ). However, these methods are not suitable for in-depth phylogenetic studies of a predefined set of proteins. Inference of subfamilies in sets of homologous proteins remains crucial in order to gain insight into their real functional and evolutionary relationships. This is usually done by collapsing internal branches of a phylogenetic tree, either manually, using a graphical tool such as the TreeView program (Page 1996 ), or in a semiautomatic way with sequence grouping guided by the reliability of the branching order. The latter method was used, for example, to define groups among receptors (Nuclear Receptors Nomenclature Committee 1999 ) and among myosin sequences (Hodge and Cope2000) . Phylogenetic trees have also been used to group sequences (Lichtarge, Bourne, and Cohen 1996 ; Corpet, Gouzy, and Kahn 1999 ), but in both cases, the user must define the maximum distance or the minimum percentage of identity required for sequences to belong to the same group. To our knowledge, only one algorithm has been proposed which addresses the problem of automatic clustering of probable functional subfamilies in a phylogenetic tree (Sjolander 1998 ). This algorithm is based on the minimization of an encoding cost of the multiple alignment of a set of proteins.
Here we present a new program called Secator which is based on a different principle and has the advantage that it is fully automatic. The first step is to create a tree from a distance matrix based on a multiple alignment using BIONJ (Gascuel 1997 ). The program assigns a dissimilarity value to each node in the tree and then collapses branches by automatically detecting the nodes joining distant subtrees (NJDSTs). The method was validated on 24 protein families and is illustrated using two well-studied protein families: the Sm proteins (Salgado-Garrido et al. 1999 ) and the nuclear receptors (Wurtz et al. 1996 ). Our automatic partitioning is in good agreement with previously defined subfamilies grouped according to biological data. In addition, the program distinguished five main subfamilies among the 233 nuclear receptors from Caenorhabditis elegans that have been predicted and aligned (J. Fagart, personal communication).
Materials and Methods
Determination of Subfamilies of Proteins from a Phylogenetic Tree of n Sequences
Given are an unrooted tree, an n-by-n distance matrix, n sequence weights either all equal to 1 (by default) or calculated by Secator using the algorithm described in Thompson, Higgins, and Gibson (1994) , and an integer resolution value (R) which enables the user to ask for more or less groups than in the original clustering if it is set to a positive or negative value, respectively. R is set to 0 by default.
- Initially, each sequence forms a different family of proteins, and a dissimilarity value between each pair (i, j) of families is calculated according to the formulawhere wseqi and wseqj are, respectively, the weights of the sequences i and j, and d(seqi, seqj) is the distance between the sequences i and j as given by the distance matrix.
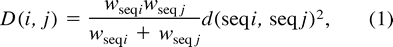
While the number of families is greater than two do
Among the pairs of families which have a common ancestor node, select the pair which has the smallest dissimilarity value. The dissimilarity value D(i, j) is assigned to the node of this pair. Families i and j are joined to form a single family (i, j). The number of families is reduced by one.
- Compute the Ward's dissimilarity value between the family (i, j) and each family k with k ≠ i, j:
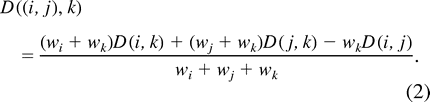
- Compute the weight of the family (i, j):\[\mathit{w}(\mathit{i,\ j})\ {=}\ \mathit{w_{i}}\ {+}\ \mathit{w_{j}}.\ (3)\]
The dissimilarity value between the two remaining families is assigned to a virtual node (node_V in fig. 1 ).
The nodes are clustered into two groups, the group with high dissimilarity values and the group with low dissimilarity values. This clustering is done by computing the partition into two groups which has the maximum interclass inertia on a subset of all possible partitions. Initially, D = {Di}i∈〈1,n〉 is the set of all the dissimilarity values sorted in decreasing order, and g is the mean of D.
For i = 2 to n do
Partition the dissimilarity values into two groups Ei and Fi, where Ei is the group of high dissimilarity values and Fi is the group of low dissimilarity values.where we = |Ei|, wf = |Fi|, and d is the usual distance.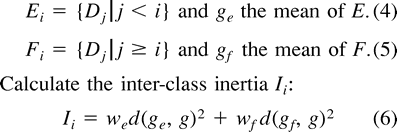
>[rf[li>The best partitioning is given by the pair (Ek, Fk), for which the corresponding Ik is the highest. This partitioning produces a threshold value of dissimilarity (TD) which is the highest value of Fk.
For |R| times do
- If R > 0 then do D is the set of all dissimilarity values less than the threshold TD.else\[\mathit{D}\ {=}\ {\{}\mathit{D_{i}},\ \mathit{i}\ {\in}\ {\langle}1,\ \mathit{n}{\rangle}{\,}{\vert}{\,}\mathit{D_{i}}\ {\leq}\ TD{\}}\ (7)\]\[\mathit{D}\ {=}\ {\{}\mathit{D_{i}},\ \mathit{i}\ {\in}\ {\langle}1,\ \mathit{n}{\rangle}{\,}{\vert}{\,}\mathit{D_{i}}\ {>}\ TD{\}}.(8)\]
Repartition D into two groups as in step 4 and compute a new TD which replaces the precedent TD.
The NJDSTs are defined as the nodes with dissimilarity values above the TD. Then, from the leaves of the tree up to the internal branches, branches are collapsed until NJDSTs are met. For example, in figure 1, a, b, and c are collapsed.
Implementation
The method presented here is implemented in the program Secator, which is written in C and should run on any UNIX machine. The program takes as input either a distance matrix in PHYLIP format or a multiple alignment in MSF or FASTA format. In the latter case, distances between the sequences are based on percentages of residue identity. A phylogenetic tree is then calculated using BIONJ (Gascuel 1997 ). Secator produces two output files: the collapsed tree in PHYLIP format and a table of the sequence groups with their mean distance (MD) scores (Thompson et al.2000) when the alignment is given. In addition to the resolution and weighting parameters, the user can also choose whether to conserve the distances in the final tree or have a multifurcate tree. A jackknife option provides an assessment of the quality of the clustering and of the number of groups. The program and multiple alignments are available by ftp at http://www-bio3d-igbmc.u-strasbg.fr/~wicker/Secator/secator.html.
Results and Discussion
The novelty of our method is the automatic clustering of the nodes of a phylogenetic tree to define probable functional subfamilies. This is realized by labeling each node with a dissimilarity value, which gives an objective estimation of the divergence of its external sub-branches. Ward's aggregative dissimilarity measure is preferred to the usual single-linkage or complete-linkage hierarchical clustering, which are also broadly used. Indeed, the progressive aspect of sequence data would tend to create oversized clusters using the single linkage. As for the complete linkage, it creates groups too compact to deal with such a sparse set of data. When an alignment is submitted, the dissimilarity is based on percentage of identity because it is the measure that is least sensitive to the physicochemical bias of the studied sequences (e.g., transmembrane sequences). However, the user has the option of providing other distances in the form of a matrix.
When dissimilarity values are above an automatically computed threshold (TD), the external subtrees are assumed to be “unmergeable,” and the nodes are designated NJDSTs. When all NJDSTs have been inferred, all branches are collapsed from the leaves up to an NJDST and the corresponding sequences are clustered into a subfamily.
The major problem is to automatically determine a suitable threshold for high dissimilarity values. In ascending hierarchical clustering, where dissimilarity values can be calculated in the same way, the threshold above which the obtained dendrogram should be cut is usually found manually by looking at the elbow of the curve of the dissimilarity values sorted in descending order (fig. 2A > and D). Many stopping rules provide an automatic threshold (Milligan and Cooper 1985 ); however, these rules are not generally suitable when the differences between clusters are fuzzy. We present a new stopping rule of geometric nature which focuses on the clustering essence of the stopping rule.
As our goal is to separate a set of high values from a set of low ones, a clustering method appears quite appropriate, particularly a squared error partitional method. This method is expected to give good results because the number of groups of dissimilarity values (two) is known and the values to be clustered are linearly separated. In addition, the exact optimum can be calculated since the number of solutions to evaluate is relatively small. Indeed, if there are n dissimilarity values to cluster, only n − 1 solutions must be estimated to find the optimal solution. These solutions are obtained by separating the dissimilarity values into two groups, where all the values of one group are higher than those of the other group. The NJDST with the lowest dissimilarity value is typically situated in the elbow of the curve.
The algorithm takes as input two parameters (resolution and weight), which enables the user to investigate the partitioning of the protein set in the light of biological knowledge. Changing the resolution parameter allows the user to select the depth of the clustering and the resulting number of subfamilies. By default, the partition method is performed only once (resolution = 0). The method is iterated on the low or high dissimilarity values for positive or negative resolutions, respectively. The absolute value of the resolution represents the number of additional iterations. The sequences are assigned equal weights by default, as this has proved to be the best setting in general. Indeed, analysis of various multiple alignments (see below) revealed that weighting of a group of highly similar sequences (typically protein sequences from organisms belonging to the same genus or closely related at the evolutionary level) frequently makes the total weight of the group negligible compared with a group of more weakly related sequences inducing an inadequate merging. Nevertheless, sequences may be weighted according to percentage of residue identity if the weighting parameter is selected. This may enhance the differentiation of a small subgroup of weakly related sequences which are topologically close to a larger group on the tree.
To assess the quality of our clustering method, we performed a jackknife on 24 structurally and manually validated multiple alignments (22 amino-acyl tRNA synthetases and the Sm and nuclear receptor protein families available at http://www-bio3d-igbmc.u-strasbg.fr/~wicker/Secator/secator.html). For each multiple alignment, 1% of the total number of sequences (n) was removed n times and the clusters were calculated for each resulting reduced multiple alignment and compared with the original clusters. A jackknife value was computed for each cluster and for the number of clusters. The jackknife value of a cluster is calculated as the percentage of the time this cluster is the same as in the result. The jackknife value of the number of groups is the percentage of the time each observed number of groups is found during the jackknife. For 82.7% of the groups, the jackknife value was >80%. Furthermore, in 75% of the examples, the jackknife value of the original number of groups was >80%. If the jackknife of a number of groups is above a certain threshold (20%), it is suggested to the user that an alternative to the original clustering exists.
At this point, it should be observed that any partition of the tree may be meaningful, as was pointed out by (Sjolander 1998 ). Indeed, there is no one criterion that is objectively better than another, and it is up to the biologist to choose the most convenient from his or her point of view or to change the criterion depending on the sequence family being analyzed. Thus, the introduction of different, complementary methods is of major importance to allow objective, reliable, and reproducible analysis.
In the next section, we illustrate our method using two well-studied protein families: the Sm proteins (Salgado-Garrido et al. 1999 ) and the ligand-binding domain of the nuclear receptors (Wurtz et al. 1996 ). These two protein families were preferred because of the presence of numerous divergent sequences from various origins and the availability of large amounts of biological, structural, and functional knowledge. In addition, these two protein families represent two extreme test cases with a family of very small proteins with percentages of identity ranging from 39% to 73% (Sm proteins), while the nuclear receptors are longer, highly variable proteins with percentages of identity ranging from 21% to 89%.
Analysis of Sm Proteins
The Sm proteins represent an important protein family involved in pre-mRNA splicing by promoting small nuclear RNA (snRNA) cap modification and targeting small nuclear ribonucleoproteins (snRNPs) to their appropriate cellular location. They are found in eukaryotes from yeast to humans, and some Sm-related proteins have recently been found in Archaea. At the structural level, a group of seven canonical Sm proteins, named B, D1, D2, D3, E, F, and G according to the corresponding human Sm proteins, forms a complex that can bind several RNAs (Kambach et al. 1999 ). At the sequence level, Sm proteins share a conserved Sm domain consisting of two blocks of weak but significant sequence similarity interrupted by a spacer region of variable length. Among the numerous proteins carrying an Sm domain, some are highly similar to the canonical Sm proteins, while others (Sm-like proteins) have no obvious counterpart in the Sm protein complex (Seraphin 1995 ).
Recently, an in-depth sequence analysis (Salgado-Garrido et al. 1999 ) showed that Sm and Sm-like proteins group into at least seven biological subtypes corresponding to the seven canonical Sm proteins with their Sm-like related proteins, while most of the archaeal proteins and two Sm-like proteins form various nonrelated groups.
We used Secator to analyze 102 sequences corresponding to 57 Sm proteins from yeast, plant, insect, and mammalian origins, as well as 45 Sm-like proteins from eukaryotic and archaeal origins. The distance matrix used as input was based on a full-length alignment, available with the definition and abbreviation of each sequence as well as the table of the sequence groups with their mean distance (MD) scores as supplementary material at http://www-bio3d-igbmc.u-strasbg.fr/~wicker/Secator/secator.html.Figure 2B shows the initial phylogenetic tree obtained with BIONJ, and figure 2C shows the resulting tree after collapsing by Secator. In figure 2A, the threshold of high dissimilarity at peak 7 corresponds to a visible disruption of the curve, implying that eight is a sensible number of subfamilies. In addition, Secator outgrouped three sequences (yLsm9, m-therm2, and aero-pern2). These sequences have features that clearly discriminate them from the rest of the family, noticeably the absence of the highly conserved dipeptide RG in the second block of conservation. Among the eight groups identified by Secator, six correspond exactly to the previously reported functional subtypes, highlighting the strong correlation between the biological grouping and the predicted subfamilies. Secator assigned the so-called group 1 of archaeal Sm-like proteins to subtype 3, which corresponds to the SmD2 canonical proteins and the Lsm3 proteins (Salgado-Garrido et al. 1999 ). At the sequence level, such a grouping may be biologically or evolutionarily relevant, since examination of the sequence conservation revealed that the SmD2, Lsm3, and archaeal sequences share two highly conserved residues (H and R at positions 45 and 90) which are absent in all other subtypes. The eighth subfamily, which was not reported in Salgado-Garrido et al. (1999) as a subtype, is composed of some Lsm1 and various Lsm1-related sequences, suggesting that these sequences might represent a new subtype.
Analysis of Nuclear Receptor Proteins
The nuclear receptor (NR) superfamily represents the single largest family of metazoan transcription factors (Tsai and O'Malley 1994 ). Most of the NRs are ligand-inducible factors that specifically regulate the expression of target genes involved in major physiological functions such as metabolism, development, and reproduction and are implicated in diseases such as cancer, diabetes, or hormone resistance syndromes (Weatherman, Fletterick, and Scanlan 1999 ). To date, more than 100 different NRs have been characterized which bind to hormones, such as sex steroids (progestins [PR], estrogens [ER], and androgens [AR]), adrenal steroids (glucocorticoids [GR] and mineralocorticoids [MR]), vitamin D3 (VDR), thyroid (TR), and retinoid (RXR 9-cis and all-trans), in addition to a variety of other metabolic and uncharacterized ligands. In general, the NRs have three structural domains: a highly variable N-terminal domain, a highly conserved DNA-binding domain (DBD), and a weakly conserved C-terminal ligand-binding domain (LBD). As the LBDs specifically bind a particular ligand type, they are the main targets for both pharmaceutical and phylogenetic studies.
The NRs (Evans 1988 ) were originally divided into three main subfamilies: the steroid receptor family, including ER, GR, MR, PR, and AR; the RXR receptor family, including the TR, VDR, RXR, and the ecdysone receptor (EcR); and a third family including the peroxisome proliferator activation receptor (PPAR), steriodogenic factor 1 (SF-1), nerve growth factor-induced receptor (NGF1), and X-linked orphan receptor DAX-1. Recently, the NRs were classified into six “subfamilies” (S1–S6) and 26 “groups” (uppercase letters) (Nuclear Receptors Nomenclature Committee 1999 ) by aligning the DNA-binding C domain and the ligand-binding E domain.
We used Secator to cluster the LBDs of 477 sequences comprising 244 classical NRs and 233 sequences from C. elegans (supplementary material is available at http://www-bio3d-igbmc.u-strasbg.fr/~wicker/Secator/secator.html). Figures 2E and 2F show the phylogenetic trees before and after collapsing. Here, the smallest dissimilarity value of NJDSTs corresponds to peak 15 (fig. 2D ).
The resulting clustering is in good agreement with the reported subfamilies even though our collapsed tree is based solely on the LBD domain, emphasizing the observed correlation existing between the DBD and LBD evolution. In addition, the method appears robust, since the inclusion of numerous highly variable C. elegans sequences does not significantly affect the clustering of the classical NRs. Secator correctly discriminates three subfamilies (fig. 2F ): S6 (composed of members related to the mouse GCNF1), S5 (including SF1 and LRH1), and S4 (including NOR and NUR).
Two Secator clusters differ slightly from the reported subfamilies. First, the highly similar groups S3A (ER) and S3B (ER-related) are clustered together. Second, group S2A (representative member: HNF4) has been excluded from subfamily 2. This result is linked to some specifically conserved residues that a large set of C. elegans sequences (Ce-2) shares with Group S2A. In fact, the major discrepancy observed between the two classifications is linked to S1. Secator clusters S1D to S1F (REV-ERB, ROR, CNR, …), but separates groups S1A (TRA and TRB), S1B (RAR), S1C (PPAR), S1H (UR, LXR, …), and S1I (VDR, ONR1, …); S1J was absent from our alignment, and S1K was merged with various orphan receptors. At the sequence level, this major difference is probably linked to the absence of the DBD in our alignment, since, as noted in Laudet (1997) , all of the S1 members share a characteristic DBD binding to direct repeat elements.
In addition, this analysis proposes for the first time a clustering of the orphan C. elegans receptors into five subfamilies (Ce-1 to Ce-5). The biological relevance of these results is strongly supported by the good agreement of our analysis with the existing functional subfamily classification. The objective subfamilies identified by Secator should prove useful in the comprehension of the evolution of this crucial protein family, and particularly in the construction of structural models of C. elegans NRs.
Further improvements and comparisons of clustering techniques in sequence analysis are clearly needed. This will require the use of a large number of well-studied test cases to compare and evaluate the different and complementary methods (work in progress). Nevertheless, Secator should prove particularly useful in a wide range of sequence analysis methods, particularly those dedicated to the identification of residues and domains indicative of structural or functional differences (Hannenhalli and Russell2000) .
William R. Taylor, Reviewing Editor
Abbreviations: NJDST, node joining distant subtrees; TD, threshold of dissimilarity.
Keywords: Secator subfamily phylogenetic tree clustering
Address for correspondence and reprints: Olivier Poch, Laboratoire de Biologie et Génomique Structurales, Institut de Génétique et de Biologie Moléculaire et Cellulaire CNRS/INSERM/ULP, BP 163, 67404 Illkirch cedex, France. poch@igbmc.u-strasbg.fr .
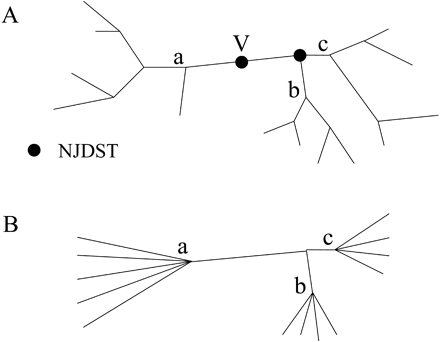
Fig. 1.—Example of a tree (A) before and (B) after collapsing. V = virtual node; branches are collapsed from the leaves up to the internal branches until nodes joining distant subtrees (dots) are met
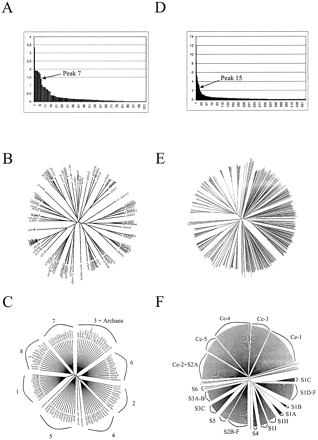
Fig. 2.—Dissimilarity value curves and phylogenetic trees before and after collapse of the Sm protein family (A, B, and C) and of the nuclear receptor family (D, E, and F). Sm subtypes are numbered from 1 to 7: subtype 1—SmB and SmN; subtype 2—SmD1 and Lsm2; subtype 3—SmD2, Lsm3, and archaeal proteins; subtype 4—SmD3 and Lsm4; subtype 5—SmE and Lsm5; subtype 6—SmF and Lsm6; subtype 7—SmG and Lsm7. The number 8 represents the subfamily including Lsm1 and Lsm1-related sequences. Subfamilies of Caenorhabditis elegans are denoted Ce-1 to Ce-5 (F), and the other subfamilies or groups are noted as in Nuclear Receptors Nomenclature Committee (1999). Full-length alignments and accession numbers are available as supplementary materials at http://www-bio3d-igbmc.u-strasbg.fr/~wicker/Secator/secator.html.
We are much indebted to Jerome Fagart and Jean-Marie Wurtz for providing their alignment of the LBD domain and to Kimmen Sjolander, who has made available her thesis and has kindly answered our questions. We are also grateful to Julie Thompson, Odile Lecompte, and Frédéric Plewniak for helpful comments.
References
Altschul S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, D. J. Lipman,
Corpet F., J. Gouzy, D. Kahn,
Enright A. J., C. A. Ouzounis,
Gascuel O.,
Hannenhalli S. S., R. B. Russell,
Kambach C., S. Walke, R. Young, J. M. Avis, E. de la Fortelle, V. A. Raker, R. Luhrmann, J. Li, K. Nagai,
Krause A., J. Stoye, M. Vingron,
Laudet V.,
Lichtarge O., H. R. Bourne, F. E. Cohen,
Milligan G. W., M. C. Cooper,
Nuclear Receptors Nomenclature Committee.
Page R. D.,
Pearson W. R.,
Salgado-Garrido J., E. Bragado-Nilsson, S. Kandels-Lewis, B. Seraphin,
Seraphin B.,
Sjolander K.,
Tatusov R. L., M. Y. Galperin, D. A. Natale, E. V. Koonin,
Thompson J. D., D. G. Higgins, T. J. Gibson,
Thompson J. D., F. Plewniak, J. Thierry, O. Poch,
Tsai M. J., B. W. O'Malley,
Weatherman R. V., R. J. Fletterick, T. S. Scanlan,
Wolf Y. I., L. Aravind, N. V. Grishin, E. V. Koonin,

{kind=link}
{kind=link}