





Abstract
Background: The influence of immune response gene variations on the development of chronic complications of Q fever is presently unclear.
Aim: To compare the frequencies of allelic polymorphisms in immune response genes in different Q fever patient groups.
Design: Genetic association study.
Methods: We measured the frequencies of immune response gene variants in: (i) an expanded group of 31 post-Q-fever fatigue patients (QFS); (ii) 22 Q fever endocarditis patients (QFE); and (iii) 22 patients who made an uncomplicated recovery from their initial attack of primary acute Q fever, comparing them with various standard control panels from the general population.
Results: There were significant differences between the three Q fever groups. QFS patients differed from both QFE and uncomplicated patients and controls in the frequency of carriage of HLA-DRB1*11 and of the 2/2 genotype of the interferon-γ intron1 microsatellite. Carriage of the HLA DRB1*11 allele was associated with reduced interferon-γ and IL-2 responses from PBMC stimulated with ligand in short-term culture. QFE showed differences in the IL-10 promoter microsatellites R and G and had higher frequencies of the TNF-α receptor II 196R polymorphism. Q fever patients who had made an uncomplicated recovery differed from those with QFS or QFE, but were not significantly different in allelic frequencies to the control panels.
Discussion: These immunogenetic differences support the concept of different immune states in chronic Q fever, determined by genetic variations in host immune responses, rather than by solely properties of Coxiella burnetii.
Introduction
Studies1,2 by our Q fever Research Group of sporadic Q fever cases in Australia, and recently in collaboration with Ayres and colleagues of those in the 1989 outbreak in Birmingham UK,3 reveal that Coxiella burnetii persists in the human host for long periods after an initial acute primary infection. In the Birmingham study, 12 years after infection from a single exposure, all clinical subgroups of Q fever patients had antibodies at essentially the same geometric mean titre.2 This was so not only in patients suffering from the post-Q-fever fatigue syndrome (QFS) (∼8%), but also in those who had recovered from the initial infection and remained asymptomatic. In addition, about the same proportion (80–90%) of bone-marrow aspirates from the symptomatic QFS patients and the asymptomatic subjects had positive PCR assays for C. burnetii DNA genomic sequences, indicating the presence of coxiella cells (probably the small-cell variant2,4). In contrast, compared with asymptomatic subjects, C. burnetii sequences were detected significantly more frequently in PBMC (as distinct from bone marrow) from the QFS patients.
Our overall conclusions from these studies are that long-term persistence of coxiella in the bone marrow is probably universal after primary infection, but coxiella numbers and viability are effectively regulated by host immune mechanisms, except in a small minority of patients, who subsequently develop QFS, Q fever endocarditis or a recrudescent infection in a tissue such as testis or bone. In addition, as the result of immunomodulation, there may be recrudescence in late pregnancy or during intense iatrogenic immunosuppression. (summarized in reference 2).
These propositions are in line with earlier qualitative statements in the literature (e.g. reference 5) that Q fever is a ‘non-sterilizing’ infection, i.e. one without final elimination of the organism from the body. It has long been accepted that there must be a focus of continuing infection to seed late-developing Q fever endocarditis or the vigorous recrudescence of infection during the last weeks of pregnancy in humans or domestic ruminants. It is also known that experimental Q fever infection in laboratory animals can be reactivated by pregnancy, X irradiation, or treatment with cortisone or chemical immunosuppressants (summary in reference 1). However, to date, the covert foci from which these recrudescent infections are seeded, or their control parameters, have not been explored systematically.
The dichotomy between the high frequency of persistent but asymptomatic infection on the one hand, and on the other, the small number of Q fever patients who remain ill with QFS (∼8–15%) or develop Q fever endocarditis (∼2–5%) after the primary infection, strongly suggests that idiosyncratic host factors determine progression to chronic Q fever.
In our first report6 we proposed (in line with the known influence of genetic factors on outcomes of infection) that such idiosyncratic factors are likely to be found in variations (polymorphisms) in the repertoire of immune response and control genes. In a preliminary test of the concept, 23 QFS patients and appropriate control panels were compared for the frequency of gene variants spanning 15 immune response genes, and also examined for HLA-B and DR frequencies.6 The QFS patients exhibited a statistically significant (p = 0.001), increased frequency of HLA-DRB1*11, compared with control panels of subjects from the general population. There were also significant (p = 0.002) differences in genotype distribution of the intron 1 interferon gamma microsatellite in the 23 QFS patients, when compared with that in a panel of Caucasian controls from the general non-Q-fever population.6
The present report extends our preliminary observations by increasing the size of the QFS group, and comparing its frequencies of immune response gene variants with subjects who had either acute Q fever and made an uncomplicated recovery, or those who had developed Q fever endocarditis.
Methods
Composition of patient groups
Recruitment of Australian Caucasian patients into this study was approved by the Royal Adelaide Hospital/IMVS ethics committee and appropriate ethics clearance was obtained for those patients from the Birmingham study.2
Post-Q-fever fatigue syndrome (QFS)
The original group (A) of 23 Australian QFS patients was supplemented with 8 QFS patients from the Birmingham study to become Group B, in total 31. Patients were classified as described previously.2,7
Q fever endocarditis (QFE)
Patients were selected on the basis of clinical evidence of endocarditis by observation of vegetations on ultrascan or on histopathological examination of the diseased valve, and a compatible serological profile, with IgG Phase I and II antibody at titres >320, low or no IgM antibody and IgA antibody to Phase I antigen at titres of ⩾160. Twenty-two patients with Q fever endocarditis were recruited from Caucasian patients, mainly from New South Wales and Queensland. Mean age was 57 years (range 29–78 years), with a mean ± SD period between primary acute Q fever and detection of endocarditis of 8.8 ± 12 years (range 2–40 years). Apart from clinically compatible features all patients had characteristic antibody patterns, supplemented by positive PCR examination of valve vegetation specimens for C. burnetii genomic sequences and in some instances by isolation of C. burnetii in cell culture or laboratory animals.
Acute Q fever with asymptomatic recovery
These comprised 22 patients from the Birmingham study who had acute primary Q fever with complete symptomatic recovery and had no QFS or other chronic sequel up to 12 years after the initial infection.
Patient samples
Blood was collected from all patients into EDTA vials, aliquotted and stored in liquid nitrogen until required.
DNA extraction
DNA was extracted from whole blood via isolation of the nuclei. Samples were mixed 1 : 1 : 3 with C1 buffer (Qiagen) and distilled water, incubated on ice for 15 min and the nuclei spun down. The pellet was washed in 250 µl C1 buffer and 750 µl distilled water, before being spun out and digested in 1 mg/ml proteinase K (Merck) for 5 h at 55°C.
HLA typing
HLA typing was performed by laboratories in Tissue Typing, Australian Red Cross Blood Service. HLA-B typing was carried out using specific oligonucleotide (SSO) probes based on the method from the 12th International Histocompatibility Workshop.8 HLA-DRB1 typing with SSOs was performed as described in the 11th International Histocompatibility Workshop.9
Microsatellite typing
All microsatellite analysis was performed using standard PCR, followed by amplicon analysis on the ABI prism 310 using GeneScan software (ABI). PCR reaction mixes contained 100 ng DNA, 20 pmol of each primer, 1 × reaction buffer (Perkin Elmer), 1 U Amplitaq gold (Perkin Elmer), 2 mM dNTPs and 4 mM MgCl2. Primers used were: IFN-γ, 5′-GCTGTCATAATAATATTCAGAC and 5′-GTACTGTGCCTTCCTGTAGGGTA; IL-10R, 5′-CCCTCCAAAATCTATTTGCATAAG and 5′-GGCACACCAACCCAGGAGAC; IL-10G, 5′-GTCCTTCCCCAGGTAGAGCAACACTCC and 5′-GTGGCTGGAGTCTAAAGTTTAAAAGATGG. All forward primers were 5′-Fam labelled, and PCR conditions were 90°C for 10 min followed by 30 cycles of 94°C for 20 s, 54°C for 40 s and 72°C for 40 s. Products were diluted 5-fold before analysis.
SNP analysis
Typing for the 196 TNF RII variant was performed using RFLP analysis. PCR conditions were as mentioned above using the primers.
5′-GAGCCCAGCCACCCCAGCCACTCTGT and 5′-TGGCTGCGTGTGTTGGGATCGTGTGG. Amplicons were digested using Nla III and analysed on 2% agarose gels. A T allele was indicated by two bands of approximately 100 bp, and a G allele was indicated by the presence of uncut amplicon.
Statistical analysis
Variability in the phenotype or allele frequencies between two populations was determined by Fisher's exact test (two sided) or by χ2 analysis. These analyses were performed on SPSS 9.0 for Windows. In similar genotype analyses, the Bonferroni adjustment of significance levels to allow for multiple comparisons is often used, advocated, or rejected as too conservative. Its relevance and its application to our data is considered in the Discussion.
Results
In our first report a wide range of immune response genes was screened for allelic variants. The present study was more restricted in approach and used published information (referenced below) about cytokine dysregulation in Q fever endocarditis as a pointer to those ‘candidate genes’ to be examined for polymorphism.
Variations in HLA allele frequencies
DNA extracted from all of the above samples was typed for HLA-A, HLA-B and HLA-DR alleles. Overall, the frequencies were closely similar in QFS and control groups (data not shown), except for a statistically significant higher frequency of HLA-DRB1*11 in the QFS A and B groups compared, respectively, with Q fever endocarditis patients, asymptomatic Q fever subjects and the control panel (Tables 1a and 1b).
Distribution of HLA-DRB1*11 carriage rates in the three clinical categories and the control panel
| Allele carriage | Controls (n = 162) | QFS A1 (n = 23) | QFS B2 (n = 23+8) | QFE3 (n = 20) | Q fever no sequelae4 (n = 22) |
|---|---|---|---|---|---|
| DRB1*11 +ve | 14 (8.6%) | 9 (39%) | 11 (35.5%) | 2 (10%) | 2 (9.1%) |
| DRB1*11 −ve | 148 (91.4%) | 14 (61%) | 20 (64.5%) | 18 (90%) | 20 (90.9%) |
| Allele carriage | Controls (n = 162) | QFS A1 (n = 23) | QFS B2 (n = 23+8) | QFE3 (n = 20) | Q fever no sequelae4 (n = 22) |
|---|---|---|---|---|---|
| DRB1*11 +ve | 14 (8.6%) | 9 (39%) | 11 (35.5%) | 2 (10%) | 2 (9.1%) |
| DRB1*11 −ve | 148 (91.4%) | 14 (61%) | 20 (64.5%) | 18 (90%) | 20 (90.9%) |
Percentages are of total in subgroup. 1Australian QFS patients. 2Australian plus Birmingham QFS patients. 3Australian Q fever endocarditis patients. 4Asymptomatic group from Birmingham study.
Distribution of HLA-DRB1*11 carriage rates in the three clinical categories and the control panel
| Allele carriage | Controls (n = 162) | QFS A1 (n = 23) | QFS B2 (n = 23+8) | QFE3 (n = 20) | Q fever no sequelae4 (n = 22) |
|---|---|---|---|---|---|
| DRB1*11 +ve | 14 (8.6%) | 9 (39%) | 11 (35.5%) | 2 (10%) | 2 (9.1%) |
| DRB1*11 −ve | 148 (91.4%) | 14 (61%) | 20 (64.5%) | 18 (90%) | 20 (90.9%) |
| Allele carriage | Controls (n = 162) | QFS A1 (n = 23) | QFS B2 (n = 23+8) | QFE3 (n = 20) | Q fever no sequelae4 (n = 22) |
|---|---|---|---|---|---|
| DRB1*11 +ve | 14 (8.6%) | 9 (39%) | 11 (35.5%) | 2 (10%) | 2 (9.1%) |
| DRB1*11 −ve | 148 (91.4%) | 14 (61%) | 20 (64.5%) | 18 (90%) | 20 (90.9%) |
Percentages are of total in subgroup. 1Australian QFS patients. 2Australian plus Birmingham QFS patients. 3Australian Q fever endocarditis patients. 4Asymptomatic group from Birmingham study.
Statistical comparison of HLA-DRB1*11 carriage rates in the groups making up Table 1a
| Group comparisons | Test | p |
|---|---|---|
| Whole table | χ2 | <0.0001 |
| QFS B vs. QFE plus Q fever no sequelae | Fisher's exact | 0.009 |
| Control panel vs. QFS (A+B) | Fisher's exact | 0.0003 |
| Control panel vs. QFE plus Q fever no sequelae | Fisher's exact | 0.76 |
| Group comparisons | Test | p |
|---|---|---|
| Whole table | χ2 | <0.0001 |
| QFS B vs. QFE plus Q fever no sequelae | Fisher's exact | 0.009 |
| Control panel vs. QFS (A+B) | Fisher's exact | 0.0003 |
| Control panel vs. QFE plus Q fever no sequelae | Fisher's exact | 0.76 |
Statistical comparison of HLA-DRB1*11 carriage rates in the groups making up Table 1a
| Group comparisons | Test | p |
|---|---|---|
| Whole table | χ2 | <0.0001 |
| QFS B vs. QFE plus Q fever no sequelae | Fisher's exact | 0.009 |
| Control panel vs. QFS (A+B) | Fisher's exact | 0.0003 |
| Control panel vs. QFE plus Q fever no sequelae | Fisher's exact | 0.76 |
| Group comparisons | Test | p |
|---|---|---|
| Whole table | χ2 | <0.0001 |
| QFS B vs. QFE plus Q fever no sequelae | Fisher's exact | 0.009 |
| Control panel vs. QFS (A+B) | Fisher's exact | 0.0003 |
| Control panel vs. QFE plus Q fever no sequelae | Fisher's exact | 0.76 |
The frequency of carriage of the HLA-DRB1*11 allele was significantly higher (35–39%) in QFS patients (Table 1a) whereas the proportions were approximately the same (8.6–10%) in Q fever endocarditis, in those who had Q fever without chronic sequelae, and in those in the control panel (Table 1b).
The possible significance of the association between HLA-DRB1*11 and QFS was previously outlined by Helbig et al.6Inter alia, it was noted that the HLA DR complex plays a part in the binding of various peptides for presentation to T lymphocytes: variable peptide binding efficiencies have been reported for HLA-DRB1*11.10 The potential effects of amino acid substitutions on peptide binding and presentation are considered in the Discussion.
Possession of DR5 molecules (e.g. HLA-DRB1*11 and HLA-DRB1*12) has been associated with lower interferon gamma production and increased humoral T helper 2 responses.11,12 To investigate this, a retrospective analysis was made of the IFN-γ responders after PHA stimulation of PBMC from the QFS patients previously studied7 by our group. This demonstrated a trend for lower production of IFN-γ from those with the HLA-DRB1*11 haplotype (Figure 1a). Earlier studies by our group13 found that, in vitro, the interaction of C. burnetii Phase 1 antigen and monocytes generates mediators that inhibit IFN-γ production by T lymphocytes. The inhibition was reversed by low doses of recombinant IL-2. A re-analysis of the Penttila et al.7 data also shows that PBMC from HLA-DRB1*11 positive carriers among the QFS group produce less IL-2 on stimulation with C burnetii antigens, compared with either HLA-DRB1*11-negative QFS patients or Q-fever-seronegative, healthy persons (Figure 1b).
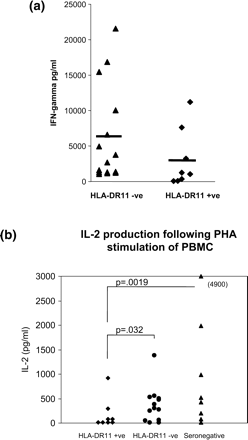
(a) INF-γ production of QFS PBMC following PHA stimulation vs. HLA-DRB1*11 carriage: p = 0.11 by Mann-Whitney U test. Data reanalysed from QFS patients examined by Penttila et al.7(b) IL-2 release from PBMC from QFS patients following C. burnetii antigen stimulation in relation to HLA-DRB1*11 carriage: p values calculated by Mann-Whitney U test. Data reanalysed from QFS patients examined by Penttila et al.7
Variation in IFN-γ intron 1 microsatellite gene in QFS and Q fever endocarditis (QFE) patients
A comparison of the distribution of genotypic variants for this gene in two forms of chronic Q fever is shown in Table 2. QFE patients had a lower proportion of the 2/2 genotype than the QFS group, although this failed to reach significance. The trend suggests that (some) QFS patients may produce IFN-γ more vigorously than QFE patients, as subjects with the 2/2 genotype are known to produce IFN-γ more effectively than do those with other genotypes.14 QFE patients are reported to have low IFN-γ responses,15 whereas QFS patients comprise two populations—responders and non-responders (see Figure 1a and reference 7). Note that significant (p = 0.002) differences in genotype distribution of the intron 1 interferon gamma microsatellite were previously observed in QFS patients when compared with a panel of Caucasian controls.6
Distribution of interferon-γ intron 1 microsatellite variants
| Genotype | QFE (n = 22) | % | QFS* (n = 30) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|
| 2/2 | 2 | 9.1 | 10 | 33.3 | 0.051 | 0.051 |
| 2/3 | 10 | 45.5 | 7 | 23.3 | NS | |
| 2/4 | 2 | 9.1 | 1 | 3.3 | NS | |
| 2/5 | 3 | 13.6 | 0 | 0.0 | 0.07 | |
| 3/3 | 3 | 13.6 | 8 | 26.7 | NS | |
| 3/4 | 2 | 9.1 | 2 | 6.7 | NS | |
| 3/5 | 0 | 0.0 | 2 | 6.7 | NS |
| Genotype | QFE (n = 22) | % | QFS* (n = 30) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|
| 2/2 | 2 | 9.1 | 10 | 33.3 | 0.051 | 0.051 |
| 2/3 | 10 | 45.5 | 7 | 23.3 | NS | |
| 2/4 | 2 | 9.1 | 1 | 3.3 | NS | |
| 2/5 | 3 | 13.6 | 0 | 0.0 | 0.07 | |
| 3/3 | 3 | 13.6 | 8 | 26.7 | NS | |
| 3/4 | 2 | 9.1 | 2 | 6.7 | NS | |
| 3/5 | 0 | 0.0 | 2 | 6.7 | NS |
*QFS values from Helbig et al.6 Paired p values calculated using Fishers exact test. Whole table χ2 df = 7, p = 0.051.
Distribution of interferon-γ intron 1 microsatellite variants
| Genotype | QFE (n = 22) | % | QFS* (n = 30) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|
| 2/2 | 2 | 9.1 | 10 | 33.3 | 0.051 | 0.051 |
| 2/3 | 10 | 45.5 | 7 | 23.3 | NS | |
| 2/4 | 2 | 9.1 | 1 | 3.3 | NS | |
| 2/5 | 3 | 13.6 | 0 | 0.0 | 0.07 | |
| 3/3 | 3 | 13.6 | 8 | 26.7 | NS | |
| 3/4 | 2 | 9.1 | 2 | 6.7 | NS | |
| 3/5 | 0 | 0.0 | 2 | 6.7 | NS |
| Genotype | QFE (n = 22) | % | QFS* (n = 30) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|
| 2/2 | 2 | 9.1 | 10 | 33.3 | 0.051 | 0.051 |
| 2/3 | 10 | 45.5 | 7 | 23.3 | NS | |
| 2/4 | 2 | 9.1 | 1 | 3.3 | NS | |
| 2/5 | 3 | 13.6 | 0 | 0.0 | 0.07 | |
| 3/3 | 3 | 13.6 | 8 | 26.7 | NS | |
| 3/4 | 2 | 9.1 | 2 | 6.7 | NS | |
| 3/5 | 0 | 0.0 | 2 | 6.7 | NS |
*QFS values from Helbig et al.6 Paired p values calculated using Fishers exact test. Whole table χ2 df = 7, p = 0.051.
Variation in IL-10 R and G promoter microsatellite in QFS and QFE patients
Overall, Q fever endocarditis patients were significantly different in their frequency of IL-10 R alleles, compared with the control panel (Table 3a, p<0.0001). In contrast, the IL-10 R allele frequencies were similar in the control panel and in the QFS patients (p = 0.39). The differences with QFE patients arise from a significantly decreased frequency of the R3 allele (p = 0.047) at the IL-10 R locus when compared to the control panel and in having three patients with the rare R1 allele (p<0.0001) (Table 3a). Although the carriage rate of the R3 allele in the QFE patients was lower in comparison with the QFS group (22.7% vs. 35.6), this difference did not reach significance (p = 0.08) (Table 3b).
Distribution of IL-10R promoter microsatellite variants
| Allele | CA repeats | Control panel (n = 181)* | % | QFE (n = 22) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| R1 | 12 | 0 | 0.0 | 3 | 6.8 | <0.0001 | <0.0001 |
| R2 | 13 | 212 | 58.6 | 30 | 68.2 | NS | |
| R3 | 14 | 139 | 38.4 | 10 | 22.7 | 0.047 | |
| R4 | 15 | 11 | 3.0 | 1 | 2.3 | NS | |
| R5 | 16 | 0 | 0.0 | 0 | 0.0 | NS | |
| QFS (n = 31) | QFE (n = 22) | ||||||
| R1 | 12 | 0 | 0.0 | 3 | 6.8 | 0.069 | 0.072 |
| R2 | 13 | 36 | 58.1 | 30 | 68.2 | NS | |
| R3 | 14 | 22 | 35.5 | 10 | 22.7 | NS | |
| R4 | 15 | 4 | 6.5 | 1 | 2.3 | NS | |
| R5 | 16 | 0 | 0.0 | 0 | 0.0 | NS |
| Allele | CA repeats | Control panel (n = 181)* | % | QFE (n = 22) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| R1 | 12 | 0 | 0.0 | 3 | 6.8 | <0.0001 | <0.0001 |
| R2 | 13 | 212 | 58.6 | 30 | 68.2 | NS | |
| R3 | 14 | 139 | 38.4 | 10 | 22.7 | 0.047 | |
| R4 | 15 | 11 | 3.0 | 1 | 2.3 | NS | |
| R5 | 16 | 0 | 0.0 | 0 | 0.0 | NS | |
| QFS (n = 31) | QFE (n = 22) | ||||||
| R1 | 12 | 0 | 0.0 | 3 | 6.8 | 0.069 | 0.072 |
| R2 | 13 | 36 | 58.1 | 30 | 68.2 | NS | |
| R3 | 14 | 22 | 35.5 | 10 | 22.7 | NS | |
| R4 | 15 | 4 | 6.5 | 1 | 2.3 | NS | |
| R5 | 16 | 0 | 0.0 | 0 | 0.0 | NS |
*Control data taken from 94 and 87 Caucasian controls from Glasgow and Oxford in the UK, respectively.16p values calculated using Fishers exact test.
Distribution of IL-10R promoter microsatellite variants
| Allele | CA repeats | Control panel (n = 181)* | % | QFE (n = 22) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| R1 | 12 | 0 | 0.0 | 3 | 6.8 | <0.0001 | <0.0001 |
| R2 | 13 | 212 | 58.6 | 30 | 68.2 | NS | |
| R3 | 14 | 139 | 38.4 | 10 | 22.7 | 0.047 | |
| R4 | 15 | 11 | 3.0 | 1 | 2.3 | NS | |
| R5 | 16 | 0 | 0.0 | 0 | 0.0 | NS | |
| QFS (n = 31) | QFE (n = 22) | ||||||
| R1 | 12 | 0 | 0.0 | 3 | 6.8 | 0.069 | 0.072 |
| R2 | 13 | 36 | 58.1 | 30 | 68.2 | NS | |
| R3 | 14 | 22 | 35.5 | 10 | 22.7 | NS | |
| R4 | 15 | 4 | 6.5 | 1 | 2.3 | NS | |
| R5 | 16 | 0 | 0.0 | 0 | 0.0 | NS |
| Allele | CA repeats | Control panel (n = 181)* | % | QFE (n = 22) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| R1 | 12 | 0 | 0.0 | 3 | 6.8 | <0.0001 | <0.0001 |
| R2 | 13 | 212 | 58.6 | 30 | 68.2 | NS | |
| R3 | 14 | 139 | 38.4 | 10 | 22.7 | 0.047 | |
| R4 | 15 | 11 | 3.0 | 1 | 2.3 | NS | |
| R5 | 16 | 0 | 0.0 | 0 | 0.0 | NS | |
| QFS (n = 31) | QFE (n = 22) | ||||||
| R1 | 12 | 0 | 0.0 | 3 | 6.8 | 0.069 | 0.072 |
| R2 | 13 | 36 | 58.1 | 30 | 68.2 | NS | |
| R3 | 14 | 22 | 35.5 | 10 | 22.7 | NS | |
| R4 | 15 | 4 | 6.5 | 1 | 2.3 | NS | |
| R5 | 16 | 0 | 0.0 | 0 | 0.0 | NS |
*Control data taken from 94 and 87 Caucasian controls from Glasgow and Oxford in the UK, respectively.16p values calculated using Fishers exact test.
IL-10 Allele R3 distribution
| QFS count (n = 31) | % | QFE count (n = 22) | % | Genotype distribution p | |
|---|---|---|---|---|---|
| With allele R3 | 19 | 61.3 | 9 | 40.9 | 0.17 |
| Without allele R3 | 12 | 38.7 | 13 | 59.1 |
| QFS count (n = 31) | % | QFE count (n = 22) | % | Genotype distribution p | |
|---|---|---|---|---|---|
| With allele R3 | 19 | 61.3 | 9 | 40.9 | 0.17 |
| Without allele R3 | 12 | 38.7 | 13 | 59.1 |
IL-10 Allele R3 distribution
| QFS count (n = 31) | % | QFE count (n = 22) | % | Genotype distribution p | |
|---|---|---|---|---|---|
| With allele R3 | 19 | 61.3 | 9 | 40.9 | 0.17 |
| Without allele R3 | 12 | 38.7 | 13 | 59.1 |
| QFS count (n = 31) | % | QFE count (n = 22) | % | Genotype distribution p | |
|---|---|---|---|---|---|
| With allele R3 | 19 | 61.3 | 9 | 40.9 | 0.17 |
| Without allele R3 | 12 | 38.7 | 13 | 59.1 |
Eskdale and colleagues16 showed that individuals with genotypes positive for the R3 allele were associated with lower IL-10 secretion on stimulation of whole blood cultures with LPS, whereas cells of the R3 negative genotype produced approximately 35% more IL-10.
An R1 allele at the IL-10 R locus is a rare occurrence with, to our knowledge, only a single literature citation of its presence in Caucasians. MacKay and colleagues17 found 3/186 rheumatoid arthritis patients were positive for R1. The unusual presence of the R1 variant in QFE suggests that this allele may have functional consequences.
All alleles at the G locus were typed, but only those at or near statistical significance level are shown in Table 4. QFE patients had a significantly increased frequency of the G14 allele at the IL-10 G locus, compared to the control panel. Once again, allele frequencies in the QFS group did not differ significantly from the those of the control panel (p = 0.89); there were differences between QFE and QFS, but these did not reach significance.
Distribution of IL-10G promoter microsatellite variants
| Allele IL-10G | CA repeats | Controls* (n = 102) | % | QFE (n = 22) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| G9 | 21 | 103 | 50.5 | 11 | 25.0 | 0.037 | 0.093 |
| G11 | 23 | 20 | 9.8 | 10 | 22.7 | 0.038 | |
| G14 | 26 | 10 | 4.9 | 6 | 13.6 | 0.044 | |
| QFS (n = 31) | QFE (n = 22) | ||||||
| G9 | 21 | 26 | 41.9 | 11 | 25.0 | 0.067 | 0.32 |
| G11 | 23 | 6 | 9.7 | 10 | 22.7 | 0.091 | |
| G14 | 26 | 2 | 3.2 | 6 | 13.6 | 0.066 |
| Allele IL-10G | CA repeats | Controls* (n = 102) | % | QFE (n = 22) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| G9 | 21 | 103 | 50.5 | 11 | 25.0 | 0.037 | 0.093 |
| G11 | 23 | 20 | 9.8 | 10 | 22.7 | 0.038 | |
| G14 | 26 | 10 | 4.9 | 6 | 13.6 | 0.044 | |
| QFS (n = 31) | QFE (n = 22) | ||||||
| G9 | 21 | 26 | 41.9 | 11 | 25.0 | 0.067 | 0.32 |
| G11 | 23 | 6 | 9.7 | 10 | 22.7 | 0.091 | |
| G14 | 26 | 2 | 3.2 | 6 | 13.6 | 0.066 |
*From reference 14: 102 UK Caucasian control patients from the Royal Infirmary tissue-typing laboratory in Glasgow. p values were calculated using Fisher's exact or χ2 test as appropriate.
Distribution of IL-10G promoter microsatellite variants
| Allele IL-10G | CA repeats | Controls* (n = 102) | % | QFE (n = 22) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| G9 | 21 | 103 | 50.5 | 11 | 25.0 | 0.037 | 0.093 |
| G11 | 23 | 20 | 9.8 | 10 | 22.7 | 0.038 | |
| G14 | 26 | 10 | 4.9 | 6 | 13.6 | 0.044 | |
| QFS (n = 31) | QFE (n = 22) | ||||||
| G9 | 21 | 26 | 41.9 | 11 | 25.0 | 0.067 | 0.32 |
| G11 | 23 | 6 | 9.7 | 10 | 22.7 | 0.091 | |
| G14 | 26 | 2 | 3.2 | 6 | 13.6 | 0.066 |
| Allele IL-10G | CA repeats | Controls* (n = 102) | % | QFE (n = 22) | % | Each genotype p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| G9 | 21 | 103 | 50.5 | 11 | 25.0 | 0.037 | 0.093 |
| G11 | 23 | 20 | 9.8 | 10 | 22.7 | 0.038 | |
| G14 | 26 | 10 | 4.9 | 6 | 13.6 | 0.044 | |
| QFS (n = 31) | QFE (n = 22) | ||||||
| G9 | 21 | 26 | 41.9 | 11 | 25.0 | 0.067 | 0.32 |
| G11 | 23 | 6 | 9.7 | 10 | 22.7 | 0.091 | |
| G14 | 26 | 2 | 3.2 | 6 | 13.6 | 0.066 |
*From reference 14: 102 UK Caucasian control patients from the Royal Infirmary tissue-typing laboratory in Glasgow. p values were calculated using Fisher's exact or χ2 test as appropriate.
A G14 allele linked to a R2 allele at the IL-10 R locus has been shown to increase IL-10 production by 43% upon LPS stimulation of whole blood cells (16). Haplotype determination cannot be performed at these loci using standard microsatellite typing techniques, as the alleles are approximately 3 kb apart. Nevertheless all QFE patients with a G14 allele also possessed a R2 allele on at least one of their chromosomes. Eskdale and colleagues have demonstrated that the G14 allele (IL-10 G locus) is exclusively paired with the R2 allele (IL-10 R locus). This suggests that QFE individuals may be predisposed to increased production of IL-10 levels in that they demonstrate a decreased frequency of IL10 R3 alleles coupled with increased IL 10 G14 allele–both leading to increased IL-10 production. This has implications for the anergic state in QFE (Discussion).
TNF-α R11 196R analysis
Ghigo and colleagues19 have shown that the 75 kDa TNF-α receptor is up-regulated in monocytes from QFE patients. Consequently polymorphic variation in this TNF-α receptor was examined. Table 5 shows analysis of TNF-α RII T and G alleles in QFS and QFE.
Distribution of TNF RII alleles at codon 196
| Gene | Genotype | Controls (n = 249)* | % | QFE (n = 22) | % | Each genotype χ2p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| TNF RII | TT | 150 | 60.0 | 9 | 40.1 | NS | 0.16 |
| TG | 85 | 34.0 | 12 | 54.5 | 0.065 | ||
| GG | 14 | 6.0 | 1 | 5.0 | NS | ||
| Phenotype | T | 235 | 0.94 | 21 | 0.95 | NS | |
| G | 99 | 0.40 | 13 | 0.59 | NS | ||
| QFS (n = 31) | QFE (n = 22) | ||||||
| TNF RII | TT | 19 | 61.3 | 9 | 40.1 | NS | 0.27 |
| TG | 10 | 32.3 | 12 | 54.5 | NS | ||
| GG | 2 | 6.5 | 1 | 5.0 | NS | ||
| Phenotype | T | 29 | 0.94 | 21 | 0.95 | NS | |
| G | 10 | 0.32 | 13 | 0.59 | NS |
| Gene | Genotype | Controls (n = 249)* | % | QFE (n = 22) | % | Each genotype χ2p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| TNF RII | TT | 150 | 60.0 | 9 | 40.1 | NS | 0.16 |
| TG | 85 | 34.0 | 12 | 54.5 | 0.065 | ||
| GG | 14 | 6.0 | 1 | 5.0 | NS | ||
| Phenotype | T | 235 | 0.94 | 21 | 0.95 | NS | |
| G | 99 | 0.40 | 13 | 0.59 | NS | ||
| QFS (n = 31) | QFE (n = 22) | ||||||
| TNF RII | TT | 19 | 61.3 | 9 | 40.1 | NS | 0.27 |
| TG | 10 | 32.3 | 12 | 54.5 | NS | ||
| GG | 2 | 6.5 | 1 | 5.0 | NS | ||
| Phenotype | T | 29 | 0.94 | 21 | 0.95 | NS | |
| G | 10 | 0.32 | 13 | 0.59 | NS |
*Data from 249 healthy UK Caucasians.18p values were calculated using Fisher's exact or χ2 test as appropriate.
Distribution of TNF RII alleles at codon 196
| Gene | Genotype | Controls (n = 249)* | % | QFE (n = 22) | % | Each genotype χ2p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| TNF RII | TT | 150 | 60.0 | 9 | 40.1 | NS | 0.16 |
| TG | 85 | 34.0 | 12 | 54.5 | 0.065 | ||
| GG | 14 | 6.0 | 1 | 5.0 | NS | ||
| Phenotype | T | 235 | 0.94 | 21 | 0.95 | NS | |
| G | 99 | 0.40 | 13 | 0.59 | NS | ||
| QFS (n = 31) | QFE (n = 22) | ||||||
| TNF RII | TT | 19 | 61.3 | 9 | 40.1 | NS | 0.27 |
| TG | 10 | 32.3 | 12 | 54.5 | NS | ||
| GG | 2 | 6.5 | 1 | 5.0 | NS | ||
| Phenotype | T | 29 | 0.94 | 21 | 0.95 | NS | |
| G | 10 | 0.32 | 13 | 0.59 | NS |
| Gene | Genotype | Controls (n = 249)* | % | QFE (n = 22) | % | Each genotype χ2p | Genotype distribution p |
|---|---|---|---|---|---|---|---|
| TNF RII | TT | 150 | 60.0 | 9 | 40.1 | NS | 0.16 |
| TG | 85 | 34.0 | 12 | 54.5 | 0.065 | ||
| GG | 14 | 6.0 | 1 | 5.0 | NS | ||
| Phenotype | T | 235 | 0.94 | 21 | 0.95 | NS | |
| G | 99 | 0.40 | 13 | 0.59 | NS | ||
| QFS (n = 31) | QFE (n = 22) | ||||||
| TNF RII | TT | 19 | 61.3 | 9 | 40.1 | NS | 0.27 |
| TG | 10 | 32.3 | 12 | 54.5 | NS | ||
| GG | 2 | 6.5 | 1 | 5.0 | NS | ||
| Phenotype | T | 29 | 0.94 | 21 | 0.95 | NS | |
| G | 10 | 0.32 | 13 | 0.59 | NS |
*Data from 249 healthy UK Caucasians.18p values were calculated using Fisher's exact or χ2 test as appropriate.
QFE patients had a noticeably higher frequency of TG haplotypes (p = 0.065, not quite significant) than controls (55% vs. 34%), whereas genotype frequencies were essentially the same in QFS and controls. Note that QFE patients’ carriage rate of the G allele was almost twice that of the QFS patients.
Discussion
The number of patients tested in our study is small, reflecting the low prevalence rates of chronic complications of Q fever. Nevertheless there appear to be significant immunogenetic differences between patients with QFS or QFE on the one hand, and on the other, those who made an uncomplicated recovery from acute Q fever, and subjects in the control panels from the general population.
QFS patients differed from the other two patient groups and the control panels in the frequency of carriage of HLA DRB1*11 and of the 2/2 genotype of the IFN-γ intron 1 microsatellite. These differences (Tables 1a and 1b) were significant by standard χ2 or Fisher's exact tests. There is some uncertainty about the validity of using the Bonferroni adjustment for multiple comparisons with our type of data.20 Applicable or not, the Bonferroni correction for Table 1b would stipulate a p value of (0.05/3) = 0.016 or less for significance. In fact, comparison of QFS B with the three other groups shows p values at 0.009 or less. Similarly, the p value of 0.002 previously recorded6 for comparison of genotype distribution in the IFN-γ intron 1 microsatellite and control panel is less than Bonferroni-corrected p value of 0.05.
QFE patients in contrast showed different allelic patterns to the control panels in the IL-10 promoter microsatellites R and G and weaker differences at these loci with QFS patients. QFE patients also had higher frequencies of the TNF RII T/G allele than the control panels, but again weaker differences were seen with the QFS group.
Overall, these differences reinforce the concept of different, polar, immune states in chronic Q fever—viz. immune hypersensitivity (QFS) and anergy (QFE)—that we have advanced elsewhere.2,7 The differences are also in line with the concept6 that chronic Q fever is determined by genetically determined variations in host immune responses to the coxiella, rather than by solely unique properties of C. burnetii as organism [cf Q fever endocarditis below and (34)].
In further support of the above interpretations, we note significant analogies between QFS and a chronic fatigue syndrome (CFS) in ∼13% of symptomatic cases of parvovirus B19 infection. Recent studies by Kerr, Tyrrell and colleagues,21 sponsored by the CFS Research Foundation UK, have shown long-term persistence of the parvovirus in cases with CFS. The persistence is accompanied by cytokine dysregulation, primarily involving IFN-γ, IL-6, MCP-1 and TNF-α, with variable alterations of IL-1, IL-2 and IL-4 levels (cf greatly raised IL-6, variably raised IFN-γ and restricted IL-2 release patterns described by Penttila et al.7 with those in Kerr et al.22). Both infections involve bone marrow cells—erythroid precursors with parvovirus,21 and as-yet unidentified marrow cells, probably of the monocyte lineage, with C. burnetii.2 In both QFS and parvovirus-associated CFS, there is allelic variation involving the HLA DRB1 subtypes, with amino acid changes in proximity to, but not an integral part of, key sequences involved in the peptide binding groove of APC and antigen presentation.21
A re-analysis of the data from Penttilla et al.7 showed that PBMC from QFS patients carrying the HLA-DRB1*11 allele produced less IFN-γ on PHA stimulation than those without the allele (Figure 1a); also that on stimulation with C. burnetii antigens the PBMC produced less IL-2 (Figure 1b). The former responses match those observed by Petrovsky and Harrison11 in PBMC from HLA-DR5-positive subjects stimulated in a mixed lymphocyte reaction. The reason for the association of the HLA-DRB1*11 and other DR5 alleles and a reduction of IFN-γ responses is not established. It is known that glutamic acid is present at amino acid 58 in all 54 HLA-DRB1*11 subtypes, whereas it is infrequently found at this position in other HLA-DRB1 alleles. Amino acid 58 is not an integral part of the peptide-presenting groove, but is directly adjacent to the P9 peptide-binding ‘pocket’ in the HLA DR molecule. The residue at this location in other HLA-DRB1 molecules is commonly alanine, a smaller and uncharged amino acid compared to the negatively charged glutamic acid. Hypothetically, a glutamic acid at amino acid 58 might influence the accommodation of C. burnetii Phase I peptides in pocket 9.
The differences in genetic variants identified in this report associated with the various chronic forms of Q fever are highly unlikely to be a complete account of all relevant polymorphisms. There are very few diseases in which a single genetic variant is associated with all cases (e.g. ankylosing spondylitis HLA B 27, narcolepsy HLA DR2). Associations are more likely to be multigenic.
The point is reinforced by a recent microarray study23 sponsored by the CFS Research Foundation UK of ‘idiopathic’ CFS patients. This showed over 16 genes up- or down-regulated in the CFS group, compared with expression rates in asymptomatic control subjects. The potential for genetic variation in QFS might be explored profitably by current Affymetrix based techniques for determination of single nucleotide polymorphisms.
In 1962, Marmion24 drew attention to a history of previous rheumatic fever and consequent cardiac valve damage in patients with the (then) recently described complication of Q fever endocarditis.25 Subsequently, numerous reports of C. burnetii infection superimposed on valves damaged by rheumatic fever, on various congenital abnormalities of the heart or on arterial prostheses led to a simple cause-and-effect view of the pathogenesis of Q fever endocarditis; namely, that anatomical distortion and endocardial damage leads to altered haemodynamics, then to fibrin deposition with trapping of infected macrophages, and finally to formation of infective vegetations.
Other explanations26–28 of the pathogenesis of Q fever endocarditis [see also (34)] suggested an association between Q fever endocarditis and coxiella plasmid genotype, or fine antigenic differences between the LPS and protein of strains isolated from endocarditis and other Q fever patients.
Our immunogenetic findings with QFE suggest that its pathogenesis may be more complex. Leaving aside the role of congenital abnormalities and inserted prostheses, it is conceivable that the variations in the immunogenetic background that conditioned an initial aberrant immunological response to Streptococcus pyogenes and subsequent rheumatic fever (see Bisno29 for summary) may also be associated with the later host failure to regulate persistent coxiella infection and development of QFE.2
Recent observations by Honstettre et al.30 are cogent in this regard. They found that patients with a pre-existing valvulopathy who later developed Q fever endocarditis had a more intense acute-phase cytokine response during their initial acute Q fever infection, compared with those patients without valvulopathy. With unstimulated PBMC from the groups, TNF-α and IL-10 levels were significantly higher in valvulopathy patients. With PBMC stimulated with C. burnetii suspensions, TNF-α and IL-10 levels were again higher in the valvulopathy group who later developed endocarditis; the acute phase levels resembled those in other patients with established Q fever endocarditis.
The longitudinal clinical and laboratory observations of Honstettre et al. fit well with the predicted effects described above for the two genetic variations we have observed in QFE—decreased frequency of IL10 R3 alleles coupled with increased IL-10 G14 allele in endocarditis patients—both leading to increased IL-10 production.
Finally, the immunogenetic findings and the comparisons (above) of fatigue syndromes after Q fever and parvovirus infection may provide some general insights into that sector of patients with ‘idiopathic’ chronic fatigue syndromes (CFS) which in reality may be post infection fatigue syndrome. It is postulated that two factors operate. First, long-term persistent infection with an agent—bacterial, viral or protozoal—and second, a genetically determined failure of homeostasis in a small minority of subjects who fail to down-regulate the continuing cytokine stimulation from the persisting infective agents or their undegraded antigens.
Cytokine dysregulation in CFS was identified in the early 1990s by Chao et al.31 in Minneapolis, who found that PBMC from CFS patients stimulated in short-term culture with LPS or PHA liberated more IL-1, 1L-6 and TNF-α than did cells from controls. At about the same time, Patarca et al.32 reported that CFS patients more often had raised serum levels of IL-1, IL-2 TNF-α and IL-2 soluble receptor than control subjects. Significantly, Keller et al.33 observed an association between HLA class 11 antigens (specifically HLA DR4, DR5 and DQ3) and CFS.
The conclusions from Q fever, parvovirus B19 infection and from the CFS studies, taken together, suggest that any group of ‘idiopathic’ CFS patients collected at random from the wider population, away from outbreaks or occupationally exposed groups, are unlikely to have laboratory evidence of infection with the same infective agent. What they are more likely to have in common is an immunogenetically determined failure of cytokine homeostasis to any of a number of infective agents with the capacity to persist for long periods in the host.
Major financial support was received from Meat and Livestock Australia Ltd and smaller grants from the Woodend Foundation Sydney (Mrs Isabel Milner, via TRUST Co of Australia Ltd) and from the NHMRC Australia via the University of New South Wales Randwick NSW and the Hanson Institute Adelaide. We are indebted to numerous physicians and pathologists, in particular Robyn Wood of Queensland Medical Laboratories for help with location and sampling of patients. We are also indebted to the subjects from the Birmingham study, who willingly participated in this project so long after the initial outbreak, and to Mrs Jayne Groves for co-ordinating sample collection in the UK.
References
Harris RJ, Storm PA, Lloyd A, Arens M, Marmion BP. Long-term persistence of Coxiella burnetii in the host after primary Q fever.
Marmion BP, Storm PA, Ayres JG, et al. Long term persistence of Coxiella burnetii after acute Q fever.
Hawker JI, Ayres JG, Blair I, et al. A large Q fever outbreak in the West Midlands: wind borne spread into a metropolitan area?
McCaul TF. The developmental cycle of Coxiella burnetii. In: Williams JC, Thompson HA, eds.
Helbig KJ, Heatley SL, Harris RJ, et al. Variation in immune response genes and chronic Q fever. Concepts: preliminary test with post-Q fever fatigue syndrome.
Penttila IA, Harris RJ, Storm P, Haynes D, Worswick DA, Marmion BP. Cytokine dysregulation in the post-Q fever fatigue syndrome.
Kennedy LJ, Poulton KV, Thompson W, et al. HLA class I DNA typing using sequence specific oligonucleotide probes (SSOP). In: Charron D, ed.
Kimura A, Sasazuki T. Eleventh International Histocompatibility Workshop reference protocol for the HLA DNA-typing technique. In: Tsuji K, Aizawa M, Sasazuki T, eds. HLA 1991.
Hurley CK, Steiner N. Differences in peptide binding of DRB1*11 and DR13 microvariants demonstrate the power of minor variation in generating DR functional diversity.
Petrovsky N, Harrison LC. HLA Class II-associated polymorphism of interferon-γ production.
Blackwell JM, Black GF, Charples C, Soo SS, Peacock CS, Miller EN. Roles of Nramp1, HLA, and a gene(s) in allelic association with IL-4, in determining T helper subset differentiation.
Izzo AA, Marmion BP. Variation in interferon-gamma responses to Coxiella burnetii antigens with lymphocytes from vaccinated or naturally infected subjects.
Pravica V, Asderakis A, Perrey C, Hajeer A, Sinnott PJ, Hutchinson IV. In vitro production of IFN-gamma correlates with CA repeat polymorphism in the human IFN-gamma gene.
Dellacasagrande J, Capo C, Raoult D, Mege JL. IFNγ mediated control of Coxiella burnetii survival in monocytes:the role of cell apoptosis and TNF.
Eskdale J, Gallagher G, Verweij C, Keijsers V, Westendorp R, Huizanga T. Interleukin-10 secretion in relation to human IL-10 locus haplotypes.
MacKay K, Milicic A, Lee D, et al. Rheumatoid arthritis susceptibility and Interleukin-10: a study of two ethnically different populations.
Allen RA, Lee EM, Roberts DH, Park BK, Pirmohamed M. Polymorphisms in the TNFα and TNF-receptor genes patients with coronary artery disease.
Ghigo E, Capo N, Amirayan D, Raoult D, Mege JL. The 75-KD tumour necrosis factor is specifically up-regulated in monocytes during Q fever endocarditis.
Proschan MA, Waclawiw MA Practical guidelines for multiplicity adjustment in clinical trials.
Kerr JR, Tyrrell DAJ. Cytokines in parvovirus B19 infection as an aid to understanding chonic fatigue syndrome.
Kerr JR, Barah F, Maffey DL, et al. Circulating tumour necrosis factor-α and interferon γ are detectable during acute and convalescent parvovirus infection and are associated with prolonged and chronic fatigue.
Kaushik NK, Fear D, Richards S CM, et al. Gene expression in peripherial blood mononuclear cells (PMBC) from Chronic Fatigue Syndrome (CFS) patients.
Marmion BP. Subacute rickettsial endocarditis; an unsual complication of Q fever.
Andrews PS, Marmion BP. Chronic Q fever 2 Morbid anatomical and bacteriological findings in a patient with endocarditis.
Samuel J, Frazier ME, Mallavia LP. Correlation of plasmid type and disease caused by
Hackstadt T, Peacock MG, Hitchcock PJ, Cole RL. Lipopolysaccharide variation in Coxiella burnetii; intrastrain variation heterogenity in structure and antigenicity.
Stein A, Raoult D. Lack of pathotype specific gene in human Coxiella burnetii isolates.
Bisno AL. Rheumatic Fever. In: Goldman L, Bennett JC, eds.
Honsettre A, Imbert G, Ghigo E, et al. Dysregulation of cytokines in acute Q fever: role of IL-10 and tumour necrosis factor in chronic evolution of Q fever.
Chao CC, Janoff EN, Hu S, Thomas K, Gallagher M, Tsang M, Peterson PE. Altered cytokine release in peripheral blood mononuclear cell culture from patients with the chronic fatigue syndrome.
Partarca R, Klimas NG, Lugtendorf S, Antoni M, Fletcher MA. Dysregulated expression of tumor necrosis factor in chronic fatigue syndrome: Interrelations with cellular sources and patterns of soluble mediator expression.
Keller RH, Lane JL, Klimas N, Reiter WM, Fletcher MA, van Reil F, Morgan R. Association between HLA class 11 antigens and the chronic fatigue immune dysfunction syndrome.
Author notes
From the 1Q fever Research Group IMVS and Hanson Institute, Adelaide, Australia, 2Department of Environmental and Occupational Medicine, University of Aberdeen, Aberdeen, UK, 3Tissue Typing, Australian Red Cross Blood Service and Research Unit of Transfusion Medicine and Immunogenetics, University of Sydney, Sydney, Australia, 4Medical Sciences, University of New South Wales, Sydney, Australia, and 5Sullivan and Nicolaides Pathology, Brisbane, Australia

{kind=link}