





Abstract
Objectives. Vasoactive intestinal peptide (VIP) has demonstrated therapeutic effects in arthritis by inhibiting both innate and acquired immune responses. We investigated the potential effects of VIP in the regulation of Toll-like receptor (TLR) expression and function in synovial fibroblasts from patients with rheumatoid arthritis (RA) and osteoarthritis (OA).
Methods. Cultured fibroblast-like synoviocytes (FLS) were obtained from patients with RA and OA. The effects of VIP on basal or TNF-α or lipopolysaccharide (LPS)-induced TLR2, TLR4 and MyD88 expression and its effects on TLR4-mediated CCL2 and CXCL8 chemokine production were studied by reverse transcription–polymerase chain reaction, western blotting and enzyme-linked immunosorbent assay.
Results. TLR2, TLR4 and MyD88 mRNA expression was increased in RA FLS compared with OA FLS. The largest increase was observed for TLR4 and there was also overexpression at the protein level in RA FLS. TLR4 and MyD88 mRNA and proteins were induced by LPS and TNF-α in RA FLS. VIP down-regulated the induced but not the constitutive expression of TLR4 and MyD88 in RA FLS. VIP treatment decreased CCL2 and CXCL8 chemokine production in response to TLR4 activation with LPS in RA FLS.
Conclusions. We demonstrate that VIP down-regulates LPS and TNF-α activation of TLR4 expression and the TLR4 functional response in terms of proinflammatory chemokine production. These studies suggest that the pleiotropic anti-inflammatory actions of VIP involve inhibitory effects on TLR4 expression and signalling.
Vasoactive intestinal peptide (VIP) is a widely distributed neuropeptide that displays potent anti-inflammatory and immunomodulatory activity by down-regulating proinflammatory and T-cell cytokines in immune-mediated diseases [1]. A beneficial effect of VIP on experimental animal models of acute and chronic inflammation, such as acute pancreatitis [2], septic shock [3], arthritis [4], inflammatory bowel disease [5] and lipopolysaccharide (LPS)-induced acute inflammation [6], has been demonstrated. In collagen-induced arthritis, VIP showed pleiotropic effects on both the innate and acquired branches of the autoimmune inflammatory response, by limiting proinflammatory cytokines production, shifting the Th1/Th2 balance [4] and showing a protective effect on joint destruction [7]. Although these mechanisms seem involved in the development of human rheumatoid arthritis (RA), cytokines and proinflammatory networks play an unequivocal role, as confirmed by the success of cytokine-targeted therapies, whereas the role of Th1/Th2 bias is less clear [8]. In human RA synovial cells, previous studies suggest that VIP can modulate cytokine and chemokine networks, particularly in TNF-α-stimulated cells [9, 10].
Toll-like receptors (TLR) are receptors expressed on the plasma membrane or in the intracellular compartment that recognize common molecules produced by pathogens, named pathogen-associated molecular patterns (PAMPs). They are conserved proteins with an extracellular leucine-rich domain and an intracellular Toll/IL-1 like receptor (TLR) domain. To date, 11 members of the TLR family have been identified in humans, TLR4 being the first to be described [11–13]. This receptor is essential for LPS signalling; this signalling also requires an additional molecule, MD-2, which associates with the extracellular portion of TLR4. In addition, TLR4 has been involved in the recognition of endogenous ligands, such as hsp60, fibronectin and multiple host proteins [11, 14]. TLR4 can signal through the adapter protein MyD88 as well as through MyD88-independent pathways, resulting in the activation of various intracellular signalling cascades, which lead to the activation of transcription factors of the nuclear transcription factor-κB (NFκB) and interferon regulatory factor (IRF) families, which ultimately induce the production of inflammatory mediators [11–13].
Although the specific ligands involved in TLR activation during autoimmune disease are unclear, TLR signalling seems involved in the pathogenesis of autoimmune inflammatory diseases, including arthritis, as revealed by genetically deficient mouse models [15–17]. In RA, TLR ligands of microbial origin, such as peptidoglycans (PGs) and double-stranded DNA, or endogenous ligands, such as heat shock proteins, fibrinogen, and hyaluronan or necrotic cell components, are commonly found in inflamed joints and have been proposed as potential TLR ligands in this milieu (for review see [18]). The expression and function of TLR in RA tissues or cells have been studied by different groups. Higher expression of TLR2 and TLR4 in synovial tissues has been found in RA patients compared with patients with osteoarthritis (OA) and healthy individuals [19]. However, previous studies have failed to demonstrate differences between RA and OA at the mRNA level of TLR2, TLR4 and TLR9 in synovial fibroblasts derived from joints, but in synovial tissues they observed weak expression of TLR2 mRNA in tissue sections of patients affected by OA compared with patients with RA [20]. Moreover, it was found that cultured synovial fibroblasts from patients with RA or OA express low levels of TLR2 and TLR9 mRNA, and no differences between RA and OA were found in this study [21]. In cultured RA and OA fibroblast-like synoviocytes (FLS), bacterial PGs and bacterial lipopeptide, but not CpG oligodeoxynucleotides, can activate, partially through TLR-2, to express integrins, metalloproteinases and proinflammatory cytokines and chemokines [21, 22]. Moreover, the expression of TLR2 was up-regulated by PGs but the expression of TLR9 was not up-regulated by CpG oligodeoxynucleotides in RA and OA FLS [17]. With regard to TLR4, data are scarce, but its involvement in the production of IFN-γ and the CXC chemokine family in synovial cells has been reported [23].
Our previous studies suggest that proinflammatory cytokine and chemokine expression is down-regulated by VIP in RA fibroblasts. To examine whether modulation of TLR-mediated signalling is involved in the anti-inflammatory response to VIP of human RA cells, we studied the effects of VIP on TLR expression and signalling in RA synovial fibroblasts.
Materials and methods
Patients and tissue samples
Synovial tissue was obtained from six patients with RA and four patients with OA at the time of surgery to replace a knee prosthesis. Informed consent was requested and obtained from every patient. All RA patients fulfilled the American College of Rheumatology 1997 criteria for the diagnosis of RA [24]. The study was performed according to the ethics recommendations of the Declaration of Helsinki and was approved by the Ethics Committee of the Hospital 12 de Octubre. FLS cultures were established from homogenized synovium in 10% fetal calf serum–Dulbecco's Modified Eagle Medium (FCS-DMEM) [25]. FLS were used between passages 3 and 8. FLS were stimulated with 20 μg/ml LPS from Salmonella enteritidis (Sigma-Aldrich, Madrid, Spain) or with 10 nM TNF-α (Genzyme, Cambridge, UK) in the presence or absence of 10 nM VIP (Neosystem, Strasbourg, France) for 24 h. Culture supernatants were harvested for determination of chemokines by enzyme-linked immunosorbent assay (ELISA). RNA was also obtained from FLS cultures.
Single-step quantitative real-time RT-PCR
Quantitative reverse transcriptase–polymerase chain reaction (RT-PCR) was performed with an ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). We used the SYBR® Green PCR Master Mix and RT-PCR kit in one-step RT-PCR, as suggested by the manufacturer (Applied Biosystems). Briefly, reactions were performed in 20 μl containing 50 ng of total RNA, 10 μl 2 × SYBR Green PCR Master Mix, 6.25 U Multi-Scribe reverse transcriptase 10 U RNase inhibitor and 0.1 mM of specific primers. The sequence of primers was; human β-actin sense, 5′-AGAAGGATTCCTATGTGGGCG-3′, antisense 5′-CATGTCGTCCCAGTTGGTGAC-3′; human TLR2, sense 5′-GGAAGAATCCTCCAATCAGGC-3′, antisense 5′-CTTCTGTGAGCCCTGAGGGA-3′; human TLR4, sense 5′-AGCCACGCATTCACAGGG–3′, antisense 5′-CATGGCTGGGATCAGAGTCC-3′; and human MyD88, sense 5′-CGGATGGTGGTGGTTGTCTC-3′, antisense 5′-CGCTTCTGATGGGCACCT-3′. The GenBank accession numbers and numbers for the 5′ and 3′ ends of nucleotides for the PCR products are: β-actin, E00829, 1435–1535; TLR2, NM 003264, 207–307; TLR4, NM138554, 112–212; and MyD88, NM 002468, 691–791. The amplification conditions were 30 min at 48°C, 10 min at 95°C, 40 cycles of denaturation at 95°C for 15 s, and annealing/extension at 60°C for 1 min.
For relative quantification, we calculated the n-fold differential expression by the ΔCt method (Ct denoting the threshold cycle of PCR amplification at which product is first detected by fluorescence), which compares the amount of target gene amplification, normalized to the β-actin endogenous reference as previously described [26]. The correct size of the amplified products was checked by electrophoresis.
Western blotting
Proteins were extracted from FLS in ice-cold lysis buffer (10 mM Tris–HCl, pH 8, 1 mM ethylenediamine tetraacetate (EDTA), 150 mM NaCl, 0.1% sodium dodecyl sulphate (SDS), 10 μg/ml of leupeptin, 10 μg/ml of aprotinin, 2 μg/ml of pepstatin A, and 0.5 mM phenylmethylsulphonyl fluoride). Protein extracts (50 μg) were subjected to 8% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were blocked with Tris-buffered saline (pH 7.6) containing 5% non-fat dry milk and 0.1% Tween-20 and then incubated overnight at 4°C with the following primary antibodies: rabbit anti-TLR4 (1:250; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-MyD88 (1:6000; Alexis Biochemicals, Lausen, Switzerland), goat anti-TLR2 (1:400; Santa Cruz Biotechnology) and mouse anti-β-actin (1:10 000; Sigma-Aldrich) as loading control. For detection, horseradish peroxidase conjugated secondary antibody at 1:5000 dilution was used. Proteins were detected using an enhanced chemiluminescence western blotting analysis system (Pierce, Rockford, IL, USA). Protein bands were analysed by densitometric analyses and normalized against the intensity of β-actin.
ELISA
The amounts of CCL2 (MCP-1) and CXCL8 (IL-8) in the supernatants of FLS cultures were determined with a human capture ELISA assay as previously described [9]. The intra-assay and inter-assay variability for chemokines determination was <5%.
Statistical analysis
Results are expressed as the mean ± s.e.m. The significance of the results was analysed using Student's two tailed t-test. P values less than 0.05 were considered significant.
Results
Expression of TLR4, TLR2 and MyD88 in RA and OA FLS
We initially studied the constitutive expression of TLR2, TLR4 and MyD88 mRNA by quantitative real-time RT-PCR in FLS from patients with RA and OA. We found a 17-fold higher expression of TLR4 mRNA in RA compared with OA FLS (Fig. 1A). The mean expression of TLR2 mRNA was also significantly higher in RA FLS, being 8-fold higher than in OA FLS, whereas the expression of MyD88 mRNA was only 2.5-fold higher in RA FLS, not reaching statistical significance (Fig. 1B and C).
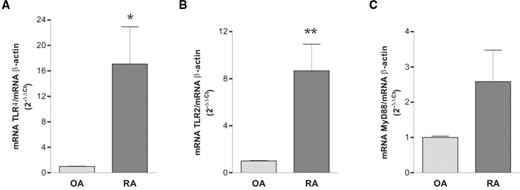
Expression of mRNA for (A) TLR4, (B) TLR2 and (C) MyD88 mRNA. Expression was studied in cultured FLS from patients with RA or OA by quantitative real-time PCR and corrected for β-actin mRNA expression level within each sample (see Materials and methods). Differences in TLR2 and TLR4 expression were statistically significant (*P<0.05; **P<0.01). Results are the mean ± s.e.m. of two experiments performed in duplicate including six FLS lines from RA patients and four from OA patients.
To analyse TLR2, TLR4 and MyD88 differences at the protein level, we performed western blot studies on FLS protein extracts. TLR2 was under the detection level in all FLS protein extracts, whereas it was readily detected in PBMC protein extracts used as positive control. A marked heterogeneity between individual FLS lines was observed for TLR4 and MyD88 protein expression. A 2-fold higher expression of TLR4 protein was observed in RA compared with OA FLS (Fig. 2). MyD88 protein levels were also heterogeneous in individual FLS lines, and although they were slightly higher in RA FLS the difference did not reach statistical significance (data not shown).
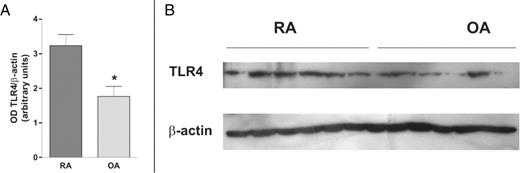
Immunoblot analysis of TLR4 protein expression. Expression of TLR4 at the protein level was studied by western blot analysis of cultured FLS cell lysates from patients with RA or OA. β-Actin was used as control. The difference between RA and OA samples was statistically significant (*P<0.01) (A). Results are the mean ± s.e.m. of three experiments including six FLS lines from different patients with RA and six from patients with OA. A representative experiment is shown (B).
Effect of VIP on TLR4 expression in FLS
We next studied whether VIP modulates constitutive or induced TLR2, TLR4 and MyD88 expression in FLS cultures from patients with RA. By quantitative real-time PCR we observed that VIP does not modify the normalized expression of TLR2, TLR4 or MyD88 mRNA under basal, non-stimulated conditions in RA FLS (Fig. 3).
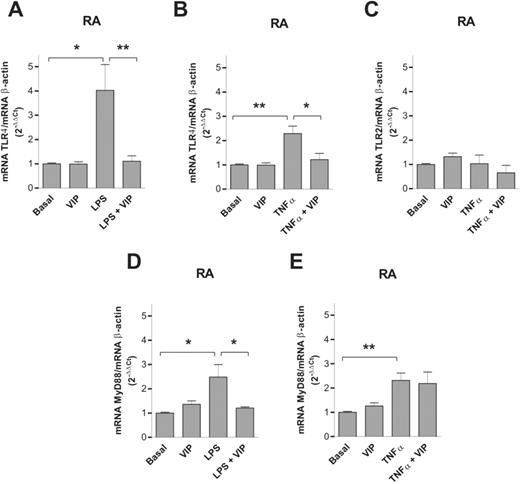
Effect of VIP on mRNA expression for TLR4, TLR2 and MyD88. The mRNA expression of TLR4, TLR2 and MyD88 was studied by quantitative real-time PCR in FLS cultures from patients with RA under basal (non-stimulated) conditions and after stimulation with 20 μg/ml LPS or 10 nM TNF-α. The results were corrected for β-actin mRNA expression within each sample. Results are mean ± s.e.m. of two experiments performed in duplicate and include three FLS lines from different patients with RA. *P<0.05, **P<0.01.
LPS and TNF-α induced a significant increase in TLR4 and MyD88 but not TLR2 mRNA expression in RA FLS. VIP significantly decreased LPS and TNF-α-induced TLR4 mRNA expression in RA FLS. VIP also significantly decreased LPS but not TNF-α-induced MyD88 expression in RA FLS.
LPS and TNF-α induction of TLR4 protein expression was also confirmed by western blot. Similar to what was observed at the mRNA level, treatment of RA FLS cultures with VIP blocked both LPS and TNF-α induction of TLR4 protein expression, whereas it did not have an effect on basal non-stimulated TLR4 protein levels (Fig. 4). In contrast, the smaller increase in MyD88 mRNA expression and its down-regulation by VIP were not confirmed at the protein level (data not shown).
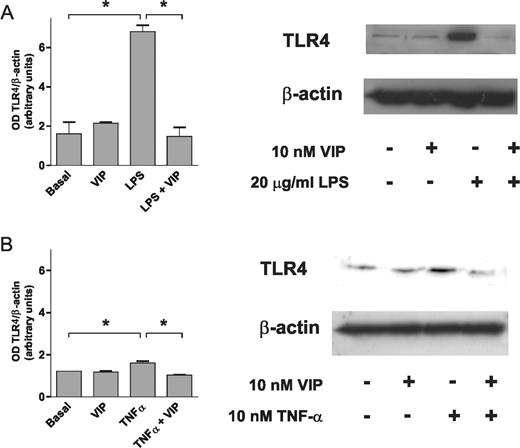
Effect of VIP on induced TLR4 expression. Western blot analysis of TLR4 protein expression in cultured FLS stimulated or not with (A) LPS or (B) TNF-α in the presence or absence of VIP at the indicated concentrations. An experiment representative of three is shown together with a densitometric analysis of TLR4 expression normalized to β-actin expression. *P<0.05.
Effect of VIP on the production of chemokines
To study whether the observed changes in TLR4 expression were accompanied by functional changes, we analysed the production of the chemokines CCL2 and CXCL8 in response to LPS stimulation. LPS induced a significant increase in CCL2 and CXCL8 release by RA FLS cultures (Fig. 5).
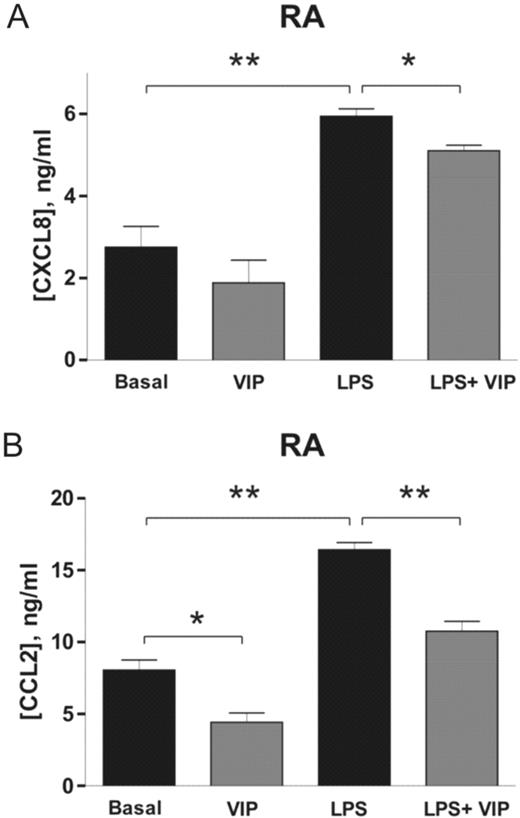
Effect of VIP on CXCL8 and CCL2 production in cultured FLS. Supernatants of FLS cultures from RA patients in the basal condition and after stimulation with 20 μg/ml LPS were collected, and levels of (A) CXCL8 and (B) CCL2 were determined by sandwich ELISA. Each result is the mean ± s.e.m. of three independent experiments performed in triplicate and includes three FLS lines from different patients with RA. *P<0.01; **P<0.001.
VIP decreased CXCL8 and CCL2 release by RA FLS cultures under non-stimulated conditions but the difference was statistically significant (P<0.01) only for CCL2. In LPS-stimulated FLS from patients with RA, VIP significantly decreased CXCL8 and CCL2 release (P<0.01 for CXCL8 and P<0.001 for CCL2).
Discussion
Activation of the immune system through TLR is an emerging facet of the pathogenesis of human chronic inflammatory diseases such as RA [27]. Crosstalk between cytokines and TLR signalling pathways has been demonstrated in RA cells in vitro, and TLR can contribute to the induction of the synthesis of a large variety of proinflammatory mediators, such as cytokines, chemokines, metalloproteinases and adhesion molecules [28]. TLR expression seems up-regulated in RA tissues and macrophages, and TLR are functional in synovial FLS. Although the mechanisms of expression and activation are not clear [20–22], several studies confirm that TLR expression is a cytokine-inducible feature [20, 28]. Our results confirm that the expression of TLR2 and MyD88 mRNA and that of TLR4 mRNA and protein are increased in cultured FLS from RA compared with OA patients, and that in the case of TLR4 it can be up-regulated in vitro by TNF-α and by its ligand LPS.
VIP has been shown to significantly reduce the incidence and severity of arthritis in an experimental model, completely abrogating joint swelling and destruction of cartilage and bone. The therapeutic effect of VIP was associated with down-regulation of both the inflammatory and the autoimmune component of the disease [4]. Recent studies suggest that VIP may represent an efficacious anti-arthritic treatment that modulates macrophage and T-cell cytokine profiles when used alongside a phosphodiesterase inhibitor [10]. Our previous studies in a murine model of inflammatory bowel disease induced by trinitrobenzene sulphonic acid suggest that VIP down-regulates TLR2 and TLR4 expression in parallel to a reduction in inflammatory and Th1-driven mediators and an improvement in the clinical and histopathological severity of the disease [29]. Our present data confirm that VIP directly interferes with TNF-α as well as with TLR4 signalling in FLS and that VIP is able to block downstream responses, such as chemokine expression and induced expression of TLR2, TLR4 and MyD88, through VIP receptors that have been described previously in FLS [30].
To date, it is known that TLR signalling is mediated through four adapter molecules: MyD88, MAL/TIRAP, TRIF and TRAM. Moreover, MyD88 is the pivotal adapter protein, being essential for the production of the inflammatory network molecules [12, 13, 31]. In this regard, it has been reported that MyD88 is involved in the streptococcal model of arthritis [32]. Our present study demonstrates that mRNA expression of MyD88 is up-regulated in LPS- and TNF-α-treated RA FLS, and that VIP is able to significantly decrease its expression after LPS stimulation at the mRNA but not at the protein level. Further studies are needed to explain this discrepancy. Recently, it has been reported that TGF-β1 inhibits the MyD88-dependent pathway in LPS-induced TLR4 signalling [33]. TLR and cytokine receptors share common intracellular pathways, in which NF-κB activation plays a pivotal role [34, 35]. Therefore, the well known negative effects of VIP signalling on NF-κB activation may underlie the observed effects [36]. In this regard, we have reported in the collagen-induced arthritis model that in vivo VIP treatment prevents NF-κB nuclear translocation through the inhibition of IκBα phosphorylation and degradation [7].
Proinflammatory chemokines are important players in RA pathology and represent an important group of mediators that can be induced in RA FLS in response to cytokines and TLR ligands. TLR stimulation by PGN induces up-regulation of proinflammatory and angiogenic chemokines in RA FLS, and this stimulation increases the chemotactic activity of FLS for PBMC [22]. Our study demonstrates that RA FLS produce CXCL8 and CCL2 chemokines upon stimulation with the TLR4 ligand LPS. Although we have previously reported that TNF-α-stimulated FLS secrete chemokines [9], the present data argues for a direct activation via TLR4 since FLS are unable to produce TNF-α and other proinflammatory cytokines such as IFN-γ and IL-1α [37]. In addition, we show that VIP down-regulates proinflammatory chemokine production in response to TLR4 stimulation. Fibroblasts have been identified as a source of chemokines in synovial tissue of RA patients [38, 39], contributing to inflammation maintenance, and chemokine blockade had beneficial effects in animal models of RA [40, 41]. Our findings demonstrate that RA FLS activated via TLR4 produce chemokines which are down-regulated by VIP, supporting its potential to modulate chemokine-mediated joint inflammation.
In conclusion, these results confirm that there is differential expression of TLR4, TLR2 and MyD88 in RA FLS compared with OA ex vivo. In addition, VIP has a regulatory effect on TLR4 and MyD88 expression and function in FLS from RA and OA patients. Although further studies are needed to evaluate the mechanisms involved, our observation supports the hypothesis that VIP down-regulates multiple proinflammatory pathways, including TLR-mediated pathways, which extends its therapeutic actions from the murine model to human RA and provides new insights into its use in therapy.

This work was supported by grants G03/152 from the Fondo de Investigación Sanitaria (FIS), BFI 2002–03489 from Ministerio de Ciencia y Tecnología, a postdoctoral fellowship from Junta de Comunidades de Castilla-La Mancha to I.G.-C. and a predoctoral fellowship from Ministerio de Ciencia y Tecnología to A.A. (Spain).
B.S., C.M., R.P.G. and J.L.P. have received a research grant from Genetrix SL, which holds a patent for the therapeutic use of VIP in RA.
References
Gomariz RP, Martinez C, Abad C, Leceta J, Delgado M. Immunology of VIP: a review and therapeutical perspectives.
Kojima M, Ito T, Oono T et al. VIP attenuation of the severity of experimental pancreatitis is due to VPAC1 receptor-mediated inhibition of cytokine production.
Delgado M, Gomariz RP, Martinez C, Abad C, Leceta J. Anti-inflammatory properties of the type 1 and type 2 vasoactive intestinal peptide receptors: role in lethal endotoxic shock.
Delgado M, Abad C, Martinez C, Leceta J, Gomariz RP. Vasoactive intestinal peptide prevents experimental arthritis by downregulating both autoimmune and inflammatory components of the disease.
Abad C, Martinez C, Juarranz MG et al. Therapeutic effects of vasoactive intestinal peptide in the trinitrobenzene sulfonic acid mice model of Crohn's disease.
Bik W, Wolinska-Witort E, Chmielowska M, Baranowska-Bik A, Rusiecka-Kuczalek E, Baranowska B. Vasoactive intestinal peptide can modulate immune and endocrine responses during lipopolysaccharide-induced acute inflammation.
Juarranz Y, Abad C, Martinez C et al. Protective effect of vasoactive intestinal peptide on bone destruction in the collagen-induced arthritis model of rheumatoid arthritis.
Skapenko A, Leipe J, Lipsky PE, Schulze-Koops H. The role of the T cell in autoimmune inflammation.
Juarranz MG, Santiago B, Torroba M et al. Vasoactive intestinal peptide modulates proinflammatory mediator synthesis in osteoarthritic and rheumatoid synovial cells.
Foey AD, Field S, Ahmed S et al. Impact of VIP and cAMP on the regulation of TNF-alpha and IL-10 production: implications for rheumatoid arthritis.
Beutler B. Interferences, questions and possibilities in toll-like receptor signalling.
Andreakos E, Foxwell B, Feldmann M. Is targeting Toll-like receptors and their signaling pathway a useful therapeutic approach to modulating cytokine-driven inflammation?
Choe JY, Crain B, Wu SR, Corr M. Interleukin 1 receptor dependence of serum transferred arthritis can be circumvented by Toll-like receptor 4 signaling.
Zhai Y, Shen XD, O’Connell R et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway.
Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes.
Brentano F, Kyburz D, Schorr O, Gay R, Gay S. The role of Toll-like receptor signalling in the pathogenesis of arthritis.
Radstake TR, Roelofs MF, Jenniskens YM et al. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma.
Seibl R, Birchler T, Loeliger S et al. Expression and regulation of Toll-like receptor 2 in rheumatoid arthritis synovium.
Kyburz D, Rethage J, Seibl R et al. Bacterial peptidoglycans but not CpG oligodeoxynucleotides activate fibroblasts by toll-like receptor signaling.
Pierer M, Rethage J, Seibl R et al. Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by Toll-like receptor 2 ligands.
Proost P, Verpoest S, Van de Borne K et al. Synergistic induction of CXCL9 and CXCL11 by Toll-like receptor ligands and interferon-gamma in fibroblasts correlates with elevated levels of CXCR3 in septic arthritis synovial fluids.
Arnett FC, Edworthy SM, Bloch DA et al.
Pablos JL, Santiago B, Galindo M et al. Synoviocyte-derived CXCL12 is displayed on endothelium and induces angiogenesis in rheumatoid arthritis.
Chang JT, Chen IH, Liao CT et al. A reverse transcription comparative real-time PCR method for quantitative detection of angiogenic growth factors in head and neck cancer patients.
Sweeney SE, Firestein GS. Rheumatoid arthritis: regulation of synovial inflammation.
Iwahashi M, Yamamura M, Aita T et al. Expression of Toll-like receptor 2 on CD16+ blood monocytes and synovial tissue macrophages in rheumatoid arthritis.
Gomariz RP, Arranz A, Abad C et al. Time-course expression of Toll-like receptors 2 and 4 in inflammatory bowel disease and homeostatic effect of VIP.
Takeba Y, Suzuki N, Kaneko A et al. Evidence for neural regulation of inflammatory synovial cell functions by secreting calcitonin gene-related peptide and vasoactive intestinal peptide in patients with rheumatoid arthritis.
Burns KF, Martinon C, Esslinger H et al. MyD88, an adapter protein involved in interleukin-1 signalling.
Joosten LA, Koenders MI, Smeets RL et al. Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88.
Naiki Y, Michelsen KS, Zhang W, Chen S, Doherty TM, Arditi M. Transforming growth factor-beta differentially inhibits MyD88-dependent, but not TRAM- and TRIF-dependent, lipopolysaccharide-induced TLR4 signaling.
Chen W, Wang J, An H, Zhou J, Zhang L, Cao X. Heat shock up-regulates TLR9 expression in human B cells through activation of ERK and NF-kappaB signal pathways.
Mansell A, Brint E, Gould JA, O’Neill LA, Hertzog PJ. Mal interacts with tumor necrosis factor receptor-associated factor (TRAF)-6 to mediate NF-kappaB activation by toll-like receptor (TLR)-2 and TLR4.
Delgado M, Munoz-Elias EJ, Kan Y et al. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit tumor necrosis factor alpha transcriptional activation by regulating nuclear factor-kB and cAMP response element-binding protein/c-Jun.
Ritchlin C. Fibroblast biology. Effector signals released by the synovial fibroblast in arthritis.
Rathanaswami P, Hachicha M, Sadick M et al. Expression of the cytokine RANTES in human rheumatoid synovial fibroblast. Differential regulation of RANTES and interleukin-8 genes by inflammatory cytokines.
Villiger PM, Terkeltaub R, Lotz M. Production of monocyte chemoattractant protein-1 by inflamed synovial tissue and cultures synoviocytes.
Podolin PL, Bolognese BJ, Foley JJ et al. A potent and selective nonpeptide antagonist of CXCR2 inhibits acute and chronic models of arthritis in the rabbit.
Author notes
Servicio de Reumatología, Hospital 12 de Octubre, 1Departamento de Biología Celular, Facultad de Biología and 2Departamento de Biología Celular, Facultad de Medicina, Universidad Complutense de Madrid, Madrid, Spain.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments