





Abstract
Rheumatoid arthritis (RA) is a chronic autoimmune-disease of unknown origin that primarily affects the joints and ultimately leads to their destruction. The involvement of immune cells is a general hallmark of autoimmune-related disorders. In this regard, macrophages, T cells and their respective cytokines play a pivotal role in RA. However, the notion that RA is a primarily T-cell-dependent disease has been strongly challenged during recent years. Rather, it has been understood that resident, fibroblast-like cells contribute significantly to the perpetuation of disease, and that they may even play a role in its initiation. These rheumatoid arthritis synovial fibroblasts (RASFs) constitute a quite unique cell type that distinguishes RA from other inflammatory conditions of the joints.
A number of studies have demonstrated that RASFs show alterations in morphology and behaviour, including molecular changes in signalling cascades, apoptosis responses and in the expression of adhesion molecules as well as matrix-degrading enzymes. These changes appear to reflect a stable activation of RASFs, which occurs independently of continuous exogenous stimulation. As a consequence, RASFs are no longer considered passive bystanders but active players in the complex intercellular network of RA.
In this review, we summarize and discuss recent research that highlights the role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis (RA). Since rheumatoid arthritis synovial fibroblasts (RASFs) mediate most relevant pathways of joint destruction, molecular insights into these cells constitute an important target for novel therapeutic approaches that inhibit the destruction of cartilage and bone in RA.
In industrialized countries, alterations in lifestyle and hygiene during the last century have shifted the spectrum of diseases from infectious to autoimmune-related disorders. It is unclear, however, whether this tendency towards autoimmunity is due to a true increase in these diseases or to an increased awareness and better diagnostic tools [1]. In this context, RA represents one of the most common autoimmune-related diseases, affecting as much as 1% of Western populations. It is a chronic polyarticular disorder that manifests primarily as a painful inflammation of the synovial tissues of joints, tendon sheaths and bursae. The progressive destruction of the articular cartilage is one of the hallmarks of the disease and determines the outcome of RA in most affected individuals.
RA is a systemic disorder, and it is commonly accepted that it emerges from a variable combination of individual genetic predisposition, environmental factors (such as potential but unproven infectious agents) and dysregulated immune responses [2–5]. While the aetiopathogenesis is only partially understood, the involvement of immune cells and their respective pro-inflammatory mediators is a common hallmark of RA as of all systemic autoimmune disorders [4, 6–8]. In addition, the rheumatoid synovium harbours a special cell population, known as activated RASFs, that is engaged in the initiation and perpetuation of RA and thus distinguishes RA from other inflammatory disorders of the joints. These cells appear to be in the centre of the local pathogenic events, and there is growing evidence that activation of RASFs (e.g. by responses of the innate immune system) is an early step in the development of RA. Once activated, RASFs produce a variety of cytokines, chemokines and matrix-degrading enzymes that mediate the interaction with neighbouring inflammatory and endothelial cells and are responsible for the progressive destruction of articular cartilage and bone. In this scenario, the production of cytokines and chemokines within the rheumatoid synovium would help to recruit T cells, macrophages and neutrophils, which, in turn, attract more inflammatory cells and, ultimately, enhance the activated state of the RASFs and of osteoclasts [9]. Taken together, various direct and indirect mechanisms contribute to the progressive destruction of articular cartilage and adjacent bone [10, 11]. Direct mechanisms consist of the attachment of fibroblasts to the underlying cartilage by up-regulation of cellular adhesion molecules (CAM) and the destruction of articular cartilage by production of matrix-degrading enzymes [10], while indirect mechanisms govern the differentiation of macrophages into osteoclasts, for example through up-regulation of receptor activator of nuclear factor kappa B ligand (RANKL) [12–14].
Synovial cell activation
RASFs are characterized by a round, large pale nucleus with prominent nucleoli, indicating very active RNA metabolism. Of interest, RASFs can be expanded in cell culture over several passages, and, in addition, they escape contact inhibition. These changes are often referred to as those of a tumour-like transformation [15] since they result in aggressive and invasive behaviour of RASFs in the adjacent cartilage and bone [16].
Recent evidence indicates the involvement of Toll-like receptors (TLRs), which are key recognition structures of the innate immune system, at an initial stage of synovial activation [17]. Hypothetically, microbial components or endogenous ligands, such as RNA from necrotic cells within the synovial fluid [18], activate RASFs through TLR signalling and lead to the up-regulated expression of pro-inflammatory cytokines and chemokines [19]. These factors would then result in the attraction and accumulation of immune cells in the synovium and, through a stimulatory loop, to chronic inflammation.
Such cytokines, together with growth factors, thus play an important role both in the continuous stimulation of RASFs towards aggressive behaviour as well as in the crosstalk between RASFs and other cell types in the synovium. Prominent examples include well-characterized cytokines like tumour necrosis factor alpha (TNFα) and interleukin (IL)-1, and also more recently described mediators like IL-15, IL-21R [20] and IL-22 [21]. One of the master cytokines that essentially trigger inflammation and joint destruction is certainly TNFα. Systemic over-expression of TNF as achieved in the TNF transgenic mouse model (hTNFtg [22]) appears to be sufficient to initiate chronic synovitis, cartilage destruction and, finally, bone erosion [23]. These findings are also confirmed by the clinical efficacy of TNF-blocking agents. However, a substantial number of patients receiving TNF blockers lack a clinical response, indicating that other, TNF-independent, pathways of inflammation and joint destruction exist in RA. Therefore it appears that the destructive properties of RASFs are not merely the response to continuous stimulation by inflammatory mediators but constitute intrinsic features of these cells. This notion has mainly been derived from studies in the severe combined immunodeficient mouse (SCID) co-implantation model [10, 11]. This animal model for RA was developed to investigate several aspects of an affected human joint under controlled conditions in vivo. Briefly, human RASF and fresh human cartilage are co-implanted together with an inert sponge under the kidney capsule of SCID mice. The SCID mice have severe defects both in cellular and humoral immune responses and, thus, are not able to reject these implants. When the implants are removed, usually after a period of 60 days, it is, therefore, possible to study the molecular mechanisms of interaction of synovial fibroblasts and the human cartilage as well as to assess the cartilage destruction histologically. As an obvious advantage of this model, both processes occur in the absence of inflammatory cells or other pro-inflammatory mediators [10, 24]. However, as with every animal model, the extrapolation of these data to human RA patients is difficult, thus limiting direct conclusions.
Angiogenesis is another histological hallmark of inflamed synovial tissue. It occurs already in early states of RA, which may be asymptomatic. Several pro-angiogenic factors are expressed by RASFs, in particular IL-8, vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF) and TGFβ [25]. In concert with RASF-derived chemokines (including MCP-1, MIP-1α, MIP-3α and RANTES), and different cytokines such as IL-15 and IL-16 (a potent chemoattractant for CD4+ cells) in the RA synovium, it has been hypothesized that T cells and macrophages expand into the synovium in an antigen-independent way. Both the up-regulation of adhesion molecules and the granzyme/perforine system of T cells could lead to diapedesis through the vascular basement membranes into the synovium [26]. Since histomorphological studies have demonstrated that neoangiogenesis and infiltrations of mononuclear cells lead to a hyperplastic lining layer, these cells are probably also involved in the disease process, showing the importance of the functional cross-talk between immune cells and RASFs. In this context, an interesting observation has been made most recently. Microparticles are small, membrane-bound vesicles, which are released from stimulated T cells and macrophages. Immune cell-derived microparticles activate synovial fibroblasts in a dose-dependent manner to release matrix metalloproteinases, pro-inflammatory cytokines and chemokines [27]. The accumulation of microparticles from various cellular origins in the synovial fluid [28] may thus contribute to the individual course of the disease, and the shedding of microparticles may represent an alternative stimulus in the complex cell–cell interaction of the pathogenesis of RA.
Attachment to and destruction of extracellular matrix
There are some direct consequences of the activation of RASFs: the up-regulation of adhesion molecules, enabling the strong interaction of fibroblasts with the extracellular matrix, which culminates in the destruction of cartilage and bone. The attachment of RASFs to the articular cartilage is the first step of synovial invasion and is mediated by the up-regulation of adhesion molecules on the surface of RASFs. Adhesion molecules are responsible for the anchoring of fibroblasts to the extracellular matrix of the articular cartilage, namely collagen type II and various glycosaminoglycans. Integrins constitute a large family of transmembrane cell-matrix adhesion molecules that are composed of two heterodimeric glycoproteins (α and β subunits). In the context of RA, integrins of the β1 subfamily play an important role. In particular, α3, α4 and α5 [formerly known as very late antigens (VLA) 3–5] are most prominently involved as the partner to β1 [29–31]. Integrins mediate the attachment of RASFs to fibronectin-rich sites of the cartilage [30] and, in addition, also to collagens and cartilage oligomeric matrix protein (COMP). The latter is a component of the hyaline cartilage that is mainly produced by chondrocytes and synovial fibroblasts. However, integrins are much more than just mechanoceptors that attach the cell to its surroundings. By activating intracellular signalling pathways, integrins mediate the contextual response of cells to the extracellular matrix. Upon adhesion of RASFs, integrins as well as other important adhesion molecules such as the vascular adhesion molecule (VCAM)-1 interact with signalling cascades that regulate the early cell cycle and the expression of matrix metalloproteinases (MMPs) [32]. In this regard, galectin-3 is up-regulated in RASFs after cell adhesion to COMP and thus contributes to inflammation and inhibition of apoptosis [33]. Similarly, c-fos [a component of the activator-protein (AP)-1 complex] and the proto-oncogene c-myc, which are both expressed within the RA synovium [34–40], were shown to be up-regulated by integrin-mediated cell adhesion [41]. Together with other key molecules, these pathways play a pivotal role in tissue destruction of articular cartilage, the most crucial event involved upon activation of RASFs. Tissue degradation essentially contributes to the progressive loss of joint function. Tissue degradation comprises the following major pathophysiological phenomena: growth, spreading and invasion of inflamed synovial tissue, and destruction of cartilage and bone. All these processes have a common underlying mechanism, namely the degradation of extracellular matrix, which is mediated by matrix-metalloproteinases (MMPs) [42], cathepsins [43] and an activated plasmin system [44].
MMPs are zinc-containing endopeptidases that are involved most prominently in tissue remodelling. Their catalytic activity is finely counter-regulated by the activity of endogenous inhibitors, the tissue inhibitors of matrix metalloproteinases (TIMPs). MMPs also act as important regulatory molecules on cytokines and adhesion molecules. Pro-inflammatory cytokines, growth factors and matrix molecules induce the expression of MMPs via transcriptional activation. The specific effects of different cytokines on the expression of individual MMPs, though, are highly variable and depend on the induced type of MMP, the cell type and the signal transduction pathway. AP-1 binding sites have been found in the promoter region of all MMPs [45], and thus AP-1 appears to play a pivotal role in the transcriptional activation of MMPs. In addition, some of the MMP promoter sites contain binding sites for NFκB [46, 47] and signal transduction and activation of transcription (STAT) [48]. Upstream of these transcription factors, all three stress- and mitogen-activated protein kinases (SAPK/MAPK), namely the extracellular regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 kinase, are highly active in chronic synovitis and also involved in the regulation of MMP expression [49]. Thus, different disease-relevant MMPs such as gelatinases (MMP-2 and MMP-9) and MT1-MMP are regulated by factors that mediate their effects through the ras proto-oncogene [50, 51]. Recently, gene transfer with dominant negative (dn) mutants of Raf-1 and dn-c-Myc demonstrated the relevance of the Ras-Raf-MAPK pathway for the activation and invasive behaviour of RASFs [52].
In addition to metalloproteases, other classes of enzymes such as cysteine and aspartyl proteases are involved in rheumatoid joint destruction. The cysteine proteases cathepsin B [43] and L [53, 54] are of special interest, and cathepsin K has also been implicated in the matrix degradation by RASFs [55]. Cathepsin B directly facilitates the degradation of ECM proteins, including fibronectin, collagen types I and IV and laminin [56]. Cathepsin B also activates other enzymes, including MMPs as well as soluble and receptor-bound forms of the serine protease urokinase plasminogen activator (uPA) [56–58]. MMPs and uPA have been shown to modulate the proteolytic cascade that mediates ECM degradation [59]. Cathepsin L cleaves collagens type I, II, IX, XI and certain proteoglycans. The expression of both cathepsin B and L has been demonstrated in RASFs at sites of invasion into cartilage and bone [43, 54]. Thus, stimulation of RASFs by pro-inflammatory cytokines, such as TNFα and IL-1 [60, 61], and the expression of proto-oncogenes lead to the release of cathepsins. For example, stable expression of constitutively active Ras resulted in increased levels of cathepsin L [62]. In addition, other proteases, such as thrombin, have been demonstrated with inflammation in RA joints induced by the secretion of IL-8 and the recruitment of leucocytes, which release cathepsin B into the synovial fluid [63].
Cell cycle, regulators and transcription factors
The ultimate cause of the activation of RASFs is still not well understood, thus limiting both our insights into the pathogenesis and the development of novel drugs. However, it has been well established that the altered morphology and the aggressive behaviour of RASFs mirror specific alterations in the transcription of disease-relevant genes and in intracellular signalling pathways, including alterations in apoptotic cascades. These changes comprise up-regulation of several proto-oncogenes as well as down-regulation or functional silencing of potentially protective tumour suppressor genes. Such events might explain the activation of the rheumatoid synovium. Figure 1 provides an overview of altered molecular players in RASFs
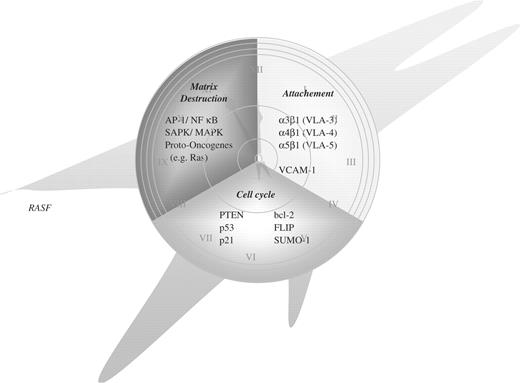
‘The time is out of joint’ (Hamlet; 1,5,188)—seen in the context of RA probably due to biological changes within the synovial fibroblast.
Synovial hyperplasia appears to be caused at least in part by the impairment of apoptosis in RASFs and synovial macrophages. Deficient apoptosis and, thus, prolonged survival of RASFs results from up-regulated anti-apoptotic molecules like bcl-2, sumo-1 (sentrin-1) and FLIP (Fas-associated death domain-like interleukin 1β converting enzyme inhibitory protein), especially at sites of synovial invasion into cartilage and bone [64–67]. In addition, alterations in the levels of expression and function of the tumour suppressor PTEN (phosphatase and tensin homologue deleted from chromosome 10) have been found in RASFs. PTEN is functionally involved in cell cycle arrest and apoptosis—and mutations in PTEN are found in a wide range of human cancers [68]. Compared with normal synovial tissue, in which PTEN is homogeneously expressed, examination of cultured RASFs showed that only 40% of cells expressed PTEN. In RASFs invading cartilage virtually no expression of PTEN was found, suggesting that the synovial hyperplasia in RA is due to defective apoptosis [69]. In this context, the tumour suppressor p53 and its downstream molecule p21 have also been investigated. The expression rate was generally found to be low (<5%) but was increased in cells that were invading the articular cartilage, suggesting that p53 could be induced in cells at sites of cartilage invasion, thus rendering the cells a selective advantage [70].
Various studies have revealed high transcription of immediate early genes in RASFs, for example egr-1 [35, 37] and fos [36, 38], as well as proto-oncogenes such as jun [36, 38] and myc [52]. The high expression of fos and jun, which are involved in the formation of the AP-1 transcription factor, appears to be mediated through upstream oncogenes like ras, scr and raf. These oncogenes, in turn, are activation molecules for mitogen-activated protein kinase (MAPK) pathways. MAP kinases, in particular p38, can be activated by inflammatory cytokines (e.g. IL-1 and TNFα) and are thought to regulate processes involved in apoptosis and proliferation. However, certain members of the MAPK family are also activated by cytokine-independent mechanisms. L1 elements are mobile genetic elements that have been shown to act as retrotransposons and are widely distributed within the human genome. In this regard, it was demonstrated that functional L1 elements induce the MAP kinase p38δ (also known as stress-activated kinase 4, SAPK4). p38δ then induces the production of MMP-1 [71], MMP-3 [72], IL-6 and IL-8 [73]. From these data, it can be concluded that RASFs are not only stimulated by pro-inflammatory cytokines but also by a cytokine-independent pathway through the activation of p38δ [74]. This notion is supported by reports on a number of RA patients who show progression of disease under TNF-blocking biologicals, even when combined with immunosuppressive drugs.
The ubiquitously expressed transcription factor nuclear factor kappa B (NFκB) is also highly activated in RASFs [75, 76]. NFκB, composed of DNA-binding heterodimers, is normally retained in the cytoplasm by its natural counterpart, IκB. In response to different factors, IκB proteins are phosphorylated, polyubiquinated and finally undergo protein shredding by the 26 proteasome [77]. This process results in the nuclear translocation of NFκB, enabling it to bind to the promoters of target genes such as IL-6, IL-8 and cyclooxygenase-2. However, NFκB not only regulates pro-inflammatory genes but also both the transcription of adhesion molecules and matrix-degrading enzymes [47]. In addition, activation of NFκB increases the synthesis of the urokinase-type plasmin activator (uPa), which has been associated with activation of some of these enzymes [59, 78–80]. Moreover, it has been suggested that NFκB negatively regulates the aforementioned tumour suppressor PTEN thus promoting cell survival [81]. Upstream of NFκB, two IκB kinases (IKK1 and -2) regulate IκB activity [82]. These enzymes are activated by the Akt serine–threonine kinase, thus decreasing the activity of pro-apoptotic proteins and increasing the activity of anti-apoptotic proteins [83].
Perspectives
Since earlier observations in arthritides of MRL-lpr/lpr mice and the description of apparently transformed synovial cells by Fassbender [15] have suggested that in the pathogenesis of RA at least two cellular mechanisms [84] are operating, it has became clear through studies in the SCID mouse model [10] that the synovial fibroblasts are important players in rheumatoid joint destruction. Subsequent work by Kuchen et al. [71] supported the concept of a cytokine-independent pathway of fibroblast-mediated joint destruction (Fig. 2). Consequently synovial fibroblasts have lost their role as innocent bystanders in the pathogenesis of RA, and it has been understood that due to their active involvement in orchestrating cellular cross-talk and mediating intracellular cascades, they represent an important target for novel therapeutic approaches to the inhibition of joint destruction.
![Functional cross-talk of cytokine-dependent and cytokine-independent pathways of joint destruction in RA. Pivotal downstream signalling cascades including the MAP kinase system and the transcription factor NFκB are activated in the synovial fibroblast through pro-inflammatory cytokines such as TNFα, and various interleukins. Similar activation of these pathways, however, is also achieved by cytokine-independent mechanisms, i.e. endogenous genetic elements (L1 retrotransposons), members of the innate immune system, namely various Toll-like receptors (reviewed in [17]) and novel mediators of cellular cross-talk such as immune cell-derived microparticles [27].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/rheumatology/45/6/10.1093_rheumatology_kel065/2/m_kel065f2.jpeg?Expires=1716350469&Signature=L0HHFmehfLLOZGCerxa2CEwGx3B65szIcH-EzRVbMES8DUbg-I-4-PxMu6APyGZweYgRy-G3ENGQUW3zrQlN49XzsVlN6kNuaNkp8QZ0CwwhJfXnis2QUkFCmZeNm4kWt54lpEZxZVpGg1PuNfNIlPgfN4E6k6IJ89NPaGJnTMXXu4QTHaMI5~u8vL1v~Gx8xSV7e4b-XgaKgQ3QuHO7lmKPp3ghsTfSXbDwKqyqo7a9g2N0uonYis~gSyXNVW981KQgHnOBCohKtX0r59jrJIASQQli3GrfQYwaEjA0fxBbuRDa6N1pRAiSRt--DOFyKClH5MeFD6x06N~~DT9PlQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Functional cross-talk of cytokine-dependent and cytokine-independent pathways of joint destruction in RA. Pivotal downstream signalling cascades including the MAP kinase system and the transcription factor NFκB are activated in the synovial fibroblast through pro-inflammatory cytokines such as TNFα, and various interleukins. Similar activation of these pathways, however, is also achieved by cytokine-independent mechanisms, i.e. endogenous genetic elements (L1 retrotransposons), members of the innate immune system, namely various Toll-like receptors (reviewed in [17]) and novel mediators of cellular cross-talk such as immune cell-derived microparticles [27].
Our insights into the molecular patterns of RA are still limited. But the exponential development in the field of molecular biology and its techniques has shed light on the different mechanisms of the disease. The development of biologicals, in particular the introduction of TNFα-blocking agents into everyday clinical practice by Maini et al. [85] was a milestone in the therapy of RA. These drugs have been supplemented by novel anti-cytokine therapies. For example, encouraging results have been achieved by early clinical trials with antibodies against IL-6R [86] and IL-15 [87], whilst application of IL-17 receptor IgG fusion constructs [88] and IL-18-binding proteins [89] are promising in animal models. Therapeutic strategies beyond cytokine targets include the recent use of CTLA4Ig, a fusion protein that interferes with the binding of CD80/CD86 to the MHC-independent T-cell surface molecule CD28 preventing its activation [90], as well as rituximab (monoclonal antibodies against CD20) that was originally applied in non-Hodgkin's lymphomas [91]. More recently, Edwards et al. [92] confirmed these promising data in patients with active RA despite methotrexate treatment, showing that two infusions of rituximab, alone or in combination with either cyclophosphamide or continued methotrexate, provided significant improvement in disease symptoms. These findings also indicate that the destructive activity of RASFs during the disease process is not completely independent of the immune system.
Moreover, the remarkable advances which have been achieved in the field of gene transfer (reviewed in [93]) towards the development of novel therapies for arthritic diseases are based both on our growing insights in the pathogenesis of RA, as well as on progress in using gene transfer methods both to validate known molecular targets and to discover novel pathways.
Finally, three principal approaches can be suggested for the future when thinking about fibroblasts as potential targets in order to abolish their potential for joint destruction: (i) interfering with the stimulation by inflammatory cytokines; (ii) modulating molecules that regulate apoptosis or proliferation; and (iii) direct inhibition of distinct matrix-degrading enzymes such as matrix metalloproteinases and cathepsins.
The main effort of research has focused on the role of pro-inflammatory cytokines, and thus virtually all biologicals in use are designed to interfere with the inflammatory pathway as described above. On the other hand, transcription factors, modulated proto-oncogenes and tumour suppressors are also of potential therapeutic interest in the case of RASFs. In this regard, using an inhibitor of proteosomal degradation (as is already in use for the treatment of multiple myeloma [94]), thus preventing activation of NFκB, as well as introducing a therapeutic vector construct over-expressing IκB, might be a feasible approach [95]. Genes regulating the cell cycle are another option for novel therapeutic strategies. For example, over-expression of cyclin-dependent kinase inhibitors such as p21 resulted in markedly reduced cellular growth of RASFs [96] and the co-transfection of RASFs with dominant negative mutations of the proto-oncogenes raf and myc reduced both the growth and invasiveness of these cells [52]. To interfere with the destructive potential of RASFs, over-expression of jun D (an antagonist of the ras proto-oncogene) inhibited the proliferation of fibroblasts by blocking the ras-mediated transformation of RASFs [97]. In the same context, jun D inhibited the formation of AP-1 thus directly down-regulating the expression of matrix-degrading enzymes. While most clinical trials of MMP inhibitors have yielded disappointing results, perhaps due to lack of selectivity [98], direct gene targeting of membrane-type (MT)-MMP-1 [99], cathepsin L [54] and the plasmin system [44] has shown promising results. So did the adenoviral over-expression of human TIMPs, in particular TIMP-3, reducing the invasiveness and bone-resorbing activity of RASFs both in vitro and in vivo [100].
Gene therapy in RA is still far from clinical use, but in order to perform functional genomics its powerful techniques should be used to explore important pathomechanisms and disease-relevant gene sequences.
The authors have declared no conflicts of interest.
References
Zinkernagel RM. Maternal antibodies, childhood infections, and autoimmune diseases.
Ermann J, Fathman CG. Autoimmune diseases: genes, bugs and failed regulation.
Marrack P, Kappler J, Kotzin BL. Autoimmune disease: why and where it occurs.
Smith JB, Haynes MK. Rheumatoid arthritis–a molecular understanding.
Silman AJ. Epidemiology and Genetics of Rheumatoid Arthritis.
Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis.
Feldmann M, Maini RN. The role of cytokines in the pathogenesis of rheumatoid arthritis.
Tarner IH, Muller-Ladner U, Gay RE, Fathman CG, Gay S. The Pathogenesis of Rheumatoid Arthritis – Gene Transfer to Detect Novel Targets for Treatment.
Muller-Ladner U, Kriegsmann J, Franklin BN, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice.
Geiler T, Kriegsmann J, Keyszer GM, Gay RE, Gay S. A new model for rheumatoid arthritis generated by engraftment of rheumatoid synovial tissue and normal human cartilage into SCID mice.
Nakano K, Okada Y, Saito K, Tanaka Y. Induction of RANKL expression and osteoclast maturation by the binding of fibroblast growth factor 2 to heparan sulfate proteoglycan on rheumatoid synovial fibroblasts.
Takayanagi H, Iizuka H, Juji T, et al. Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis.
Fassbender HG. Histomorphological basis of articular cartilage destruction in rheumatoid arthritis.
Lafyatis R, Remmers EF, Roberts AB, Yocum DE, Sporn MB, Wilder RL. Anchorage-independent growth of synoviocytes from arthritic and normal joints. Stimulation by exogenous platelet-derived growth factor and inhibition by transforming growth factor-beta and retinoids.
Brentano F, Kyburz D, Schorr O, Gay R, Gay S. The role of Toll-like receptor signalling in the pathogenesis of arthritis.
Brentano F, Schorr O, Gay RE, Gay S, Kyburz D. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3.
Pierer M, Rethage J, Seibl R, et al. Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by Toll-like receptor 2 ligands.
Jungel A, Distler JH, Kurowska-Stolarska M, et al. Expression of interleukin-21 receptor, but not interleukin-21, in synovial fibroblasts and synovial macrophages of patients with rheumatoid arthritis.
Ikeuchi H, Kuroiwa T, Hiramatsu N, et al. Expression of interleukin-22 in rheumatoid arthritis: potential role as a proinflammatory cytokine.
Keffer J, Probert L, Cazlaris H, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis.
Zwerina J, Hayer S, Tohidast-Akrad M, et al. Single and combined inhibition of tumor necrosis factor, interleukin-1, and RANKL pathways in tumor necrosis factor-induced arthritis: effects on synovial inflammation, bone erosion, and cartilage destruction.
Muller-Ladner U, Pap T, Gay RE, Gay S. Gene transfer as a future therapy for rheumatoid arthritis.
Bodolay E, Koch AE, Kim J, Szegedi G, Szekanecz Z. Angiogenesis and chemokines in rheumatoid arthritis and other systemic inflammatory rheumatic diseases.
Muller-Ladner U, Kriegsmann J, Tschopp J, Gay RE, Gay S. Demonstration of granzyme A and perforin messenger RNA in the synovium of patients with rheumatoid arthritis.
Distler JH, Jungel A, Huber LC, et al. The induction of matrix metalloproteinase and cytokine expression in synovial fibroblasts stimulated with immune cell microparticles.
Berckmans RJ, Nieuwland R, Tak PP, et al. Cell-derived microparticles in synovial fluid from inflamed arthritic joints support coagulation exclusively via a factor VII-dependent mechanism.
Rinaldi N, Schwarz-Eywill M, Weis D, et al. Increased expression of integrins on fibroblast-like synoviocytes from rheumatoid arthritis in vitro correlates with enhanced binding to extracellular matrix proteins.
Ishikawa H, Hirata S, Andoh Y, et al. An immunohistochemical and immunoelectron microscopic study of adhesion molecules in synovial pannus formation in rheumatoid arthritis.
Kriegsmann J, Keyszer GM, Geiler T, Brauer R, Gay RE, Gay S. Expression of vascular cell adhesion molecule-1 mRNA and protein in rheumatoid synovium demonstrated by in situ hybridization and immunohistochemistry.
Neidhart M, Zaucke F, von Knoch R, et al. Galectin-3 is induced in rheumatoid arthritis synovial fibroblasts after adhesion to cartilage oligomeric matrix protein.
Trabandt A, Aicher WK, Gay RE, et al. Expression of the collagenolytic and Ras-induced cysteine proteinase cathepsin L and proliferation-associated oncogenes in synovial cells of MRL/I mice and patients with rheumatoid arthritis.
Trabandt A, Aicher WK, Gay RE, Sukhatme VP, Fassbender HG, Gay S. Spontaneous expression of immediately-early response genes c-fos and egr-1 in collagenase-producing rheumatoid synovial fibroblasts.
Dooley S, Herlitzka I, Hanselmann R, et al. Constitutive expression of c-fos and c-jun, overexpression of ets-2, and reduced expression of metastasis suppressor gene nm23-H1 in rheumatoid arthritis.
Grimbacher B, Aicher WK, Peter HH, Eibel H. Measurement of transcription factor c-fos and EGR-1 mRNA transcription levels in synovial tissue by quantitative RT-PCR.
Kontny E, Ziolkowska M, Dudzinka E, Filipowicz-Sosnowska A, Ryzewska A. Modified expression of c-Fos and c-Jun proteins and production of interleukin-1 beta in patients with rheumatoid arthritis.
Asahara H, Fujisawa K, Kobata T, et al. Direct evidence of high DNA binding activity of transcription factor AP-1 in rheumatoid arthritis synovium.
Qu Z, Garcia CH, O'Rourke LM, Planck SR, Kohli M, Rosenbaum JT. Local proliferation of fibroblast-like synoviocytes contributes to synovial hyperplasia. Results of proliferating cell nuclear antigen/cyclin, c-myc, and nucleolar organizer region staining.
Dike LE, Farmer SR. Cell adhesion induces expression of growth-associated genes in suspension-arrested fibroblasts.
Pap T, Gay S, Schett G. Matrix Metalloproteinases. In: Smolen J, Lipsky J, Dunitz M, eds.
Trabandt A, Gay RE, Fassbender HG, Gay S. Cathepsin B in synovial cells at the site of joint destruction in rheumatoid arthritis.
van der Laan WH, Pap T, Ronday HK, et al. Cartilage degradation and invasion by rheumatoid synovial fibroblasts is inhibited by gene transfer of a cell surface-targeted plasmin inhibitor.
Benbow U, Brinckerhoff CE. The AP-1 site and MMP gene regulation: what is all the fuss about?
Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3.
Barchowsky A, Frleta D, Vincenti MP. Integration of the NF-kappaB and mitogen-activated protein kinase/AP-1 pathways at the collagenase-1 promoter: divergence of IL-1 and TNF-dependent signal transduction in rabbit primary synovial fibroblasts.
Li WQ, Dehnade F, Zafarullah M. Oncostatin M-induced matrix metalloproteinase and tissue inhibitor of metalloproteinase-3 genes expression in chondrocytes requires Janus kinase/STAT signaling pathway.
Schett G, Tohidast-Akrad M, Smolen JS, et al. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis.
Gum R, Juarez J, Allgayer H, Mazar A, Wang Y, Boyd D. Stimulation of urokinase-type plasminogen activator receptor expression by PMA requires JNK1-dependent and -independent signaling modules.
Korzus E, Nagase H, Rydell R, Travis J. The mitogen-activated protein kinase and JAK-STAT signaling pathways are required for an oncostatin M-responsive element-mediated activation of matrix metalloproteinase 1 gene expression.
Pap T, Nawrath M, Heinrich J, et al. Cooperation of Ras- and c-Myc-dependent pathways in regulating the growth and invasiveness of synovial fibroblasts in rheumatoid arthritis.
Keyszer GM, Heer AH, Kriegsmann J, et al. Comparative analysis of cathepsin L, cathepsin D, and collagenase messenger RNA expression in synovial tissues of patients with rheumatoid arthritis and osteoarthritis, by in situ hybridization.
Schedel J, Seemayer CA, Pap T, et al. Targeting cathepsin L (CL) by specific ribozymes decreases CL protein synthesis and cartilage destruction in rheumatoid arthritis.
Hummel KM, Petrow PK, Franz JK, et al. Cysteine proteinase cathepsin K mRNA is expressed in synovium of patients with rheumatoid arthritis and is detected at sites of synovial bone destruction.
Guinec N, Dalet-Fumeron V, Pagano M. “In vitro” study of basement membrane degradation by the cysteine proteinases, cathepsins B, B-like and L. Digestion of collagen IV, laminin, fibronectin, and release of gelatinase activities from basement membrane fibronectin.
Kobayashi H, Ohi H, Sugimura M, Shinohara H, Fujii T, Terao T. Inhibition of in vitro ovarian cancer cell invasion by modulation of urokinase-type plasminogen activator and cathepsin B.
Kobayashi H, Moniwa N, Sugimura M, Shinohara H, Ohi H, Terao T. Effects of membrane-associated cathepsin B on the activation of receptor-bound prourokinase and subsequent invasion of reconstituted basement membranes.
Lakka SS, Gondi CS, Yanamandra N, et al. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis.
Lemaire R, Huet G, Zerimech F, et al. Selective induction of the secretion of cathepsins B and L by cytokines in synovial fibroblast-like cells.
Huet G, Flipo RM, Colin C, et al. Stimulation of the secretion of latent cysteine proteinase activity by tumor necrosis factor alpha and interleukin-1.
Collette J, Ulku AS, Der CJ, Jones A, Erickson AH. Enhanced cathepsin L expression is mediated by different Ras effector pathways in fibroblasts and epithelial cells.
Mishiro T, Nakano S, Takahara S, et al. Relationship between cathepsin B and thrombin in rheumatoid arthritis.
Franz JK, Pap T, Hummel KM, et al. Expression of sentrin, a novel antiapoptotic molecule, at sites of synovial invasion in rheumatoid arthritis.
Perlman H, Liu H, Georganas C, et al. Differential expression pattern of the antiapoptotic proteins, Bcl-2 and FLIP, in experimental arthritis.
Schedel J, Gay RE, Kuenzler P, et al. FLICE-inhibitory protein expression in synovial fibroblasts and at sites of cartilage and bone erosion in rheumatoid arthritis.
Catrina AI, Ulfgren AK, Lindblad S, Grondal L, Klareskog L. Low levels of apoptosis and high FLIP expression in early rheumatoid arthritis synovium.
Goberdhan DC, Wilson C. PTEN: tumour suppressor, multifunctional growth regulator and more.
Pap T, Franz JK, Hummel KM, Jeisy E, Gay R, Gay S. Activation of synovial fibroblasts in rheumatoid arthritis: lack of Expression of the tumour suppressor PTEN at sites of invasive growth and destruction.
Seemayer CA, Kuchen S, Neidhart M, et al. p53 in rheumatoid arthritis synovial fibroblasts at sites of invasion.
Kuchen S, Seemayer CA, Kuenzler P, et al. Cytokine-Independent up-Regulation of Matrix Metalloproteinase 1 mRNA Expression by the Stress Activated Protein Kinase 4.
Ospelt C, Neidhart M, Michel BA, Simmen B, Gay RE, Gay S. p38delta induces MMP-3 in activated rhuematoid arthritis synovial fibroblasts.
Suzuki M, Tetsuka T, Yoshida S, et al. The role of p38 mitogen-activated protein kinase in IL-6 and IL-8 production from the TNF-alpha- or IL-1beta-stimulated rheumatoid synovial fibroblasts.
Neidhart M, Rethage J, Kuchen S, et al. Retrotransposable L1 elements expressed in rheumatoid arthritis synovial tissue: association with genomic DNA hypomethylation and influence on gene expression.
Miagkov AV, Kovalenko DV, Brown CE, et al. NF-kappaB activation provides the potential link between inflammation and hyperplasia in the arthritic joint.
Marok R, Winyard PG, Coumbe A, et al. Activation of the transcription factor nuclear factor-kappaB in human inflamed synovial tissue.
Palombella VJ, Conner EM, Fuseler JW, et al. Role of the proteasome and NF-kappaB in streptococcal cell wall-induced polyarthritis.
Abraham E, Gyetko MR, Kuhn K, et al. Urokinase-type plasminogen activator potentiates lipopolysaccharide-induced neutrophil activation.
Hasegawa T, Sorensen L, Dohi M, Rao NV, Hoidal JR, Marshall BC. Induction of urokinase-type plasminogen activator receptor by IL-1 beta.
Sidenius N, Blasi F. The urokinase plasminogen activator system in cancer: recent advances and implication for prognosis and therapy.
Vasudevan KM, Gurumurthy S, Rangnekar VM. Suppression of PTEN expression by NF-kappa B prevents apoptosis.
Aupperle K, Bennett B, Han Z, Boyle D, Manning A, Firestein G. NF-kappa B regulation by I kappa B kinase-2 in rheumatoid arthritis synoviocytes.
Gustin JA, Ozes ON, Akca H, et al. Cell type-specific expression of the IkappaB kinases determines the significance of phosphatidylinositol 3-kinase/Akt signaling to NF-kappa B activation.
Gay S, Gay RE, Koopman WJ. Molecular and cellular mechanisms of joint destruction in rheumatoid arthritis: two cellular mechanisms explain joint destruction?
Maini R, St Clair EW, Breedveld F, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group.
Choy EH, Isenberg DA, Garrood T, et al. Therapeutic benefit of blocking interleukin-6 activity with an anti-interleukin-6 receptor monoclonal antibody in rheumatoid arthritis: a randomized, double-blind, placebo-controlled, dose-escalation trial.
Baslund B, Tvede N, Danneskiold-Samsoe B, Peterson J, Peterson L, Schuurmann J. A novel human monoclonal antibody against IL-15 (humax-IL-15) in patients with active rheumatoid arthritis (RA): results of a double-blind, placebo-controlled phase I/II trial.
Bush KA, Farmer KM, Walker JS, Kirkham BW. Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein.
Plater-Zyberk C, Joosten LA, Helsen MM, et al. Therapeutic effect of neutralizing endogenous IL-18 activity in the collagen-induced model of arthritis.
Kremer JM, Westhovens R, Leon M, et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig.
Edwards JC, Szczepanski L, Szechinski J, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis.
Huber LC, Pap T, Muller-Ladner U, Gay RE, Gay S. Gene targeting: roadmap to future therapies.
Braun T, Carvalho G, Coquelle A, et al. NF-{kappa}B constitutes a potential therapeutic target in high-risk myelodysplastic syndromes.
Tak PP, Gerlag DM, Aupperle KR, et al. Inhibitor of nuclear factor kappaB kinase beta is a key regulator of synovial inflammation.
Nonomura Y, Kohsaka H, Nasu K, Terada Y, Ikeda M, Miyasaka N. Suppression of arthritis by forced expression of cyclin-dependent kinase inhibitor p21(Cip1) gene into the joints.
Wakisaka S, Suzuki N, Saito N, Ochi T, Sakane T. Possible correction of abnormal rheumatoid arthritis synovial cell function by jun D transfection in vitro.
Rutkauskaite E, Zacharias W, Schedel J, et al. Ribozymes that inhibit the production of matrix metalloproteinase 1 reduce the invasiveness of rheumatoid arthritis synovial fibroblasts.
Author notes
Center of Experimental Rheumatology, University Hospital Zurich, Switzerland, 1Department of Internal Medicine and Rheumatology, Justus-Liebig University Giessen, Kerckhoff-Clinic, Bad Nauheim and 2Division of Molecular Medicine of Musculoskeletal Tissue, University Hospital Munster, Germany.

{kind=link}
{kind=link}
Comments