




Abstract
Histopathological reports of multiple sclerosis and its animal models have shown evidence of a link between axonal injury in active lesions and impaired glutamate metabolism. Mature oligodendrocytes play a role in glutamate uptake to maintain glutamate homeostasis but in multiple sclerosis white matter the loss of expression of glutamate transporters in the lesion vicinity results in ineffective glutamate removal. Using a magnetic resonance spectroscopy technique that isolates the glutamate resonance at 3 T, we compared glutamate levels between normal subjects and multiple sclerosis patients in different brain areas. Metabolite concentrations (glutamate, glutamine, N-acetyl-aspartate, myo-inositol, choline, creatine) were derived from LCmodel and corrected for T1 relaxation time. Glutamate concentrations were found to be elevated in acute lesions (P = 0.02) and normal-appearing white matter (P = 0.03), with no significant elevation in chronic lesions (P = 0.77). The N-acetyl-aspartate level in chronic lesions was significantly lower (P < 0.001) than in acute lesions and normal-appearing white matter. The choline level in acute lesions was significantly higher (P < 0.001) than in chronic lesions. Evidence was also found for increased glial activity in multiple sclerosis, with significantly higher (P < 0.001) myo-inositol levels in acute lesions compared with control white matter. These in vivo results support the hypothesis that altered glutamate metabolism is present in brains of multiple sclerosis patients.
Introduction
Glutamate and glutamine are two important neurochemicals in the central nervous system. Glutamate is metabolized in the neurons and released in the synaptic space, where it binds to its postsynaptic receptors. To clear it from the synaptic space and prevent neurotoxicity due to continued signalling, glutamate is internalized into astrocytes (Shousboe et al., 1993; Rothstein et al., 1994) and converted into glutamine (Berl and Clarke, 1983). Glutamine then leaves the astrocytes and is taken up by neurons, where glutaminase regenerates glutamate, which completes the normal functioning of the well-known glutamate/glutamine (Shousboe et al., 1993) cycle. Under pathological conditions an excess of glutamate in the synaptic space can trigger a toxic cascade leading to cell death (Lipton and Rosenberg, 1994; Rothstein, 1996). This neuroexcitotoxicity cascade might have a key role linking the presence of inflammation to axonal injury in multiple sclerosis. Recent histopathological studies (Piani et al., 1991) of brain tissues have linked, for the first time, white matter axonal injury to the presence of inflammatory glutamate-producing immune cells in proximity to active multiple sclerosis lesions. Moreover, in multiple sclerosis white matter tissue outside of active plaques, the expression of glutamate receptors (EAAT-1 and EAAT-2) on the surface of oligodendrocytes (glial cell) and the glutamate reuptake are defective (Pitt et al., 2003). Supporting these results in multiple sclerosis, the blockade of glutamate receptors was shown to ameliorate oligodendrocyte survival and neurological sequelae, and to reduce dephosphorylation of axonal neurofilament in experimental allergic encephalomyelitis (EAE) (Pitt et al., 2000; Smith et al., 2000). Based on these important findings, distant Wallerian degeneration and/or retrograde degeneration of neurons and neuronal processes derived from multiple sclerosis lesions (Simon et al., 2000) can possibly be related, at least in part, by glutamate excitotoxic events. Excess glutamate in active lesions could therefore be an important predictor of axonal injury, brain atrophy and long-term accumulation of clinical disability. Neurotoxic properties of excess glutamate have been associated with a wide range of other degenerative diseases, such as amyotrophic lateral sclerosis, Huntington disease and Alzheimer disease (Doble, 1999; Auger and Attwell, 2000). A non-invasive method of monitoring changes in levels of glutamate will have a significant impact in understanding the pathogenesis of multiple sclerosis and other neurological diseases.
TE-averaged MRS
Even at 3 T, in conventional short echo spectra, the N-acetyl group of N-acetyl aspartate (NAA) (2.02 p.p.m.) overlaps with the C3 protons of glutamate. The C4 protons of glutamate at 2.35 p.p.m. are contaminated by C3 protons of NAA, as well as C3 and C4 protons of glutamine. The co-resonant signals from the C2 protons of glutamate and glutamine at 3.78 p.p.m. are partly obscured by myo-inositol. The overlapping peaks make it difficult to investigate the individual levels of glutamate and glutamine. Therefore, studies (Hattori et al., 2002) have focused on the evaluation of the total glutamate+glutamine levels under pathological conditions. Several prior studies (Lee et al., 1995; Pan et al., 1996; Thompson and Allen, 1998) have used the simplification of strong coupling patterns at high field strengths to devise spectral editing methods to resolve the glutamate resonance. Using a 4.1 T magnet, Pan and colleagues reported that glutamate had higher concentrations (2–4 mM higher) in grey matter compared with white matter (Pan et al., 1996). With most commercial manufacturers now offering magnets at 3 T, patient studies are becoming increasingly common at this field strength. It is therefore valuable to have a technique for measuring glutamate that overcomes the problem of overlapping spectral resonance associated with conventional point-resolved spectroscopy (PRESS) acquisitions at 3 T. Recent studies have shown differences in glutamate levels at 3 T between the angular cingulate cortex and hippocampus using a single-echo PRESS sequence at echo time (TE) = 80 ms (Schubert et al., 2004). This single echo acquisition scheme results in a mixed-phase glutamate detection. Mixed phased resonances are susceptible to cancellation due to field inhomogeneities and single-echo sequences are sensitive to biological and pathological variations in T2.
Recently a method was developed by our group (Hurd et al., 2004) which provides an unobstructed single-line resonance detection for glutamate at 2.35 p.p.m. at 3 T that is distinct from glutamine and NAA. In this method the conventional PRESS localization technique was modified to collect data over multiple echo times at fixed increments. The optimum echo times and increments were determined using simulations and phantom acquisitions. Using this scheme it was shown that the zeroth component of this multi-echo data set, obtained by averaging the different TE acquisitions, results in a spectrum that isolates the glutamate resonance. Hereafter, this acquisition scheme will be referred to as TE-averaged PRESS.
In this study we used the single-voxel TE-averaged PRESS technique to estimate the levels of glutamate and glutamine in addition to NAA, choline, creatine and myo-inositol in contrast-enhancing acute lesions, chronic lesions and normal-appearing white matter (NAWM) brain areas of multiple sclerosis patients. These were compared with metabolite levels in normal control white matter. Metabolite concentrations are estimated using LCmodel (Provencher, 1993) and corrected for relaxation effects.
Methods
Data acquisition
Data were acquired on a 3 T Signa scanner (Menlo Park, CA, USA) using the standard quadrature head coil and PROBE/SVQ™ (automated PRESS). This sequence was modified (Hurd et al., 2004) to collect data in 64 increments of 2.5 ms starting at TE = 35 ms with a number of averages (NEX) of 4. TR was set at 2s for all studies. The scan time for this acquisition was ∼9 min. In multiple sclerosis patients spectra were derived from an 8cc single voxel in a left or right parietal NAWM region, surrounding an acute gadolinium (Gad) enhancing lesion or non-enhancing chronic hypointense T1 lesion. These results were compared with data from a single voxel in the mostly white matter region in controls. A limited data set was obtained from the occipital grey matter in controls to determine glutamate levels in the grey matter.
Data processing
After each examination, the images and raw spectra data were transferred to a Sun workstation (Sun Microsystems, CA, USA). Time domain data were extracted from the single-voxel raw files using General Electric's spectroscopy processing tool Sage™. The different echo time water-suppressed acquisitions were averaged to generate a single TE-averaged free induction decay (FID). This TE-averaged FID was presented for LCmodel processing without any spectral apodization or zero-filling.
Spectral quantification
The metabolite concentrations were obtained using LCmodel quantification algorithm. LCmodel analyses the in vivo spectrum as a linear combination of individual in vitro metabolite spectra that constitute a basis set. Hence, LCmodel requires an in vitro basis set of individual metabolite spectra acquired with similar conditions as in vivo spectra. In general, the basis set is acquired once for each acquisition protocol. The basis set used in this study consisted of spectra from the following metabolites: NAA, choline, creatine, glutamate, glutamine and myo-inositol. To allow for differences in machine characteristics between the acquisition of the basis set and the in vivo spectra, LCmodel uses a calibration factor to scale the in vitro basis set to the in vivo spectra. The calibration factor was obtained by using LCmodel to estimate the known concentration of NAA in the MRS sphere using the same basis set as that used for in vivo quantification. The same calibration factor was then used for all data sets. The spectral analysis window was confined to 3.85–1.8 p.p.m. for 3 T data to prevent variability in the results from lipid artefacts. Spectra were included in the final analysis based on quality criteria defined by objective output parameters from the LCmodel analysis: sufficient spectral resolution (full width at half minimum ≤0.07 p.p.m.), sufficient information (signal-to-noise ratio ≥4) and residuals that were randomly scattered about zero to indicate a reasonable fit.
Error estimates in LCmodel
Errors in metabolite quantification in LCmodel are expressed in percent standard deviation (%SD) of the estimated concentrations and represent the 95% confidence intervals of the estimated concentration values. A percentage standard deviation >50% indicates that the metabolite concentration could range from zero to twice the estimated concentration. Hence metabolite concentrations estimated with percentage standard deviation greater than 50% were taken to be unreliable and the associated metabolites were assumed to be undetectable. In this study, quantification estimates for NAA, choline, creatine, glutamate and myo-inositol were included only if their percentage standard deviation was within 20% and glutamine concentrations were used only if its percentage standard deviation was constrained to within 30%.
Correction for MR relaxation times
To estimate T1 relaxation the following acquisition scheme was used: TE-averaged PRESS acquisition was used to collect data in 16 increments of 5.0 ms starting at 35 ms at repetition time (TR) 2 s with the number of excitations (NEX) = 8 and then repeated with a TR of 8 s with NEX = 2. From the longitudinal MR relaxation equation, if the TR is ∼5 times the anticipated T1 then a complete recovery of longitudinal magnetization is expected at TR = 8 s. If the anticipated T1 is ∼2 s, then at a TR of 2 s there is ∼60% recovery of signal. Hence the chosen TRs provide a robust estimate of T1. The T1 relaxation times were obtained from a two-point fit to the metabolite spectra at TR = 2 s and TR = 8 s after the respective data sets had been normalized for the number of excitations.
Within LCmodel the estimated concentration of each metabolite was corrected for T1 relaxation effects using the correction factor fTR = (1 − exp[−TR/T1vitro])/(1 − exp[−TR/T1vivo]). Here ‘vitro’ refers to the relaxation values of the metabolites in solution and were obtained from the work of Ethofer and colleagues (Ethofer et al., 2003). In this study, the in vitro glutamate relaxation value was 0.92 s and the glutamine T1 relaxation values were taken to be the same as that of glutamate. A relaxation correction factor fTR was derived for each metabolite based on its relaxation value. Since the TE-averaged acquisition collects data at different echo times, the measurement of T2 relaxation times was readily obtained for each data set simultaneously with the metabolite information. The peak heights of the respective metabolite peaks at the different echo times were fitted to a single exponential fit using SAGE™ (GE Medical Systems, Milwaukee, WI) T2 fitting to determine the T2 relaxation times of NAA, choline and creatine.
Correction for oedema
Acute lesions are expected to contain significant levels of oedema. The importance of correcting the volume of interest for oedema was shown by Helms et al. (2001). In that study, increases in glutamate+glutamine in acute lesions were apparent only after volumes were corrected for oedema. In the TE-averaged scheme the largest echo time data are collected at ∼185 ms, which is not sufficient to measure the T2 of water adequately. To estimate the T2 of oedematous water within an acute enhancing lesion a separate acquisition was used in which data were collected at eight different echo times, with the largest echo time at 1.5 s with a scan time of ∼1 min. A biexponential fit was used to fit this in vivo decay curve to extract the short and long components. It has been shown that the relative size of these components correspond to intra- and extracellular compartments (Barnes et al., 1987). The long component, in a region of interest (VOI) surrounding an acute lesion, was used to estimate the volume changes due to oedematous extracellular water (Helms et al., 2001). This volume change was obtained by converting the long component into the corresponding volume fraction (slong) by comparison of 100% water content obtained from a calibration experiment using a water phantom under similar conditions as the in vivo experiment. Using the relation Vcorr = (1 − slong) × VVOI, where slong is the volume fraction associated with the long component, the corrected volume (Vcorr) can be obtained from the prescribed VVOI. The LCmodel analysis was then used to estimate metabolite concentrations using the Vcorr as the VOI for acute enhancing lesions. The effect of this correction is to increase the metabolite estimates relative to their uncorrected values.
Statistical comparisons
Statistical comparisons between two samples were made using the Wilcoxon rank sum test. A P value of 0.05 or less was regarded as significant. The coefficient of variation (CV) was computed as (RMSE/Σ (meani)/n)×100, where RMSE = square root [Σ(variancei/n)]. Here i represents the ith data set from a total number of n controls.
Study population
Sixteen controls (eight males and eight females) and 25 multiple sclerosis patients (13 males and 12 females) were scanned for this study. The mean age of the controls and patients was 38 ± 12 and 42 ± 12 years respectively. The patients had a mean disease duration of 8 ± 7 years with a mean EDSS of 3.5 ± 2.1 Of the 25 multiple sclerosis patients, the number and regions of interest investigated for each patient were as follows: seven patients (NAWM only), five patients (NAWM and one chronic voxel), one patient (NAWM + two chronic), four patients (NAWM + acute lesion), two patients (two acute lesions each), one patient (acute only), one patient (chronic + acute) and four patients (chronic only). The diagnosis of multiple sclerosis was based on the McDonald criteria (McDonald et al., 2001). Sixteen relapsing–remitting, four secondary progressive and seven primary progressive multiple sclerosis patients from the University of California San Francisco Multiple Sclerosis Center were asked to participate. All relapsing–remitting patients and one secondary progressive patient were treated with either interferon β or glatiramer acetate. Primary progressive multiple sclerosis patients were not treated except for one who was on minocycline therapy.
All multiple sclerosis patients are followed by clinicians from our multiple sclerosis Center and are scanned regularly using either routine clinical MRI scans or as part of ongoing longitudinal MRI studies. Patients were asked to participate in this MRS study if their last scan showed an acute contrast-enhancing lesion or a non-enhancing T1-hypointense lesion present for at least more than 6 months. For patients with acute contrast-enhancing lesions, all MRS research scans were performed between 48 h and 2 weeks after the gadolinium-enhancing lesions had been detected. The University of California at San Francisco Committee on Human Research approved the study and all subjects gave their informed written consent.
Results
Differences between conventional and TE-averaged PRESS
In a conventional 3 T PRESS spectrum at TE 35, glutamate at 2.35 p.p.m. overlaps with the N-acetyl group of NAA (2.02 p.p.m.) and glutamine signals, making it difficult to isolate the glutamate resonance (Fig. 1A). In comparison, TE-averaged PRESS (Fig. 1B) fully resolves the glutamate at 2.35 p.p.m. from overlap from NAA and glutamine, resulting in its unobstructed detection (Hurd et al., 2004). Figure 2A and B illustrate a TE-averaged spectrum from an acute enhancing and chronic lesion respectively.
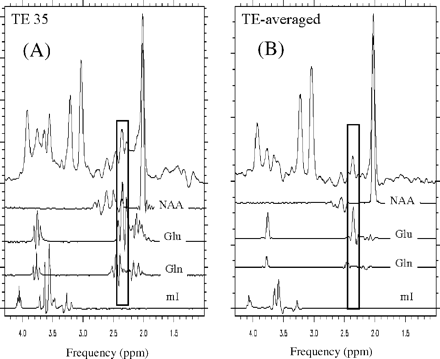
Comparison of glutamate detection with conventional PRESS and TE-averaged PRESS at 3 T in an in vivo spectrum. In a conventional 3 T PRESS spectrum at TE 35 ms (A), glutamate at 2.35 p.p.m. overlaps with the N-acetyl group of NAA (2.02 p.p.m.) and glutamine signals, making it difficult to isolate the glutamate resonance. In comparison, TE-averaged PRESS (B) fully resolves the glutamate at 2.35 p.p.m. from overlap from NAA and glutamine, resulting in its unobstructed detection. In this figure each trace represents a TE-averaged spectrum of the given metabolite, i.e. NAA, glutamate, glutamine and myo-inositol, obtained from phantoms that only contained each respective metabolite.
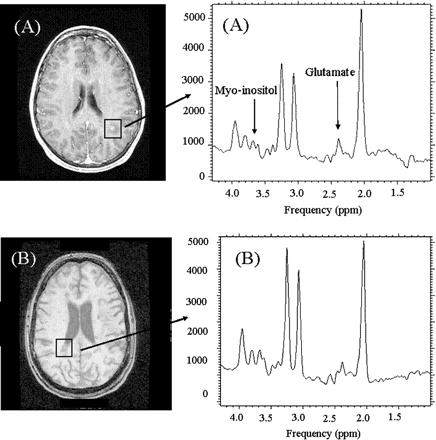
Representative TE-averaged PRESS spectrum from an 8 ml single voxel (A) surrounding an acute enhancing lesion and (B) chronic lesion.
Corrections for MR relaxation time
The relaxation values for the metabolites are shown in Table 1. As reported in previous studies, the T1 relaxation values (Table 1A) decrease as NAA > creatine > choline > myo-inositol and the T2 values (Table 1B) as NAA > choline > creatine. It was observed that the water T1 relaxation values in NAWM (1.19 ± 0.2 s) were longer than those of controls (0.83 ± 0.4 s) but the metabolite T1 relaxation values in NAWM were shorter than control white matter.
Metabolite (A) T1 and (B) T2 relaxation times (mean ± SD) in control white matter and normal-appearing white matter (NAWM). The T1 values of water in controls and NAWM are also indicated
| Water | NAA | Choline | Creatine | Myo-inositol | Glutamate | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (A) | ||||||||||||
| White matter | 0.83 ± 0.2 | 1.86 ± 0.2 | 1.32 ± 0.2 | 1.74 ± 0.1 | 1.03 ± 0.2 | 1.23 ± 0.2 | ||||||
| NAWM | 1.19 ± 0.4 | 1.68 ± 0.1 | 1.14 ± 0.1 | 1.59 ± 0.2 | 1.00 ± 0.1 | 1.14 ± 0.1 | ||||||
| (B) | ||||||||||||
| White matter | 0.23 ± 0.05 | 0.18 ± 0.03 | 0.14 ± 0.01 | |||||||||
| NAWM | 0.25 ± 0.05 | 0.20 ± 0.04 | 0.14 ± 0.02 | |||||||||
| Water | NAA | Choline | Creatine | Myo-inositol | Glutamate | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (A) | ||||||||||||
| White matter | 0.83 ± 0.2 | 1.86 ± 0.2 | 1.32 ± 0.2 | 1.74 ± 0.1 | 1.03 ± 0.2 | 1.23 ± 0.2 | ||||||
| NAWM | 1.19 ± 0.4 | 1.68 ± 0.1 | 1.14 ± 0.1 | 1.59 ± 0.2 | 1.00 ± 0.1 | 1.14 ± 0.1 | ||||||
| (B) | ||||||||||||
| White matter | 0.23 ± 0.05 | 0.18 ± 0.03 | 0.14 ± 0.01 | |||||||||
| NAWM | 0.25 ± 0.05 | 0.20 ± 0.04 | 0.14 ± 0.02 | |||||||||
Metabolite (A) T1 and (B) T2 relaxation times (mean ± SD) in control white matter and normal-appearing white matter (NAWM). The T1 values of water in controls and NAWM are also indicated
| Water | NAA | Choline | Creatine | Myo-inositol | Glutamate | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (A) | ||||||||||||
| White matter | 0.83 ± 0.2 | 1.86 ± 0.2 | 1.32 ± 0.2 | 1.74 ± 0.1 | 1.03 ± 0.2 | 1.23 ± 0.2 | ||||||
| NAWM | 1.19 ± 0.4 | 1.68 ± 0.1 | 1.14 ± 0.1 | 1.59 ± 0.2 | 1.00 ± 0.1 | 1.14 ± 0.1 | ||||||
| (B) | ||||||||||||
| White matter | 0.23 ± 0.05 | 0.18 ± 0.03 | 0.14 ± 0.01 | |||||||||
| NAWM | 0.25 ± 0.05 | 0.20 ± 0.04 | 0.14 ± 0.02 | |||||||||
| Water | NAA | Choline | Creatine | Myo-inositol | Glutamate | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (A) | ||||||||||||
| White matter | 0.83 ± 0.2 | 1.86 ± 0.2 | 1.32 ± 0.2 | 1.74 ± 0.1 | 1.03 ± 0.2 | 1.23 ± 0.2 | ||||||
| NAWM | 1.19 ± 0.4 | 1.68 ± 0.1 | 1.14 ± 0.1 | 1.59 ± 0.2 | 1.00 ± 0.1 | 1.14 ± 0.1 | ||||||
| (B) | ||||||||||||
| White matter | 0.23 ± 0.05 | 0.18 ± 0.03 | 0.14 ± 0.01 | |||||||||
| NAWM | 0.25 ± 0.05 | 0.20 ± 0.04 | 0.14 ± 0.02 | |||||||||
The T2 relaxation values of NAA, choline and creatine were similar in NAWM relative to control white matter. Due to the strong J coupling of the glutamate, glutamine and myo-inositol resonances, their signal does not follow the single exponential decay behaviour with multiple echo times, as shown for glutamate in Fig. 3. This figure was obtained using GAMMA simulation software (Smith et al., 1994). This software uses the Bloch equations to determine how spins behave with the components and timing relationships of the pulse sequence. It was also difficult to find a set of echo times in this curve at which the glutamate signal was at its maximum phase and not overlapping with other metabolites. Hence, the T2 relaxation of glutamate, glutamine and myo-inositol could not be determined with the TE-averaged method. Since it was not possible to determine the T2 relaxation of glutamate, glutamine and myo-inositol, data were not corrected for T2 relaxation to allow adequate comparison between all metabolite concentrations.
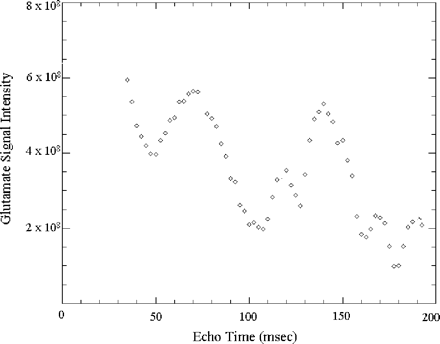
Simulation of the glutamate resonance with multiple echo times using GAMMA (Smith et al., 1994) software.
Reliability of metabolite quantification
The reproducibility of the estimated in vivo metabolite concentrations with LCmodel is illustrated in Table 2. These values incorporate both methodological and biological variations. The non-uniform B1 field due to the dielectric effect at 3 T is also a factor contributing to the variability of LCmodel concentrations. This is expected to affect both control and multiple sclerosis scans. The reproducibility of strongly coupled resonances of glutamate and myo-inositol is similar to singlet resonances, namely NAA and creatine. It is noted that the resulting differences in metabolite levels reported in the present study are much greater than the inherent reproducibility of the technique. Using the TE-averaged PRESS scheme we found that the glutamate levels in control white matter was ∼7 mM compared with ∼9 mM in grey matter. This is consistent with prior studies (Pan et al., 1996) which have reported 2 mM higher glutamate levels in grey matter compared with white matter.
Estimated concentration (mM) and coefficient of variation (CV) from white matter of normal controls obtained by TE-averaged PRESS acquisition
| NAA | Choline | Creatine | Glutamate | Myo-inositol | |
|---|---|---|---|---|---|
| Concentration (mM) | 12.8 ± 0.9 | 2.0 ± 0.2 | 6.9 ± 0.4 | 7.1 ± 0.7 | 3.9 ± 0.3 |
| CV (%) | 9.3 | 3.2 | 8.8 | 10.3 | 9.2 |
| NAA | Choline | Creatine | Glutamate | Myo-inositol | |
|---|---|---|---|---|---|
| Concentration (mM) | 12.8 ± 0.9 | 2.0 ± 0.2 | 6.9 ± 0.4 | 7.1 ± 0.7 | 3.9 ± 0.3 |
| CV (%) | 9.3 | 3.2 | 8.8 | 10.3 | 9.2 |
The CV was obtained by scanning five controls at least twice on different days.
Estimated concentration (mM) and coefficient of variation (CV) from white matter of normal controls obtained by TE-averaged PRESS acquisition
| NAA | Choline | Creatine | Glutamate | Myo-inositol | |
|---|---|---|---|---|---|
| Concentration (mM) | 12.8 ± 0.9 | 2.0 ± 0.2 | 6.9 ± 0.4 | 7.1 ± 0.7 | 3.9 ± 0.3 |
| CV (%) | 9.3 | 3.2 | 8.8 | 10.3 | 9.2 |
| NAA | Choline | Creatine | Glutamate | Myo-inositol | |
|---|---|---|---|---|---|
| Concentration (mM) | 12.8 ± 0.9 | 2.0 ± 0.2 | 6.9 ± 0.4 | 7.1 ± 0.7 | 3.9 ± 0.3 |
| CV (%) | 9.3 | 3.2 | 8.8 | 10.3 | 9.2 |
The CV was obtained by scanning five controls at least twice on different days.
Metabolite levels in multiple sclerosis
Table 3 shows the comparison of metabolite levels of control white matter, NAWM and regions surrounding acute and chronic lesions.
Concentration of metabolites in control white matter, NAWM and regions surrounding acute and chronic lesions (mM; mean ± SD)
| NAA | Choline | Creatine | Myo-inositol | Glutamate | Glutamine | |
|---|---|---|---|---|---|---|
| Control white matter | 13.4 ± 1.7 | 2.0 ± 0.4 | 7.1 ± 1.2 | 4.3 ± 1.2 | 6.9 ± 0.8 | 5.5 ± 2.0 |
| (n = 16) | (n = 13)* | (n = 7)* | ||||
| NAWM | 12.8 ± 2.5 | 2.4 ± 0.4 | 8.0 ± 1.5 | 5.6 ± 1.6 | 8.1 ± 1.6 | 6.1 ± 1.2 |
| P = 1.0 | P = 0.03 | P = 0.13 | P = 0.04 | P = 0.03 (n = 10)* | P = 0.52 (n = 7)* | |
| (n = 17) | (n = 10)* | (n = 7)* | ||||
| Contrast-enhancing lesions | 12.7 ± 1.3 | 2.6 ± 0.3 | 8.1 ± 0.7 | 6.1 ± 0.9 | 8.0 ± 1.0 | 6.9 ± 0.9 |
| P = 0.44 | P = 0.002 | P = 0.02 | P < 0.001 | P = 0.02 | P = 0.12 | |
| (n = 10) | (n = 8)* | (n = 7)* | ||||
| Chronic lesions | 10.8 ± 2.0 | 2.0 ± 0.5 | 7.3 ± 1.2 | 5.9 ± 1.8 | 6.7 ± 1.0 | 5.8 ± 1.3 |
| P < 0.001 | P = 0.94 | P = 0.56 | P = 0.02 | P = 0.77 | P = 0.61 | |
| (n = 12) | (n = 11)* | (n = 6)* | (n = 7)* |
| NAA | Choline | Creatine | Myo-inositol | Glutamate | Glutamine | |
|---|---|---|---|---|---|---|
| Control white matter | 13.4 ± 1.7 | 2.0 ± 0.4 | 7.1 ± 1.2 | 4.3 ± 1.2 | 6.9 ± 0.8 | 5.5 ± 2.0 |
| (n = 16) | (n = 13)* | (n = 7)* | ||||
| NAWM | 12.8 ± 2.5 | 2.4 ± 0.4 | 8.0 ± 1.5 | 5.6 ± 1.6 | 8.1 ± 1.6 | 6.1 ± 1.2 |
| P = 1.0 | P = 0.03 | P = 0.13 | P = 0.04 | P = 0.03 (n = 10)* | P = 0.52 (n = 7)* | |
| (n = 17) | (n = 10)* | (n = 7)* | ||||
| Contrast-enhancing lesions | 12.7 ± 1.3 | 2.6 ± 0.3 | 8.1 ± 0.7 | 6.1 ± 0.9 | 8.0 ± 1.0 | 6.9 ± 0.9 |
| P = 0.44 | P = 0.002 | P = 0.02 | P < 0.001 | P = 0.02 | P = 0.12 | |
| (n = 10) | (n = 8)* | (n = 7)* | ||||
| Chronic lesions | 10.8 ± 2.0 | 2.0 ± 0.5 | 7.3 ± 1.2 | 5.9 ± 1.8 | 6.7 ± 1.0 | 5.8 ± 1.3 |
| P < 0.001 | P = 0.94 | P = 0.56 | P = 0.02 | P = 0.77 | P = 0.61 | |
| (n = 12) | (n = 11)* | (n = 6)* | (n = 7)* |
The highlighted results are significantly (P < 0.05) different from controls. Concentrations are reported only if their error estimate with LCmodel were within 20% for NAA, choline, creatine, glutamate and myo-inositol (see text) and within 30% for glutamine. Hence, in some cases metabolite estimates were obtained only for a subset of voxels and are so indicated. Statistics were evaluated using the Wilcoxon rank-sum test. The distribution of the regions of interest investigated among the different patients is outlined in the text.
Reliable metabolite estimates were obtained only from a subset of the total voxels indicated.
Concentration of metabolites in control white matter, NAWM and regions surrounding acute and chronic lesions (mM; mean ± SD)
| NAA | Choline | Creatine | Myo-inositol | Glutamate | Glutamine | |
|---|---|---|---|---|---|---|
| Control white matter | 13.4 ± 1.7 | 2.0 ± 0.4 | 7.1 ± 1.2 | 4.3 ± 1.2 | 6.9 ± 0.8 | 5.5 ± 2.0 |
| (n = 16) | (n = 13)* | (n = 7)* | ||||
| NAWM | 12.8 ± 2.5 | 2.4 ± 0.4 | 8.0 ± 1.5 | 5.6 ± 1.6 | 8.1 ± 1.6 | 6.1 ± 1.2 |
| P = 1.0 | P = 0.03 | P = 0.13 | P = 0.04 | P = 0.03 (n = 10)* | P = 0.52 (n = 7)* | |
| (n = 17) | (n = 10)* | (n = 7)* | ||||
| Contrast-enhancing lesions | 12.7 ± 1.3 | 2.6 ± 0.3 | 8.1 ± 0.7 | 6.1 ± 0.9 | 8.0 ± 1.0 | 6.9 ± 0.9 |
| P = 0.44 | P = 0.002 | P = 0.02 | P < 0.001 | P = 0.02 | P = 0.12 | |
| (n = 10) | (n = 8)* | (n = 7)* | ||||
| Chronic lesions | 10.8 ± 2.0 | 2.0 ± 0.5 | 7.3 ± 1.2 | 5.9 ± 1.8 | 6.7 ± 1.0 | 5.8 ± 1.3 |
| P < 0.001 | P = 0.94 | P = 0.56 | P = 0.02 | P = 0.77 | P = 0.61 | |
| (n = 12) | (n = 11)* | (n = 6)* | (n = 7)* |
| NAA | Choline | Creatine | Myo-inositol | Glutamate | Glutamine | |
|---|---|---|---|---|---|---|
| Control white matter | 13.4 ± 1.7 | 2.0 ± 0.4 | 7.1 ± 1.2 | 4.3 ± 1.2 | 6.9 ± 0.8 | 5.5 ± 2.0 |
| (n = 16) | (n = 13)* | (n = 7)* | ||||
| NAWM | 12.8 ± 2.5 | 2.4 ± 0.4 | 8.0 ± 1.5 | 5.6 ± 1.6 | 8.1 ± 1.6 | 6.1 ± 1.2 |
| P = 1.0 | P = 0.03 | P = 0.13 | P = 0.04 | P = 0.03 (n = 10)* | P = 0.52 (n = 7)* | |
| (n = 17) | (n = 10)* | (n = 7)* | ||||
| Contrast-enhancing lesions | 12.7 ± 1.3 | 2.6 ± 0.3 | 8.1 ± 0.7 | 6.1 ± 0.9 | 8.0 ± 1.0 | 6.9 ± 0.9 |
| P = 0.44 | P = 0.002 | P = 0.02 | P < 0.001 | P = 0.02 | P = 0.12 | |
| (n = 10) | (n = 8)* | (n = 7)* | ||||
| Chronic lesions | 10.8 ± 2.0 | 2.0 ± 0.5 | 7.3 ± 1.2 | 5.9 ± 1.8 | 6.7 ± 1.0 | 5.8 ± 1.3 |
| P < 0.001 | P = 0.94 | P = 0.56 | P = 0.02 | P = 0.77 | P = 0.61 | |
| (n = 12) | (n = 11)* | (n = 6)* | (n = 7)* |
The highlighted results are significantly (P < 0.05) different from controls. Concentrations are reported only if their error estimate with LCmodel were within 20% for NAA, choline, creatine, glutamate and myo-inositol (see text) and within 30% for glutamine. Hence, in some cases metabolite estimates were obtained only for a subset of voxels and are so indicated. Statistics were evaluated using the Wilcoxon rank-sum test. The distribution of the regions of interest investigated among the different patients is outlined in the text.
Reliable metabolite estimates were obtained only from a subset of the total voxels indicated.
Glutamate levels were higher in acute enhancing lesions (P = 0.02) and NAWM (P = 0.03) relative to control white matter. No significant differences (P = 0.77) were observed in glutamate levels in chronic lesions relative to control white matter. The NAA level in chronic lesions was significantly different (P < 0.001) from that in control matter. However, no significant differences were found between NAA levels in NAWM and acute lesions relative to normal control white matter. Reliable estimates for glutamine were obtained only for a subset of the total number of voxels in each case and are indicated as such in the table. Relative to control white matter, myo-inositol levels were significantly different in NAWM (P = 0.04), acute lesions (P < 0.001) and chronic lesions (P = 0.02). Choline levels were significantly different in NAWM (P = 0.03) and acute lesions (P < 0.001) but not in chronic lesions (P = 0.94). Creatine levels were found to be somewhat variable across multiple sclerosis regions of interest, with the most difference in acute lesions (P = 0.02) relative to control white matter.
Oedema correction
Estimates for extracellular water content were ∼7% in control white matter and ∼14% for oedematous acute lesions. Volume correction for water in a voxel enhances the difference in glutamate levels between control white matter and acute lesions from ∼10% before correction to ∼19% after correction (Fig. 4).
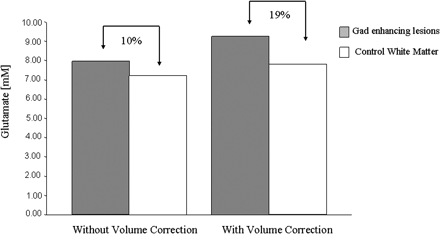
Glutamate levels without and with volume correction for oedema in Gad enhancing lesions and water content in control white matter. n = 4 in each case.
Discussion
With a recently developed TE-averaged PRESS MR spectroscopic method we were able to unambiguously detect the glutamate resonance and evaluate its levels in multiple sclerosis brain in association with other metabolites, NAA, choline, creatine and myo-inositol. The TE-averaged PRESS acquisition provides unobstructed detection of glutamate at 3 T at 2.35 p.p.m. and can be considered analogous to the singlet NAA detection. Besides enhancing the glutamate estimation in quantification schemes, the singlet detection of glutamate also improves the estimation of glutamine, which co-resonates with glutamate at 3.75 p.p.m. since the glutamate estimate has been constrained with its estimate at 2.35 p.p.m. Using this method we have also reported levels of myo-inositol, NAA, choline and creatine.
Corrections for MR relaxation time
Since the concentration estimates are sensitive to metabolite relaxation values, which can vary between normal and diseased states, we correct for these effects before making comparisons of metabolite concentration levels between different pathologies. All metabolites in this study were corrected for T1 relaxation, which was obtained from the same region as the spectra in a separate acquisition. This ensured that the T1 relaxation value adequately represented the same brain region as spectra. The T1 relaxation value of NAA, choline and creatine was lower in multiple sclerosis NAWM relative to white matter in the control. The water T1 relaxation is greater in the NAWM relative to control white matter, consistent with our previous studies (Srinivasan et al., 2003) at 1.5 T. The water T1 relaxation values are higher at 3 T relative to 1.5 T, as expected with increasing field strength. This difference in relaxation time between water and metabolites possibly follows the results in Ethofer et al. (2003), suggesting that since metabolites are mainly located inside the cells their relaxation behaviour depends more on the differences in the microenvironment, such as cell size, than on tissue integrity and composition, which determines the T1 relaxation of water protons. This behaviour of metabolite T1s in multiple sclerosis NAWM needs further investigation.
The T2 values of NAA, choline and creatine were not significantly different between multiple sclerosis and control white matter. The difference in T2 values between the present and our previous study (Hurd et al., 2004) is due to the fact that in the prior study only echo times greater than 60 ms were used in the T2 fitting procedure. It was believed that shorter echo times also contain a component of macromolecular signal that is not present at the longer echo times due to their short T2 values. Due to the limitations in the number of echo times, we were not able to estimate this macromolecular signal and hence used all the echo times from 35 to 195 ms to fit the T2 value. These results are consistent with other studies that have used similar echo times in their fitting procedure (Mlynarik et al., 2001). While it was not possible to measure the T2 of glutamate with the TE-averaged technique, we tried to determine the effect of a T2 relaxation correction by assuming that the T2 relaxation values for glutamate had the same relationship and order of magnitude difference between controls and multiple sclerosis as its T1 relaxation value. When we incorporated a T2 correction within LCmodel based on this assumption, we found that the statistical significance of the results became even stronger. Hence, without the T2 relaxation correction the results presented in this study are conservative estimates.
Metabolite levels in multiple sclerosis
Acute lesions
The inflammatory infiltrate within active multiple sclerosis lesions is composed of different populations of activated T cells, plasma cells, macrophages containing myelin debris and resident microglia cells, the last two populations being the most abundant (Traugott et al., 1983; Bruck et al., 1995). The most important finding we report in this in vivo study is an elevation of glutamate in contrast-enhancing areas compared with white matter areas of normal controls. This is consistent with active inflammatory infiltrates, where large quantities of glutamate are produced and released by activated leucocytes, macrophages and microglial cells (Piani et al., 1991). It can be seen as a new in vivo marker of inflammation and its relevance to neuro-axonal injury in multiple sclerosis will require further investigation. Such elevation was suspected by Helms and colleagues (Helms et al., 2001), but glutamate and glutamine could not be studied separately and unambiguously.
The extracellular water content in acute enhancing lesions was approximately twice what was found in normal control white matter, consistent with the presence of oedema in those lesions. Using a T2 relaxation method, volume correction for the oedematous content enhances the differences in glutamate levels and we recommend using it in the future when studying metabolites in acute lesions.
Our NAA results (Table 3) in contrast-enhancing areas are consistent with recent single-voxel studies of contrast-enhancing lesions using LCmodel (Mader et al., 2000), where NAA levels did not differ significantly from control white matter areas. We also found an increase in creatine consistent with high-energy metabolism from hypercellularity (Burtis and Ashwood, 1994). Taken together, these results certainly have an implication in multiple sclerosis when interpreting NAA/creatine ratios as a marker of neuronal loss or dysfunction within contrast-enhancing areas. Choline was significantly elevated, possibly due to an increase in cell turnover and/or myelin debris, as reported previously (Narayana et al., 1998; Mader et al., 2000; Helms, 2001).
Chronic lesions
Inactive or ‘chronic’ multiple sclerosis plaques are characterized by hypocellularity, absence of myelin breakdown, prominent fibrillary gliosis and reduced axonal density (Lassmann, 1998). In our study, glutamate levels were not different in non-enhancing chronic T1-hypointense areas compared with control white matter areas. We believe this is due to the absence or quasi-absence of activated immune cells in chronic plaques. However, NAA was significantly reduced in chronic T1-hypointense areas with marked elevation of myo-inositol. Both of these findings are consistent with pathological evidence of reduced axonal density and gliosis. Since NAA is consistently reduced in chronic lesions (van Walderveen et al., 1999) it would be important to find predictors of this decrease in the early enhancing phase of lesions. We suspect that glutamate is such a candidate and certainly deserves careful longitudinal investigation.
Normal-appearing white matter
Several histopathological studies have identified abnormalities in NAWM in multiple sclerosis, such as an increase in the activity or number of glial cells (Allen et al., 2001), changes in myelin layer organization and membrane proteins (Chia et al., 1984; Wood and Moscarello, 1984; Newcombe et al., 1986), reduced axonal density (Evangelou et al., 2000) and the presence of perivascular T cells, B cells and macrophages (Prineas and Wright, 1978). Aside from creatine, metabolite findings in NAWM in our study followed closely those found in contrast-enhancing areas. In particular, the excess of glutamate found in NAWM should mainly relate to the presence of inflammatory cells and astrocytosis. One has to assume that the glutamate MRS signal detected is mainly derived from the intracellular compartment, approximately between 5 and 10 mmol/l, with very little or no contribution from the extracellular compartment, which under normal conditions is around 0.5–1.0 µmol/l (Leighton et al., 2001) and below current sensitivity of in vivo MRS. Interestingly, however, recent studies on human white matter found that key enzymes of glutamate metabolism and glutamate transporters were present predominantly on oligodendrocytes and to a much lesser extent on astrocytes, implying a major role for oligodendrocytes in the maintenance of glutamate homeostasis in white matter (Pitt et al., 2003). In multiple sclerosis white matter, oligodendrocytes lost expression of receptors in the lesion vicinity, resulting in reduced glutamate uptake by >75% (Pitt et al., 2003) and in ineffective glutamate removal. This pathological process can only contribute to the detection of increased glutamate levels in NAWM areas. Glutamate in NAWM areas will be further investigated to test its relationship with brain atrophy and clinical disability.
NAA in NAWM was not statistically different from control white matter in this present study, which contrasts with previous results (Fu et al., 1998; Sarchielli et al., 1999). This discrepancy could possibly be due to the location of the single voxel, the volume of NAWM studied, the sample size and the use of relaxation corrections in quantification. We believe NAA in NAWM should be investigated further from larger volumes of brain tissue using multivoxel spectroscopic imaging and T1 corrections.
Our data also suggest a measurable increase in glial activity, with statistically significant differences in myo-inositol levels in NAWM relative to control white matter areas. These findings are consistent with other recent MRS findings (Fernando et al., 2004) that have shown increases in myo-inositol levels in NAWM brain areas of patients presenting with a clinically isolated syndrome. Myo-inositol levels were found to correlate with functional impairment measures using the MSFC scale in clinically definite RRMS (Chard et al., 2002) and disability in primary progressive multiple sclerosis patients (Sastre-Garriga et al., 2003), in whom brain myo-inositol is synthesized primarily in glial cells and does not cross the blood–brain barrier (Brand et al., 1993).
Conclusions
Using single voxel TE-averaged PRESS we have acquired and quantified in vivo brain metabolites from healthy controls and multiple sclerosis patients. This technique is optimized for glutamate detection and measurement. This is the first time glutamate, glutamine and myo-inositol have been investigated along with NAA, choline and creatine in the same cohort of multiple sclerosis patients. Glutamate was found to be elevated in acute contrast-enhancing lesions and NAWM. We will investigate whether increase in glutamate relates to neurotoxicity by predicting axonal injury and brain atrophy using cross-sectional and longitudinal studies.
The authors would like to thank Jean Brittain from GE Medical Systems, Menlo Park, for her support in using the 3 T GE scanner. This work was supported by the UC Industry–University Cooperative Research Program LSIT01-10107.
References
Allen IV, McQuaid S, Mirakhur M, Nevin G. Pathological abnormalities in the normal-appearing white matter in multiple sclerosis.
Auger C, Attwell D. Fast removal of synaptic glutamate by post synaptic transporters.
Barnes D, McDonald WI, Johnson C, Tofts PS, Landon DN. Quantitative nuclear magnetic resonance imaging: characterization of experimental cerebral edema.
Berl S, Clarke DD. The metabolic compartmentation concept. In: Hertz L, Kvamne EM, McGeer G, Schousboue A, editors. Glutamine, glutamate and GABA in the central nervous system. New York: Alan R. Liss;
Bezzi P, Volterra A, A neuron-glia signaling network in the active brain.
Brand A, Richter-Landsberg C, Leibfritz D. Multinuclear NMR studies on the energy metabolism of glial and neuronal cells.
Bruck W, Porada P, Poser S, Rieckmann P, Hanefeld F, Kretzschmar HA, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions.
Burtis CA, Ashwood ER. Tietz textbook of clinical chemistry. Philadelphia: W.B. Saunders;
Chard DT, Griffin CM, McLean MA, Kapeller P, Kapoor R, Thompson AJ, et al. Brain metabolite changes in cortical grey and normal-appearing white matter in clinically early relapsing-remitting multiple sclerosis.
Chia LS, Thompson JE, Moscarello MA. Alteration of lipid-phase behavior in multiple sclerosis myelin revealed by wide-angle x-ray diffraction.
Doble A. The role of excitotoxicity in neurodegenrative disease: implications for therapy.
Ethofer T, Mader I, Seeger U, Helms G, Erb M, Grodd W, Ludolph A, Klose U, Comparison of longitudinal metabolite relaxation times in different regions of the human brain at 1.5 and 3 tesla.
Evangelou N, Esiri MM, Smith S, Palace J, and Matthews PM. Quantitative pathological evidence for axonal loss in normal appearing white matter in multiple sclerosis.
Fernando KT, McLean MA, Chard DT, MacManus DG, Dalton CM, Miszkiel KA, et al. Elevated white matter myo-inositol in clinically isolated syndromes suggestive of multiple sclerosis.
Fu L, Matthews PM, De Stefano N, Worsley KJ, Narayanan S, Francis GS, et al. Imaging axonal damage of normal-appearing white matter in multiple sclerosis.
Hattori N, Abe K, Sakoda S, Sawada S. Proton MR spectroscopic study at 3 tesla on glutamate/glutamine in Alzheimer's disease.
Helms G. Volume correction for edema in single-volume proton MR spectroscopy of contrast-enhancing multiple sclerosis lesions.
Hurd R, Napapon S, Srinivasan R, Vigneron D, Pelletier D, Nelson S. Measurement of brain glutamate using TE-averaged PRESS at 3 T.
Leighton, MP, Prost RW, Ulmer JL, Smith MM, Daniels DL, Strottmann JM, et al. Pictorial review of glutamate excitotoxicity: fundamental concepts for neuroimaging.
Lassmann H. Pathology of multiple sclerosis. In: Compston A, editor, McAlpine's multiple sclerosis. London: Churchill Livingstone;
Lee HK, Yaman A, Nalcioglu O. Homonuclear J-refocused spectral editing technique for quantification of glutamine and glutamate by 1H NMR spectroscopy.
Lipton S, Rosenberg P. Excitatory amino acids as a final common pathway for neurologic disorders.
Mader I, Roser W, Kappos L, Hagberg G, Seelig J, Radue EW, et al. Serial proton MR spectroscopy of contrast-enhancing multiple sclerosis plaques: absolute metabolic values over 2 years during a clinical pharmacological study.
McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis.
Mlynarik V, Gruber S, Moser E. Proton T1 and T2 relaxation times of human brain metabolites at 3 T.
Narayana PA, Doyle TJ, Lai D, Wolinsky JS. Serial proton magnetic resonance spectroscopic imaging, contrast-enhanced magnetic resonance imaging, and quantitative lesion volumetry in multiple sclerosis.
Newcombe J, Woodroofe MN, Cuzner ML, Distribution of glial fibrillary acidic protein in gliosed human white matter.
Pan J, Mason, Pohost G, Hetherington H. Spectroscopic imaging of human brain glutamate by water-suppressed J-refocused coherence transfer at 4.1T.
Piani D, Frei K, Do KD, Cuenod M, Fontana A. Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate.
Pitt D, Werner P, Raine C. Glutamate excitotoxicity in a model of multiple sclerosis.
Pitt D, Nagelmeier IE, Wilson HC, Raine CS. Glutamate uptake by oligodendrocytes: Implications for excitotoxicity in multiple sclerosis.
Prineas JW, Wright RG. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis.
Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra.
Rothstein JD, Martin L, Levey AL, Dyke-Hoberg M, et al. Localization of neuronal and glial glutamate transporters.
Sarchielli P, Presciutti O, Pelliccioli GP, Tarducci R, Gobbi G, Chiarini P, et al. Absolute quantification of brain metabolites by proton magnetic resonance spectroscopy in normal-appearing white matter of multiple sclerosis patients.
Sastre-Garriga J, Ingle GT, Chard DT, Ramio LL, McLean MA, Miller DH, et al. Brain metabolite changes in clinically early primary progressive multiple sclerosis.
Schubert F, Gallinat J, Seifert F, Rinneberg H. Glutamate concentrations in human brain using single voxel proton magnetic resonance spectroscopy at 3 tesla.
Shousboe U, Westergaard N, Sonewald U, Petersen SB, et al. Glutamate and glutamine metabolism and compartmentation in astrocytes.
Smith SA, Levante TO, Meier BH, Ernst RR. Computer simulations in magnetic resonance. An object oriented programming approach.
Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists.
Simon J, Kinkel R, Jacobs L, Bub L, Simonian N. A Wallerian degeneration pattern in patients at risk for MS.
Srinivasan R, Henry R, Pelletier, D, Nelson S. Standardized, reproducible, high resolution global measurements of T1 relaxation metrics in multiple sclerosis.
Traugott U, Reinherz EL, Raine CS. Multiple sclerosis. Distribution of T cells, T cells subsets and Ia-positive macrophages in lesions of different stages.
Thompson RB, Allen PS. A new multiple quantum filter design procedure for use on strongly coupled spin systems found in vivo: its application to glutamate.
van Walderveen MA, Barkhof F, Pouwels PJ, van Schijndel RA, Polman CH, Castelijns JA. Neuronal damage in T1-hypointense multiple sclerosis lesions demonstrated in vivo using proton magnetic resonance spectroscopy.
Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage.
Author notes
1Department of Radiology, University of California-San Francisco, San Francisco, 2GE Medical Systems, Menlo Park, CA, and 3Department of Neurology, University of California-San Francisco, San Francisco, USA


{kind=link}
{kind=link}
{kind=link}
{kind=link}