




Abstract
Arsenic-induced carcinogenesis is a worldwide problem for which there is currently limited means for control. Recently, we showed that arsenite in drinking water greatly potentiates solar ultraviolet radiation (UVR) induced skin cancer in mice, at concentrations as low as 1.25 mg/l. In this study, we examined the protective efficacy of vitamin E and 1,4-phenylene bis (methylene)selenocyanate ( p -XSC) against tumors induced by UVR and UVR + arsenite. Hairless mice were exposed to UVR alone (1.0 kJ/m 2 × 3 times weekly) or UVR + sodium arsenite (5 mg/l in drinking water) and fed lab chow supplemented or not with vitamin E (RRR-α-tocopheryl acetate, 62.5 IU/kg diet) or p -XSC (10 mg/kg) for 26 weeks. The tumor yield for mice receiving UVR alone was 3.6 tumors/mouse and the addition of arsenite to the drinking water increased the yield to 7.0 tumors/mouse ( P < 0.005). Vitamin E and p -XSC reduced the tumor yield in mice given UVR + arsenite by 2.1-fold ( P < 0.001) and 2-fold ( P < 0.002), respectively. Vitamin E, but not p-XSC, reduced the tumor yield induced by UVR alone by 30% ( P < 0.05). No significant difference in tumor types or grade of malignancy was observed in mice treated with or without chemopreventives. Immunostaining of mouse skin for 8-oxo-2′-deoxyguanosine (8-oxo-dG) revealed a significant reduction of 8-oxo-dG formation in mice treated with vitamin E or p -XSC compared with those treated with UVR + arsenite. These results show that vitamin E and p -XSC protect strongly against arsenite-induced enhancement of UVR carcinogenesis.
Introduction
People drinking water containing high arsenic exhibit increased risk of cancer (mostly skin, and also lung, bladder and possibly liver and prostate cancers) ( 1 ). Although arsenic in the form of arsenite + arsenate in drinking water is a major public health problem worldwide, the problem is especially severe in parts of Asia where control measures are limited. This laboratory developed the first animal model for arsenic-induced skin carcinogenesis and showed that non-toxic concentrations of sodium arsenite in drinking water significantly enhance solar-spectrum ultraviolet radiation (UVR)-induced skin cancers ( 2 , 3 ) that are almost all squamous cell carcinomas. This animal model for arsenic-related cancer induction provides a framework for understanding and developing chemopreventive measures against arsenic-induced skin cancers.
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are produced in cells exposed to arsenic and can cause damage to lipids, proteins and DNA ( 4 – 6 ). The addition of various antioxidants to the culture medium protects cells against arsenite toxicity and genotoxicity ( 7 , 8 ). Arsenite increases 8-oxo-2′-deoxyguanosine (8-oxo-dG) in DNA ( 9 ), induces the oxidative stress-related proteins hemeoxygenase ( 10 ) and cyclooxygenase-2 ( 11 ), and activates NF-kappa B ( 12 ). Moreover, these events can be blocked by antioxidants. Arsenite and its trivalent methylated metabolites inhibit glutathione reductase and thioredoxin reductase (TR), and this inhibition plays a role in the pro-oxidative activity of the arsenicals ( 13 , 14 ).
Vitamin E is well known for providing protection to biological membranes through its ability to acts as an antioxidant. Vitamin E strengthens epidermal antioxidant defenses against UVR-induced oxidative damage ( 15 ). Dietary supplement of vitamin E reduces UVR-induced skin tumor in mice ( 16 , 17 ). Topical vitamin E also prevents UV-induced skin photodamage and tumor in mice ( 18 ). Since treatment with arsenite causes increased oxidative DNA damage and enhances UVR carcinogenesis in mouse skin, the antioxidant activity of vitamin E was chosen as an inhibitor against arsenite-enhanced UVR carcinogenesis.
Selenium is an essential trace element and acts as an antioxidant at least in part through its incorporation as selenocysteine in selenoproteins ( 19 , 20 ). The human genome contains 25 genes that are expected to encode over 30 selenoproteins due to alternative splicing ( 21 ). These include the glutathione peroxidase (GPX) and TR families, which have antioxidant actions ( 19 , 20 ). Dietary selenium supplementation protects against oxidative lipid and DNA damage in keratinocytes ( 22 ). Selenium protects primary human keratinocytes from apoptosis induced by UVR ( 23 ). Lower than normal levels of dietary selenium are associated with up to a 4-fold increased risk of developing skin tumor in people exposed to environmental UVR ( 22 ). 1,4-Phenylene bis (methylene)selenocyanate ( p -XSC), a synthetic organoselenium compound, has been shown to be the most effective and the least toxic selenium compound in several experimental cancer models ( 24 – 26 ). p -XSC inhibited NNK and DMBA-induced 8-oxo-dG formation in rodent lung DNA ( 24 ). Selenomethionine, the predominant form of selenium in most selenium rich diets, protected cells with wild-type (but not null) p53 alleles from UVR-induced oxidative DNA damage ( 27 ). Se-methylselenocysteine, an active selenium metabolite (the predominant form of organic selenium in Brazil nuts), also exhibits anti-carcinogenic effects ( 28 ). Organic selenium in the form of selenized yeast, given to arsenic-exposed individuals for 14 months, caused significantly greater decreases in clinical signs and symtoms and a greater decrease in the arsenic content of the body, compared with controls ( 29 ). Recently, we showed that organoselenium compounds prevent spontaneous and arsenite-induced mutagenesis in human osteosarcoma (HOS) cells ( 30 ). Most spontaneous mutagenesis appears to be caused by endogenous oxidative processes and can be blocked by dietary antioxidant supplements ( 31 , 32 ).
Epidemiological data suggest that susceptibility to arsenic's carcinogenicity may differ from area to area due to various confounding factors, and nutritional status is considered an important modulator of arsenite toxicity ( 33 ). Many areas of the world that are chronically exposed to arsenic have low nutritional status, especially low dietary selenium in food ( 34 , 35 ). For example, the diets of people within the delta region of Bangladesh and West Bengal are very low in essential nutrients and selenium ( 35 ).
To determine whether arsenite enhancement of UVR carcinogenesis can be blocked by dietary vitamin E or p -XSC, mice exposed to UVR alone or to UVR+ arsenite were provided with dietary supplements, and the effects on tumor yield, histopathological type of tumor and an oxidative biomarker were evaluated. The results show that both vitamin E and selenium compounds selectively protect against arsenite-potentiated UVR-induced skin tumor, and suggest that the mechanism is at least in part via their antioxidant action.
Materials and methods
Mice
Weanling female hairless mice (CrL: SK1-hrBD) were purchased from Charles River Laboratories (Portage, MI). They were housed in plastic cages with mesh covers, and fed with autoclaved loose mouse chow and water ad libitum . Room illumination was an automated cycle of 12 h light and 12 h dark, and room temperature was maintained within the range 22–25°C. Experiments were conducted according to protocols approved by the Institutional Animal Care and Use Committee of the New York University School of Medicine. A total of 135 mice were randomized into six experimental groups as follows: Group 1, UVR + standard diet (30 mice); Group 2, UVR + supplemental vitamin E (30 mice); Group 3, UVR + supplemental p -XSC (30 mice); Group 4, UVR + arsenite + standard diet (15 mice); Group 5, UVR + arsenite + supplemental vitamin E (15 mice); and Group 6: UVR + arsenite + supplemental p -XSC (15 mice). Groups 1–3 tested the effect of vitamin E or p -XSC on UVR carcinogenesis. Groups 4–6 examined effects of the supplements on carcinogenesis induced by UVR + arsenite. Because skin tumors have never been reported in untreated mice or in mice treated with either arsenite alone, or vitamin E alone, or selenium compounds alone, such controls were not included in this study in order to reduce animal usage. Since previous studies ( 2 , 3 ) showed that tumor yields were lower with UVR alone compared with UVR + arsenite, larger sample sizes were used for Groups 1–3. A sample size of 30 animals per group was calculated to give 80% power to detect a 40% decrease in mean tumor yield in the groups receiving UVR + chemopreventive agents compared with mice treated with UVR alone. Similarly, sample sizes of 15 mice per group were used for Groups 4–6 to achieve 80% power to detect a 30% decrease in mean tumor yield in groups receiving the UVR + arsenite + chemopreventives compared with mice treated with UVR + arsenite.
Diets
Two different powdered diets containing 62.5 IU/kg supplemental vitamin E (RRR-α-tocopheryl acetate, Sigma Biochemical, St Louis, MO) or 10 mg/kg p -XSC (synthesized and provided by Dr Karam El-Bayoumy of Pennsylvania State University, PA, USA) were formulated by Harlem Teklad (Madison, WI). The PMI 5001 diet from the same company, which contained all base nutrients for rodents including vitamin E (49.0 IU/kg) and selenium (0.2 mg/kg), was used as the control diet. According to Dr El-Bayoumy p -XSC has >99% purity based on chromatographic and spectral characteristics and has stability in the PMI 5001 powdered meal used in this experiment. Diets were stored in a −20°C freezer. Mice were fed each diet 3 weeks prior to beginning of UVR exposures. Food consumption was recorded at weekly intervals. The weight of each mouse was determined at 4-week intervals.
Arsenite
Mice in Groups 4–6 received sodium arsenite (Sigma Biochemical) in the drinking water at a concentration of 5.0 mg/l starting at 3 weeks of age. This concentration causes optimal cocarcinogenesis with UVR ( 3 ). Water consumption was determined at weekly intervals.
UVR exposure
Three weeks after arsenite and supplemental diet exposures began, mice were irradiated with a bank of four parallel Westinghouse solar FS-20 lamps three times a week at a dose of 1.0 kJ/m 2 for ∼6 months as described previously ( 2 , 3 ). The UVR dose rates were measured with an IL 1400A digital radiometer/photometer equipped with a SEL240 UVB-1 detector (International Light, Newburgport, MA, USA). The UVR dose of 1.0 kJ/m 2 was ∼30% of the minimal erythemic dose (MED) for these mice.
Tumor count
The mice were monitored and photographed digitally at various times during the experiment for the presence of tumor-like lesions visible to the naked eye. The number of tumors of diameter ≥1 mm and persisting at least 2 weeks after appearance was recorded for each animal and were included in the calculations of total number of tumors, tumor yield per mouse, percentage of mice with tumor, and distribution of time-to-first tumor.
Histopathology
A total of 60 tumors were randomly selected (10 tumors from each group), fixed with formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin for histopathological diagnosis. The tumor types were classified as described previously ( 2 ). Briefly, the designations were (i) papillomas, tumors with papules of epidermal cells without cellular atypicality, (ii) precancerous lesions, small tumors consisting of acanthotic and papillomatous growth of epidermis of cells with some degree of atypicality but no local invasive growth into dermis and (iii) squamous cell carcinomas (SCC), tumours with nests of atypical cells invading into the dermis or deeper subcutaneous tissue.
Statistics
Differences in the total number of tumors were tested by the Wilcoxon rank-sum test and differences in their histological grades were analyzed by Fisher's exact test. Differences in the distribution of tumor volumes were analyzed by the χ 2 -test. The distribution of the time-to-first tumor was analyzed by the Kolmogarov–Smirnov test.
8-Oxo-dG detection
Normal appearing skin taken from the dorsal area of 10 mice in each group was used. For staining purposes, paraffin sections were deparaffinized in xylene and absolute ethyl alcohol. Sections were pretreated with proteinase K (10 µg/ml) in PBS for 40 min. The DNA was denatured by incubating the slide in 4 N HCl for 7 min and then neutralized by incubating in 50 mM Tris–base for 5 min at room temperature. After blocking with non-immune serum the sections were incubated overnight at 40°C with mouse monoclonal anti-8-oxo-dG antibody (Trevigen, cat no. 4355-MC-100) at a dilution of 1:300 in PBS. Sections were then treated for 10 min with biotinylated anti-mouse link antibody, and then with the streptavidin-biotin peroxidase system (Ultravision Detection System kit, Labvision, CA). Sections were then treated with 1% hydrogen peroxide in methanol to inhibit endogenous peroxidase. 3-Amino-9-ethyl carbazol was used as the chromogen. A section incubated with normal serum instead of anti-8-oxo-dG was used as a negative control.
Results
Effect of vitamin E or p-XSC treatment on health of mice
Previously, we reported that there were no adverse effects on mice that were given up to 10 mg/l arsenite in drinking water with or without UVR ( 2 ). Vitamin E or p -XSC treatment also did not affect the growth of these mice. There were no differences in food and water consumption among the treatment groups during the time periods measured. Mice in the various treatment groups were equally active and healthy. There were no deaths in any group of mice for the duration of the experiment.
Tumorigenesis
Skin cancers developed only in the UVR-exposed regions of skin of mice ( Figure 1 ). The first tumor appeared in mice exposed to UVR + arsenite at 10 weeks after beginning UVR exposure, whereas the first tumor in mice exposed to UVR alone appeared after 12 weeks of UVR exposure. After a lag period, the number of tumors per mouse increased steadily for the duration of the experiment ( Figure 2 ). The final tumor number and tumor yield per mouse in each group is shown in Table I and Figure 3 . As seen in previous experiments ( 2 , 3 ) mice exposed to UVR plus arsenite exhibited an enhanced tumor yield (1.94-fold) compared with mice exposed to UVR alone.
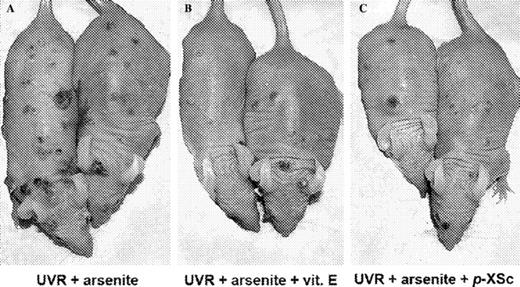
Representative skin tumor distributions in mice treated with ( A ) UVR + arsenite, ( B ) UVR + arsenite + vitamin E and ( C ) UVR + arsenite + p -XSC. Hairless mice were subjected to UVR plus arsenite exposure with or without vitamin E or organoselenium p -XSC in the diet, as described in Materials and methods. The photograph was taken after 26 weeks of UVR exposure. See online Supplementary material for a color version of this figure.
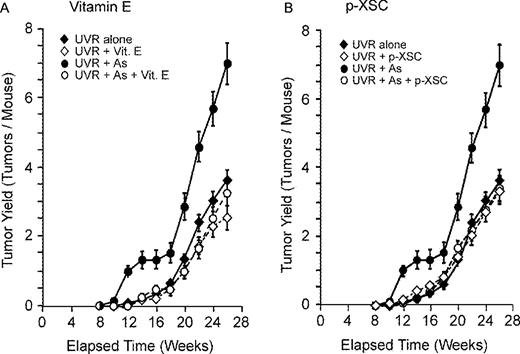
Effect of vitamin E or p -XSC on UVR and UVR + arsenite tumorigenesis. Hairless mice exposed to UVR or UVR + arsenite (see Materials and methods) and were fed diets with or without 62.5 IU/kg RRR-α-tocopheryl acetate ( A ) or 10 mg/kg p -XSC ( B ). Tumors were scored at each time period and number of tumors per mouse was calculated. Error bars are standard deviations from the mean.
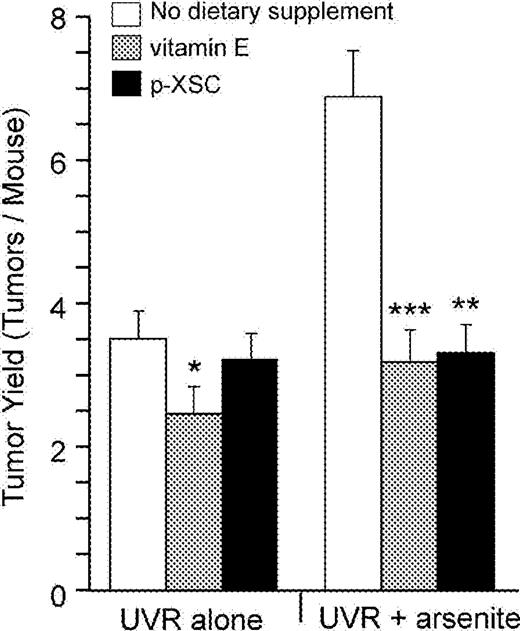
Final tumor yield after 26 weeks of UVR exposure. Both vitamin E and organoselenium ( p -XSC) significantly inhibited the arsenite-enhanced UVR-induced tumors. Vitamin E, but not p -XSC, also decreased tumors in mice treated with UVR alone. Error bars are standard deviations from the mean. *P < 0.05 for UVR + vitamin E vs UVR alone, ***P < 0.001 for UVR + arsenite + vitamin E versus UVR + arsenite and **P < 0.002 for UVR + arsenite + p -XSC versus. UVR + arsenite. In each of the six groups 95% of the tumors were SCC with variable degrees of invasiveness and differentiation.
Protocol and final tumor yield
| Groups | No. of mice | Total tumors a | Average tumor/mouse (SE) | P -value * | Reduction * (%) |
|---|---|---|---|---|---|
| 1 UVR alone (1 kJ/m 2 ) | 30 | 108 | 3.60 (0.340) | — | — |
| 2 UVR + vitamin E (62.5 IU/kg) | 30 | 76 | 2.53 (0.280) | 0.032 | 30 |
| 3 UVR + p -XSC (10 mg/kg) | 30 | 99 | 3.33 (0.330) | 0.321 | 7 |
| 4 UVR + arsenite (5 mg/kg) | 15 | 105 | 7.00 (0.600) | — | — |
| 5 UVR + arsenite + vitamin E | 15 | 49 | 3.27 (0.420) | <0.001 | 53 |
| 6 UVR + arsenite + p -XSC | 15 | 51 | 3.40 (0.360) | <0.002 | 52 |
| Groups | No. of mice | Total tumors a | Average tumor/mouse (SE) | P -value * | Reduction * (%) |
|---|---|---|---|---|---|
| 1 UVR alone (1 kJ/m 2 ) | 30 | 108 | 3.60 (0.340) | — | — |
| 2 UVR + vitamin E (62.5 IU/kg) | 30 | 76 | 2.53 (0.280) | 0.032 | 30 |
| 3 UVR + p -XSC (10 mg/kg) | 30 | 99 | 3.33 (0.330) | 0.321 | 7 |
| 4 UVR + arsenite (5 mg/kg) | 15 | 105 | 7.00 (0.600) | — | — |
| 5 UVR + arsenite + vitamin E | 15 | 49 | 3.27 (0.420) | <0.001 | 53 |
| 6 UVR + arsenite + p -XSC | 15 | 51 | 3.40 (0.360) | <0.002 | 52 |
In each of the six groups 95% of the tumors were SCC with variable degrees of invasiveness and differentiation.
For P -value and tumor yield reduction, mice in Groups 2 and 3 were compared with those in Group 1 and mice in Groups 5 and 6 were compared with those in Group 4. One-tailed P -values were estimated by Fisher's t -statistics.
Protocol and final tumor yield
| Groups | No. of mice | Total tumors a | Average tumor/mouse (SE) | P -value * | Reduction * (%) |
|---|---|---|---|---|---|
| 1 UVR alone (1 kJ/m 2 ) | 30 | 108 | 3.60 (0.340) | — | — |
| 2 UVR + vitamin E (62.5 IU/kg) | 30 | 76 | 2.53 (0.280) | 0.032 | 30 |
| 3 UVR + p -XSC (10 mg/kg) | 30 | 99 | 3.33 (0.330) | 0.321 | 7 |
| 4 UVR + arsenite (5 mg/kg) | 15 | 105 | 7.00 (0.600) | — | — |
| 5 UVR + arsenite + vitamin E | 15 | 49 | 3.27 (0.420) | <0.001 | 53 |
| 6 UVR + arsenite + p -XSC | 15 | 51 | 3.40 (0.360) | <0.002 | 52 |
| Groups | No. of mice | Total tumors a | Average tumor/mouse (SE) | P -value * | Reduction * (%) |
|---|---|---|---|---|---|
| 1 UVR alone (1 kJ/m 2 ) | 30 | 108 | 3.60 (0.340) | — | — |
| 2 UVR + vitamin E (62.5 IU/kg) | 30 | 76 | 2.53 (0.280) | 0.032 | 30 |
| 3 UVR + p -XSC (10 mg/kg) | 30 | 99 | 3.33 (0.330) | 0.321 | 7 |
| 4 UVR + arsenite (5 mg/kg) | 15 | 105 | 7.00 (0.600) | — | — |
| 5 UVR + arsenite + vitamin E | 15 | 49 | 3.27 (0.420) | <0.001 | 53 |
| 6 UVR + arsenite + p -XSC | 15 | 51 | 3.40 (0.360) | <0.002 | 52 |
In each of the six groups 95% of the tumors were SCC with variable degrees of invasiveness and differentiation.
For P -value and tumor yield reduction, mice in Groups 2 and 3 were compared with those in Group 1 and mice in Groups 5 and 6 were compared with those in Group 4. One-tailed P -values were estimated by Fisher's t -statistics.
Dietary supplement with vitamin E significantly inhibited skin tumor formation both in UVR alone and UVR + arsenite-treated mice. As shown in Figures 2A and 3 and Table I , mice treated with supplemental vitamin E (Groups 2 and 5) had fewer tumors per mouse compared with mice without vitamin E (Groups 1 and 4) for the entire duration of the experiment. Vitamin E reduced the final UVR-induced tumor yield by 30% (3.60 versus 2.53 tumors/mouse) and had greater effect (53% reduction) against UVR + arsenite-induced tumors (7.00 versus 3.27 tumors/mouse) ( Figure 3 ). As shown in Figure 4A , vitamin E caused a slight delay in tumor incidence (percentage of mice with tumor) in mice given UVR alone and a stronger delay in incidence of mice given UVR plus arsenite. At 20 weeks of UVR exposure, 100% of mice given UVR + arsenite developed at least one tumor compared with 70, 63.3 and 46.7% of mice given UVR alone, UVR + vitamin E and UVR + arsenite + vitamin E, respectively.
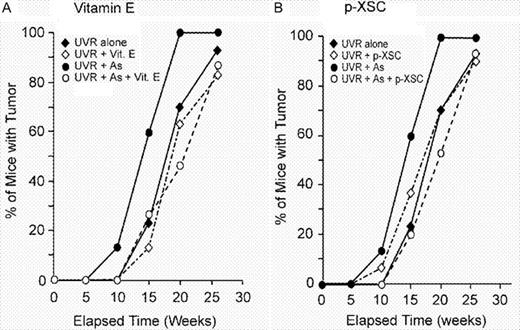
Effect of vitamin E ( A ) or p -XSC ( B ) on tumor incidence. Mice were exposed to UVR alone (30 mice) or UVR + arsenite (15 mice) with or without chemopreventives, as described in Materials and methods. Tumor of diameter ≥1 mm and persisting at least 2 weeks after appearance was recorded for each animal separately, and percentage of mice with tumor was calculated in each week.
In contrast with vitamin E, p -XSC had no significant effect on UVR-induced tumorigenesis ( Figures 2B and 3 and Table I ). However, p -XSC significantly inhibited the arsenite-induced enhancement of UVR carcinogenesis ( Figures 2B and 3 , and Table I ). p -XSC reduced the tumor yield in mice treated with UVR + arsenite from 7.0 tumors/mouse to 3.40 tumors/mouse (a 52% reduction). A similar effect of p -XSC was seen with tumor incidence. At 20 weeks, 53.3% mice treated with UVR + arsenite + p -XSC developed tumors compared with 100% of mice treated with UVR + arsenite ( Figure 4B ).
Histopathology of the tumors
A total of 60 tumors were randomly selected (10 tumors from each group) and examined histopathologically. There were no significant differences in the histopathological characteristics of tumors among these six groups (data not shown). In each of the six groups ∼95% of the tumors were SCC with variable degrees of invasiveness and differentiation, confirming previous studies ( 2 , 3 ). A few papillomas, fibrosarcomas and premalignant hyperplasias were also seen.
Epidermal thickness
Epidermal hyperplasia was examined on skin samples obtained from the mice undergoing carcinogenesis. The sampling was performed by biopsy on one day after the final UVR exposure. Epidermal hyperplasia was seen in all groups. Mice receiving both UVR + arsenite had greater skin thickness than those given UVR or arsenite alone, as seen in previous experiments ( 1 , 2 ). However, there was no significant effect of either vitamin E or p -XSC on skin thickness (data not shown).
8-Oxo-dG formation in mouse skin
Skin from untreated (control) mice and from mice exposed to arsenite alone (10 mg/l) were obtained from our previous experiment ( 2 ). Epidermis from untreated mice showed no 8-oxo-dG nuclear staining ( Figure 5A ). Both UVR and arsenite treatment increased the 8-oxo-dG level in mouse skin ( Figure 5B and C ). The combined UVR + arsenite-treated mice exhibited the greatest 8-oxo-dG levels, with strongly positive cells located throughout the epidermis ( Figure 5D ). Vitamin E or p -XSC treatment had little or no effect on 8-oxo-dG formation in skin of mice treated with UVR alone (data not shown). In contrast, both vitamin E and p -XSC significantly reduced the 8-oxo-dG formation in skin treated with UVR + arsenite ( Figure 5E and F ).
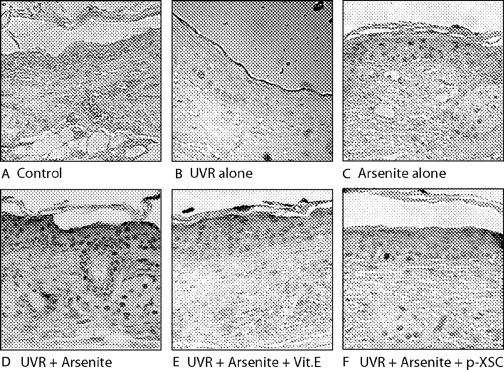
Vitamin E and p -XSC significantly inhibit 8-oxo-dG levels in skin of mice treated with UVR + arsenite. ( A ) Control; ( B ) UVR alone; ( C ) arsenite alone; ( D ) UVR + arsenite; ( E ) UVR + arsenite + vitamin E; and ( F ) UVR + arsenite + p -XSC. Each picture is representative of 10 samples. Magnification: 220× original. See online Supplementary material for a color version of this figure.
Discussion
This study demonstrates that arsenite enhancement of UVR carcinogenesis in mouse skin can be blocked by non-toxic dietary levels of both vitamin E and p -XSC. In addition, vitamin E, but not p -XSC, was ∼30% protective against UVR (alone) carcinogenesis. The overall preventive action of vitamin E exhibited two components, one acting directly on the UVR response and a second acting on the arsenite enhancement response. Vitamin E inhibited the arsenite enhancement by 23%, after taking into account the 30% effect on UVR alone. Tumor onset was also delayed by vitamin E in mice treated with UVR alone and by vitamin E and p -XSC in UVR + arsenite exposed mice.
DNA damage induced by UVR is the most common cause of skin cancer in humans. Direct DNA photoproducts (cyclobutane pyrimidine dimers and 6–4 photoproducts) as well as oxidative DNA products such as 8-oxo-dG occur in normal human epidermis after UVR ( 36 ). Mutations of the p53 gene at dipyrimidine sites (mostly C→T transitions) in human cutaneous SCC have the exact features of mutations caused by UV photoproducts ( 37 ). The same is true of human basal cell carcinoma ( 38 ) and of UVB-induced SCC in hairless mice ( 39 ). On the other hand, an appreciable contribution of oxidative DNA damage to UVR mutagenesis has been reported in sunlight-exposed mouse epidermis ( 40 ). In mouse skin, acute UVR exposure modestly depletes α-tocopherol ( 18 ), while chronic UVR exposure triggers an adaptive response involving epidermal vitamin E accumulation ( 41 ). Vitamin E significantly improves cell viability and restores GSH after UVR exposure, and preincubation of mouse keratinocytes with a water-soluble vitamin E analog, Trolox, significantly decreased the UVB-induced 8-oxo-dG formation in DNA ( 42 ). In UVB-exposed human keratinocytes, the increased level of 8-oxo-dG could be blocked by the hydroxyl radical scavenger mannitol ( 43 ). Topically applied natural vitamin E (α-tocopherol) as well as esterified vitamin E (α-tocopherol acetate and α-tocopherol succinate, which are less photoprotective than natural α-tocopherol), have previously been shown to reduce the induction of skin tumors in UVB irradiated mice ( 44 ). Topical α-tocopherol inhibits DNA dimer formation in a dose-dependent manner, and this form of vitamin E is important for cellular uptake and distribution for their optimal photoprotection ( 45 , 46 ). Dietary supplementation with α-tocopheryl acetate has also been reported to significantly inhibit UVR-induced skin tumor in mice ( 16 ).
p -XSC in the present study showed no inhibition of UVR carcinogenesis but a complete inhibition of the arsenite enhancement. The former finding appears to be at variance with a previous report. Pence et al . ( 47 ) showed that selenium deficiency enhanced the tumor yield in UVB-exposed mice compared with a mere adequate level of 0.1 mg/kg selenium (as selenite), and 0.5 mg/kg was protective. However, our control mice received 0.2 mg/kg selenium and additional selenium (in the form of p -XSC) was not protective against UVR alone.
The data presented here suggest that p -XSC may be acting by a mechanism different from that of vitamin E. Although selenium compounds are known to have antioxidant activity, the concentration used in the present study was insufficient to inhibit UVR carcinogenesis. In contrast, vitamin E was able to inhibit 30% of the UVR-induced tumor yield. P -XSC was more effective as an inhibitor of the arsenite enhancement of UVR carcinogenesis. In addition to its antioxidant activity, selenium compounds may act as an antagonist to arsenical compounds and decrease their toxicity ( 48 ). In vivo , selenium and arsenic form seleno-bis ( S -glutathionyl) arsinium ion ( 49 ). This complex is excreted via the hepato-biliary route and fecal elimination. Also, selenium counteracts arsenic toxicity by forming very insoluble selenides (arsenic hemiselenide) directly in tissues. Whether p -XSC forms these complexes is not known, but it is possible that p -XSC (or a metabolite) may indirectly reduce arsenite levels in the skin. Conversely, selenium deficiency could adversely affect arsenite excretion or formation of insoluble selenide, thereby increasing arsenite concentration in the skin. Selenite blocks arsenite methylation in liver ( 50 , 51 ). While this would have the effect of reducing excretion (since methylated forms are more rapidly excreted), it might also reduce the levels of trivalent methylated arsenic species that are reported to be more toxic and genotoxic than arsenite ( 52 ). It is not known whether p -XSC affects arsenic methylation.
In this experiment, neither vitamin E nor p-XSC showed any significant effects on epidermal hyperplasia. In mice, arsenite had a synergistic effect on UVR-induced proliferation and epidermal thickness ( 53 ). However, this effect was not dose-related and occurred even at the lowest dose (1.25 mg/l) of arsenite ( 2 ). We conclude that arsenite-induced increases in epithelial proliferation may play only a minor role in cocarcinogenesis with UVR because increased proliferation of skin by arsenite alone does lead to skin cancer, and vitamin E and p -XSC block arsenite enhancement of UVR carcinogenesis without blocking the effects on skin proliferation. These results confirm the suggestion by Berton et al . ( 54 ) that vitamin E mitigates initial events associated with UVR, such as DNA damage and p53 expression, but has limited potential in preventing UVR-induced proliferation.
The immunohistochemical results reported here indicate that vitamin E and p -XSC have nearly identical effects on 8-oxo-dG formation. Both vitamin E and p -XSC significantly reduced the levels of 8-oxo-dG, a biomarker for oxidative DNA damage, in mice exposed to UVR + arsenite and had little effect on 8-oxo-dG levels in mice exposed to UVR alone. Increased 8-oxo-dG levels have been found in human skin and in tumors resulting from exposure to arsenic as well as in normal tissues adjacent to the arsenic-related Bowen's disease lesions ( 9 ). We have recently shown that two organic selenium compounds, seleno- l -methionine and Se-(methyl)selenocysteine, block not only arsenite-induced mutagenesis but also spontaneous mutagenesis in HOS cells ( 30 ), presumably by inhibition of spontaneous oxidative DNA damage ( 31 ). In the present study, the close correlation between 8-oxo-dG levels and cancer yield suggest that reducing oxidative damage is a possible mechanism. Of course, the effects of vitamin E or p -XSC on arsenic metabolism ( 50 ) or on arsenite-induced effects, such as altered DNA methylation ( 55 ) and DNA repair ( 5 ), could contribute to the anti-carcinogenic action of these compounds. As mentioned above, p -XSC and other selenium compounds may act as arsenic antagonists by other mechanisms ( 48 – 51 ). More studies will be needed to establish whether other mechanisms contribute to the anti-carcinogenic activities of vitamin E and p -XSC.
Soil and plants in an arsenic-contaminated area of Bangladesh were found to have low levels of selenium, suggesting that low dietary selenium may increase the risk of arsenic carcinogenesis ( 35 ). In this area, 92% of the water samples with detectable concentrations of arsenic did not have detectable concentrations of selenium ( 56 ). Meanwhile, people are already exposed to arsenic, and signs and symptoms of this crisis are very visible in affected areas of the world. The results of this study suggest the possibility of a cost-effective dietary intervention to prevent arsenic-related cancers. Arsenic can be precipitated from water with iron filings or alum, and the precipitate can be filtered. However, a filter that works in the laboratory may not work in the rural areas of developing countries, and adapting a large-scale technology to individual homes is likely to be problematic, expensive and time consuming ( 57 ). The recommended dietary allowance of selenium for adult North Americans is 55 µg/day, and the safe upper limit of intake is considered to be 400 µg/day ( 58 ). Administration of 100–200 µg/day selenium (as selenium yeast) for 14 months significantly reduced the severity of symptoms in people in Mongolia with arsenicosis, and the arsenic concentration in blood, urine and hair declined with selenium treatment over time compared with control subjects ( 29 ). However, since there are concerns about toxicity of chronic intake of elevated levels of selenium compounds, well-controlled human trials are necessary to determine the dose, efficacy and safety of selenium supplementation in humans to ameliorate the deleterious effects of exposure to inorganic arsenic.
Our results show that vitamin E and p -XSC significantly inhibit arsenite enhancement of UVR carcinogenesis in mice. If the same is true for humans, both vitamin E and organic selenium (in the form of selenized yeast or pure compounds) could be supplemented to human diets, either separately or as a combination of vitamin E plus selenium to reduce the dose of each.
Supplementary material
Supplementary material can be found at: http://www.carcin.oxfordjournals.org/
Authors are listed in the order of their contribution to the work.
The authors thank Dr Karam El-Bayoumy of Pennsylvania State University, PA, USA for generously supplying the p -XSC compound. This work was supported by National Institute of Environmental Health Sciences (NIEHS) grants ES09252 and ES10344 to T.R. and is part of the Nelson Institute of Environmental Medicine and the Kaplan Cancer Center programs supported by grant CA16087 from the National Cancer Institute and center grant ES00260 from the NIEHS. A.N.U. was supported by a postdoctoral fellowship from the Cancer Research and Prevention Foundation, formerly known as the Cancer Research Foundation of America.
Conflict of Interest Statement : None declared.
References
National Research Council (
Rossman,T.G., Uddin,A.N., Burns,F.J. and Bosland,M.C. (
Burns,F.J., Uddin,A.N., Wu,F., Nadas,A. and Rossman,T.G. (
Kitchin,K.T. and Ahmad,S. (
Rossman,T.G. (
Shi,H., Shi,X. and Liu,K.J. (
Nordenson,I. and Beckman,L. (
Hei,T.K., Liu,S.X. and Waldren,C. (
Matsui,M., Nishigori,C., Toyokuni,S., Takada,J., Akaboshi,M., Ishikawa,M., Imamura,S. and Miyachi,Y. (
Keyse,S.M. and Tyrrell,R.M. (
Tsai,S.H., Liang,Y.C., Chen,L., Ho,F.M., Hsieh,M.S. and Lin,J.K. (
Barchowsky,A., Dudeck,E.J., Treadwell,M.D. and Wetterhahn,K.E. (
Styblo,M., Serves,S.V., Cullen,W.R. and Thomas,D.J. (
Lin,S., Cullen,W.R. and Thomas,D.J. (
Shindo,Y., Witt,E. and Packer,L. (
Gerrish,K.E. and Gensler,H.L. (
Burke,K.E., Clive,J., Combs,G.F., Commisso,J., Keen,C.L and Nakamura,R.M. (
Krol,E.S., Kramer-Stickland,K.A. and Liebler,D.C. (
Behne,D. and Kyriakopoulos,A. (
McKenzie,R.C., Arthur,J.R. and Beckett,G.J. (
Kryukov,G.V., Castellano,S., Novoselov,S.V., Lobanov,A.V., Zehtab,O., Guigó,R. and Gladyshev,V.N. (
McKenzie,R.C. (
Rafferty,T.S., Beckett,G.J., Walker,C., Bisset,Y.C. and McKenzie,R.C. (
El-Bayoumy,K. (
El-Bayoumy,K., Rao,C.V. and Reddy,B.S. (
Meuillet,E., Stratton,S., Cherukuri,D.P., Goulet,A-C., Kagey,J., Porterfield,B. and Nelson,M.A. (
Seo,Y.R., Kelly,M.R. and Smith,M.L. (
Medina,D., Thompson,H., Ganther,H. and Ip,C. (
Wuyi,W., Linsheng,Y., Shaofan,H., Jian,T. and Hairong,L. (
Rossman,T.G. and Uddin,A.N. (
Rossman,T.G. and Goncharova,E.I. (
Mure,K. and Rossman,T.G. (
Gabel,T. (
Schoen,A., Beck,B., Sharma,R. and Dube,E. (
Spallholz,J.E., Boylan,L.M., Palace,V., Chen,J., Smith,L., Rahman,M.M. and Robertson,J.D. (
Ahmed,N.U., Ueda,M., Nikaido,O., Osawa,T. and Ichihashi,M. (
Brash,D.E., Rudolph,J.A., Simon,J.A. et al . (
Ziegler,A., Leffell,D.J., Kunala,S. et al . (
Dumaz,N., van Kranen,H.J., de Vries,A., Berg,R.J.W., Wester,P.W., van Kreijl,C.F., Sarasin,A., Daya-Grosjean,L. and de Gruijl,F.R. (
Ikehata,H., Nakamura,S., Asamura,T. and Ono,T. (
Liebler,D.C. and Burr,J.A. (
Stewart,M.S., Cameron,G.S. and Pence,B.C. (
Pelle,E., Huang,X., Mammone,T., Marenus,K., Maes,D. and Frenkel,K. (
Gensler,H.L. and Magdaleno,M. (
McVean,M. and Liebler,D.C. (
McVean,M. and Liebler,D.C. (
Pence,B.C., Delver,E. and Dunn,D.M. (
Kenyon,E.M., Hughes,M.F. and Levander,O.A. (
Gailer,J.G., George,G.N., Pickering,I.J. et al . (
Styblo,M. and Thomas,D.J. (
Walton,F.S., Waters,S.B., Jolley,S.L., LeCluyse,E.L., Thomas,D.J. and Styblo,M. (
Styblo,M., DelRazo,L.M., Vega,L., Germolec,D.R., LeCluyse,E.L., Hamilton,G.A., Reed,W., Wang,C., Cullen,W.R. and Thomas,D.J. (
Rossman,T.G., Uddin,A.N. and Burns,F.J. (
Berton,T.R., Conti,C.J., Mitchell,D.L., Aldaz,C.M., Lubet,R.A. and Fischer,S.M. (
Chen,H,, Li,S., Liu,J., Diwan,B.A., Barrett,J.C. and Waalkes,M.P. (
Frisbie,S.H., Ortega,R., Maynard,D.M. and Sarkar,B. (
Halim,N.S. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}