Article Text

Abstract
Patients with biallelic mutations in BRCA2 are in Fanconi anaemia group D1. We analysed the severity of the mutations in 27 cases, classified according to their association with breast cancer in heterozygotes, and their predicted functional effect. Twenty mutations were frameshifts or truncations, three involved splice sites, five were missense variants of unknown severity and two were benign polymorphisms. Five patients had VACTERL-H association. Leukaemia was reported in 13 patients, and solid tumours in 15; 6 patients had two or more malignancies. The cumulative probability of any malignancy was 97% by age 5.2 years. IVS7+1G→A and IVS7+2T→G were associated with AML, and 886delGT and 6174delT with brain tumours. However, patients with other alleles remained at very high risk of these events. Missense mutations formed a distinct cluster in a highly conserved region of the BRCA2 protein.
The small group of patients with biallelic mutations in BRCA2 is distinctive in the severity of the phenotype, and early onset and high rates of leukaemia and specific solid tumours, and may comprise an extreme variant of Fanconi anaemia. Several of the alleles were not associated with cancer in presumed carriers, and thus counselling presents more uncertainties than usual.
- AML, acute myelogenous leukaemia
- BIC, Breast Cancer Information Core; HBOC, hereditary breast/ovarian cancer
- VACTERL-H (V, vertebral; A, anal atresia; C, cardiac; TE, tracheoesophageal fistula, oesophageal atresia; R, renal or radial; L, limb anomalies; H, hydrocephalus)
- tumour
Statistics from Altmetric.com
- AML, acute myelogenous leukaemia
- BIC, Breast Cancer Information Core; HBOC, hereditary breast/ovarian cancer
- VACTERL-H (V, vertebral; A, anal atresia; C, cardiac; TE, tracheoesophageal fistula, oesophageal atresia; R, renal or radial; L, limb anomalies; H, hydrocephalus)
Fanconi anemia is a genetically heterogeneous and predominantly autosomal recessive disorder characterised by defective DNA repair with high rates of birth defects, aplastic anaemia, leukaemia and solid tumours.1 Fanconi anaemia is associated with homozygotic or biallelic mutations in at least 12 genes: FANCA, FANCB (X-linked recessive), FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCI (not yet cloned), FANCJ, FANCL and FANCM.2 In response to DNA damage, the protein products of all of these genes (except for FANCD1, FANCD2 and FANCJ) form a complex, which is required for the ubiquitination of the protein product of FANCD2, a key component of the DNA damage repair pathway.3
More than 1000 unique mutations in BRCA1 or BRCA24 are associated with familial breast/ovarian cancer.5 The molecular bases of these seemingly different genetic disorders converged with detection of BRCA1 proteins in Fanconi anaemia-related DNA damage-response foci,6 and in the discovery of biallelic mutations in BRCA2 in patients classified as Fanconi anaemia complementation group FA-D1.7 Furthermore, patients with a Fanconi anaemia clinical phenotype were recently found to have germline mutations in two genes which act downstream of the D2 ubiquitination step, namely BRCA2 and FANCJ/BRIP1/BACH1, the latter a gene whose product may interact with BRCA1.8,9 The precise roles of these downstream genes in Fanconi anaemia have not been entirely defined.
In view of the mechanistic implications of this nexus between two distinct cancer susceptibility syndromes and molecular pathways, we analysed the clinical and molecular features associated with the BRCA2 mutations identified in patients classified as FA-D1, using data from 26 previously reported cases and findings from a newly diagnosed case. Five patients had components of VACTERL-H (V, vertebral; A, anal atresia; C, cardiac; TE, tracheoesophageal fistula, oesophageal atresia; R, renal or radial; L, limb anomalies; H, hydrocephalus) association (and its variants (MIM 192350 and MIM 27695010). We evaluated genotype/cancer correlations between specific mutations in BRCA2 and specific types of malignancies. Homozygotic mutations in BRCA2 are embryonic lethal in mice, and it had been assumed that humans carrying homozygotic mutations would also fail to come to term.11,12 Therefore, we evaluated whether the reported FANCD1/BRCA2 mutations led to functional inactivation of the BRCA2 gene or a physiologically meaningful diminution of the BRCA2 gene product. Finally, we mapped the intragenic location of each of the Fanconi anaemia-related BRCA2 mutations, and quantitatively compared this distribution with that among heterozygotes reported to the Breast Cancer Information Core (BIC) database (http://research.nhgri.nih.gov/bic/).13
SUBJECTS AND METHODS
We identified published cases with biallelic mutations in FANCD1/BRCA2 from a PubMed search using combinations of the terms “Fanconi Anemia”, “FANCD1”, “BRCA2” and “VATER”. We diagnosed a previously unreported case in our NCI Inherited Bone Marrow Failure Syndromes Cohort.14 The protocol for this cohort was approved by the Institutional Review Board of the National Cancer Institute (Rockville, Maryland, USA).
Analysis of BRCA2 alleles
Information regarding the functional consequences of each allele reported in patients with FA-D1 with regard to breast cancer was obtained from the BIC database.13 Mutations were classified as “deleterious” (truncating or splice-site mutations predicted to disrupt the function of the protein, which are associated with a greatly increased risk of breast cancer), “probably deleterious” (likely to disrupt normal splicing), “unknown” or “unclassified variants” (missense mutations of unknown clinical significance with regard to breast cancer risk), or “benign” (not associated with increased risk of breast or ovarian cancer). We then compared the phenotypes of the patients with the prior classification of the severity of the mutations. We were surprised to find that five of the Fanconi anaemia-related BRCA2 mutations were categorised as unclassified variants and two as benign polymorphisms. Alleles classified as benign polymorphisms are commonly found in control populations and have not been associated with breast cancer in case–control studies. As the mutation data for this review were derived from the literature, we cannot exclude the possibility of additional undiscovered deleterious mutations in cis with those reported, such as large deletions, which might lead to reclassification of the severity of the mutations that we have called “benign” or “of unknown significance”.
Positional analysis
Positional analysis considered whether the Fanconi anaemia-associated missense mutations had an “unusual” distribution along the protein sequence, as described below. As a reference, we considered the location of all unclassified BRCA2 missense mutations reported to the BIC. We asked whether the Fanconi anaemia-associated missense mutations were distributed uniformly, non-uniformly but with the same frequency curve as other missense mutations in the BIC, or non-randomly (ie, in a cluster). We used exact and non-parametric statistical tests because of the small sample size.
The BRCA2 protein contains 3418 amino acids. If the n = 5 Fanconi anaemia-related missense mutations were distributed randomly, then 1/3 each should fall in positions 1:1139, 1140:2279 and 2280:3418. Under this hypothesis, the observed frequency counts (N1, N2, N3) constitute a sample from a multinomial distribution with equal frequencies, and the Pearson’s χ2 statistic is:

This equation provides a measure of the discrepancy between observed and expected values. We computed the exact distribution of this statistic under the null hypothesis and used this distribution to obtain an exact p value.
To determine whether Fanconi anaemia missense mutations have the same distribution as other missense mutations reported to the BIC, we used the Wilcoxon rank sum test to compare the median position of Fanconi anaemia missense mutations with the median position of 733 distinct BRCA2 missense mutations found in the BIC. To specifically test for a “cluster”, we considered the distance between the most NH2 and -COOH proximal positions of the Fanconi anaemia missense mutations—that is, the observed range statistic—compared with the expected distribution of the range statistic computed from 100 000 Monte Carlo simulations. The reported p value equals the probability of observing a range statistic as small as, or smaller than, what was observed. The expected distribution of the range was computed in two ways, assuming that the positions of the Fanconi anaemia missense mutations were a sample from a uniform distribution, and sampling five mutations with replacement from the 733 BIC missense mutations.
We performed similar analyses of the Fanconi anaemia-associated truncation or frameshift mutations.
Genotype/phenotype correlations
The correlation between the presence of a specific mutation and a specific outcome was assessed using the odds ratio (OR) and its 95% confidence interval (CI); significance was tested with the Fisher exact test. The relative hazard of an event in those with or without the mutation was estimated using the Cox proportional hazards model. Statistical analyses were performed using Stata9.
Cancer history in families
Data on the types and ages of cancer in family members were obtained from the published case reports, and from the pedigree obtained by interviewing family members of our new case.
RESULTS
Case report
A 2100-g 37-week female (NCI 1) had been identified in utero with hydrocephalus, fused kidneys and growth retardation. At birth, she had intrauterine growth retardation, corneal opacities (diagnosed as Peter’s anomaly, MIM 604229), an anteriorly placed anus, small kidneys and long thumbs with increased laxity; this constellation led to a later diagnosis of VACTERL-H (MIM 276950). Other physical findings included microcephaly, facial dysmorphia, microphthalmia, esotropia, growth failure, café-au-lait spots and malposition of the kidneys. Surgical procedures during the first year included a ventriculoperitoneal shunt, anoplasty and repair of a tethered spinal cord due to a filum terminale lipoma.
Her karyotype was 46,XX, with “structural chromosome changes that may reflect chromosome instability, an acute viral infection, or artefact of culture”. The chromosome breakage test for Fanconi anaemia was not done until age 20 months, when an astute geneticist observed her birth defects and suggested the diagnosis of Fanconi anaemia. Chromosome breakage studies of peripheral blood lymphocytes with both diepoxybutane and mitomycin C were diagnostic of Fanconi anaemia. Direct DNA sequencing (Myriad Genetics, Salt Lake City, Utah, USA) showed biallelic mutations in BRCA2, 6174delT and Q3066X, thereby identifying her as belonging to group FA-D1. At age 3.1 years, she was diagnosed with a medulloblastoma.
Genotype/birth defect associations in patients with BRCA2 mutations
Table 1 summarises the published cases and our new case.7,15–24 The series includes 27 patients from 21 families. One family was not genotyped, and there were 30 unique alleles among the 40 alleles that were typed. Twenty of the Fanconi anaemia-related BRCA2 mutations were frameshift or truncating mutations; three involved splice sites, five were missense variants of unknown clinical severity and two were benign polymorphisms. Pathogenicity of the specific mutations is described below.
Patient characteristics, malignancies, and BRCA2 mutations
Several recurrent birth defects were reported, including hyperpigmentation and/or café-au-lait spots, short stature, microcephaly, abnormal thumbs, and renal anomalies. Although small numbers make it difficult to judge whether these features occurred more frequently than expected, five patients (1A, 1B, SB1690CB, PT2, and NCI 1) had three or more of the VATER association anomalies. In these patients, both mutations in BRCA2 were considered to be deleterious or probably deleterious (see below). We detected no significant association between specific BRCA2 mutations and VATER association. Among the five patients with VATER association, two cousins had brain tumours (cousins 1A and 1B); one had AML (SB1690CB); another had Wilms’ tumour, neuroblastoma and brain tumour (PT2); and NCI 1 had a brain tumour at age 3.1 years. The distribution of tumour types did not differ between the patients with and without VATER features.
Two patients (HSC62 and PT2) carried homozygotic mutations that are probably deleterious or deleterious, IVS19-1G→A and 1548del4, respectively, both of whom had birth defects. The homozygotic IVS19-1G→A patient had no tumour by age 30 years; the homozygotic 1548del4 patient had VATER features, Wilms’ tumour, neuroblastoma and brain tumour, all by age 3 years.
The spectrum of physical findings reported in all five of the patients with one allele of unknown clinical relevance seemed to be similar to those in patients with two deleterious alleles; it is interesting (but not statistically significant) that none of these patients met the criteria for VATER association.
Genotype/cancer associations in FANCD1/BRCA2 patients
Table 1 summarises the types and ages of cancers reported in patients with FA-D1. Five of the 10 patients with acute myelogenous leukaemia (AML) had mutations in IVS7 (two were IVS7+1G→A, and three were IVS7+2T→G), and five had a variety of other mutations. One additional patient with IVS7+2T→G died at <1 year of age from stem cell transplant complications, without developing AML. Although the OR for the association of AML with IVS7 mutations compared with other BRCA2 mutations was 15 (95% CI 1.1 to 750, p = 0.02), patients with other BRCA2 mutations also had a high risk of AML.25 The OR for AML in 10 of 27 patients with FA-D1 compared with 9 of 145 patients with Fanconi anaemia (all other complementation groups combined) whose mutations were not classified26 is 9 (95% CI 2.8 to 28, p<0.001).
Reid et al22 suggested a correlation between BRCA2 886delGT and brain tumours. In our review, we found 12 patients who had brain tumours, five of whom had one 886delGT allele, an allele observed only in patients with brain tumour (OR ∞, p = 0.006). The 12 patients with brain tumours were from nine families and included three pairs of siblings; only three of the nine families had the 886delGT allele. Thus, the observed association might be due to other familial risk factors for brain tumours, since the number of families is small. Seven patients with brain tumours from six families did not carry 886delGT, indicating that patients with other mutations are also at risk of brain tumours. For example, 6174delT occurred in four patients, all of whom had brain tumours (OR ∞, p = 0.03). One patient had both 886delGT and 6174delT. Seven of the brain tumours were medulloblastomas, and the others were described, respectively, as either medulloblastoma or astrocytoma, astrocytoma, glioblastoma multiforme, posterior fossa tumour and brain tumour.
Wilms’ tumour was commonly reported in these patients. Three of the seven cases had the 886delGT mutation (OR 6.8, 95% CI 0.5 to 98, p = 0.09), two of whom were siblings; the association between this allele and Wilms’ tumour may be familial rather than BRCA2-specific. Comparing the 15 patients with FA-D1 who had solid tumours with 14 patients with tumours among 145 patients with unclassified Fanconi anaemia in our previous report26 yielded an OR = 12 (95% CI 4 to 22, p<0.001) for patients with FA-D1 and solid tumours.
Two of the 27 patients had no history of cancer at the time they were reported (ages 2 and 30 years): one was homozygotic for a probably deleterious allele (HSC62), and the other had a benign BRCA2 allele with a mutation in FANCB (HSC230). The other patient with one benign BRCA2 allele had T cell acute lymphoblastic leukaemia, which is atypical for Fanconi anaemia, in which AML is the usual subtype. The OR for any malignancy in 25 patients with FA-D1 compared with 23 of 145 patients with Fanconi anaemia in our earlier study26 was 66 (95% CI 14 to 594, p<0.001).
In the current series, the cumulative probability of developing leukaemia was 79% by age 10 years;25 the probability of developing a solid tumour reached a plateau of 83% by age 6.7 years (fig 1A). The most common solid malignancy was a brain tumour: the probability among all patients was 85% by age 9 years (fig 1B). All five patients with the 886delGT allele developed a brain tumour by 9 years (median age 4.5), while 7 of the 22 patients with other alleles developed their brain tumours by 4.9 years of age (fig 1C). The cumulative probability of developing a Wilms’ tumour was 63% by age 6.7 years (data not shown), and of any malignancy was 97% by age 5.2 years (fig 1D).

Solid tumours in patients with Fanconi anaemia due to mutations in FANCD1/BRCA2. (A) Probability of any solid tumour. (B) Probability of a brain tumour. (C) Probability of a brain tumour according to the presence or absence of the 886delGT mutation. Dashed line: mutations other than 886delGT (n = 22); solid line: 886delGT mutation (n = 5). (D) Probability of any malignancy. The plateaus in all of the figures correspond to patient HSC62, who reached age 30 years at the last report. This patient was homozygotic for IVS19-1G→A, which results in an in-frame deletion of exon 19, an allele for which severity is inferred but not proved.
Severity of BRCA2 mutations in patients
In all, 20 of the 30 unique alleles among the 40 alleles were classified as “deleterious”, on the basis of reports in the BIC database and their structure (nonsense or frame-shift mutations leading to early protein truncation, table 1). Three alleles that involve splice sites (IVS19-1G→A, IVS7+2T→G, and IVS7+1G→A) were designated as “probably deleterious”. According to the BIC database, both mutations in IVS7 in BRCA2 have been found in families with breast and ovarian cancer. These mutations alter a splice site, which results in skipping of exon 7. In one patient with breast cancer and the IVS7+2T→G mutation, no RNA was produced in vitro from the chromosome carrying the mutation.27 Five missense mutations were of “uncertain significance”, as they were not reported to be significantly associated with breast or ovarian cancer in the BIC.
In addition, in two patients one allele was classified as a benign polymorphism by the BIC. K3326X in patient HSC230 is known to be a polymorphic stop codon.28 This patient was originally and correctly classified as FA-B, not FA-D1, and had a frameshift mutation in exon 8 of FANCB (1838insT) which leads to a premature stop codon.15 He had been screened for mutations in BRCA2 before the FANCB gene was identified. To complicate matters further, HSC230 was recently found to be a BRCA2 mosaic: fibroblasts heterozygotic for 3033/delAAAC and wild-type BRCA2, and lymphoblasts that contain both mutant BRCA2 alleles.29 Thus, the Fanconi anaemia phenotype of HSC230 is due to the mutation in FANCB, and not due to the combination of a deleterious mutation and a benign polymorphism in BRCA2. This patient had not developed cancer when reported at age 2 years. We have retained this patient on our list of “FANCD1/BRCA2” subjects to emphasise the caution required in interpretation of the causal role of mutations in BRCA2 in Fanconi anaemia.
Case 900/1 also had both a deleterious allele and a benign polymorphism (N372H) in BRCA2, and was reported to have a mild phenotype, with café-au-lait spots and short stature. The N372H allele was not associated with increased sporadic breast cancer risk in the Nurses’ Health Study cohort.30 This male patient was reported not to belong to Fanconi anaemia groups A, C, D2, E, F or G, but was apparently not tested for B, L, M or J, and thus classification of this case as FA-D1 might also be uncertain.
Severity of BRCA2 mutations in families of patients with FANCD1
Family histories were examined to determine whether the types of cancers seen in breast/ovarian cancer families5 were also reported in the Fanconi anaemia families. This analysis is limited, as several articles did not mention family history, and the ages of other family members at risk of cancer were not provided. If both BRCA2 alleles in patients with FA-D1 have deleterious mutations, it might be expected that breast and/or ovarian cancer would be a frequent occurrence in both parental bloodlines, as both parents of affected children would be obligate carriers of a single copy of a deleterious allele, as would half of each parent’s first-degree relatives. Although the dataset is small and subject to ascertainment bias due to the known association between BRCA2 and breast/ovarian cancer, it is noteworthy that cancers associated with carriers of mutations in BRCA2 were reported in 11 of the 21 families (table 2).
Cancers in families of FANCD1/BRCA2 Probands
Among the 11 families in which cancer was reported, the breast cancer occurred on the side of the family carrying a “benign” allele (N372H) in family 900/1; in four families cancer was noted on only one side of the family (357/1, 800/1, 900/1 and SB1690CB); in four families the cancer history is not extensive; and in only two families was there a strong family history of breast cancer or breast cancer-associated cancers (1A and 1B, and NCI 1). In five families, breast cancer was reported in both maternal and paternal relatives, whereas in four there was a breast cancer history on only one side of the family, and in only two non-breast cancers were mentioned, one of which (brain) is not part of the breast/ovarian cancer spectrum (K2, 800/1). In all, 27 female relatives (13 <50 years of age) and one male relative had breast cancer. In the parental generation, there were one prostate cancer at age 65 years in a carrier of C3069X, and two breast cancers at ages 38 and 45 years in carriers of Y1894X and S1882X; these are all known deleterious mutations. At the level of the grandparents of the probands, six grandmothers (out of 22 in the eleven families reported) had breast cancer at ages 29, 40, 46, 48, 65 and 70 years. The BRCA2 alleles were not specifically identified in the grandparents, although the daughter of the grandmother with breast cancer at age 29 years carried the benign N372H allele. The male breast cancer developed in a grandfather at age 60 years; his daughter carried the deleterious mutation Y1894X.
No ovarian cancer was reported, which may not be surprising given the small number of families and the fact that ovarian cancer penetrance is significantly lower in carriers of mutations in BRCA2 compared with BRCA1. There were four cases of prostate cancer: one in the father of a case and three in grandfathers (one at age 48 years); prostate cancer is part of the BRCA2 cancer susceptibility spectrum.5 There were also three cases of colon cancer, two cases of bladder cancer, one melanoma, and a small number of other, not clearly BRCA2-related, cancers. In one family maternal cancer history was not available, and in three other families the paternal family history did not report any cancers (two of these involved deleterious alleles). Although the currently available data are not sufficiently robust to deal with this question definitively, the family history information does suggest that a single copy of some of the deleterious BRCA2 alleles which are found in patients with FA-D1 confers a pattern of cancer susceptibility similar to that described for this gene in the context of hereditary breast/ovarian cancer. However, the data also suggest that several of the alleles reported in patients with FANCD1/BRCA2 are not associated with increased rates of cancer in family members who are presumed to be carriers of one of the alleles.
Location of the FANCD1/BRCA2 mutations
The FANCD1/BRCA2 mutations were distributed throughout the length of the BRCA2 gene (fig 2), and the two benign polymorphisms, N372H and K3326X, were at the N-terminal and C-terminal, respectively. However, the Fanconi anaemia-associated missense mutation-related amino acid changes were not uniformly distributed across the BRCA2 protein (p = 0.012, exact Pearson’s statistic). The median locations for the 5 Fanconi anaemia and 733 BIC-associated missense mutations were dissimilar, but not significantly so (median positions 2510 and 1889, respectively, Wilcoxon p = 0.10). However, the Fanconi anaemia-associated missense mutations were tightly clustered along the BRCA2 protein between positions 2336 and 2729 inclusive (394 residues, ∼11% of the total). The range test was highly significant (p<0.001 and <0.001, respectively, using uniform and BIC-derived null distributions).

{kind=link}
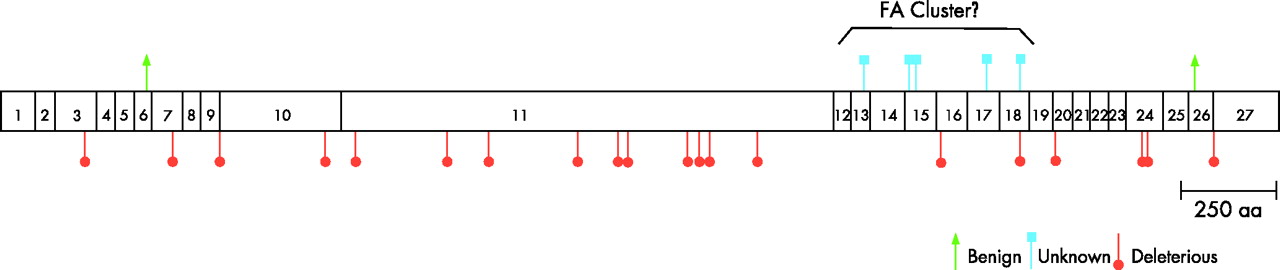{kind=link}
Schematic of the BRCA2 protein-coding region. The location of each exon is shown (numbered boxes). The location of mutant alleles found in patients with Fanconi anaemia is indicated. Orange circles: deleterious (frameshift/nonsense mutations leading to truncation; blue squares: unknown (missense mutations); green arrows: benign (not associated with cancer risk). The classification of the mutant alleles is defined in Subjects and methods.
The 20 Fanconi anaemia-associated truncation or frameshift mutations were distributed uniformly across the BRCA2 protein (fig 2; p = 0.613, exact Pearson’s statistic), with no evidence of clustering (p = 0.312, uniform range statistic).
DISCUSSION
The functional effects of all of the FANCD1/BRCA2 mutations need to be examined more closely, to determine whether those with two mutant alleles have truly Fanconi anaemia or variant Fanconi anaemia, and to provide appropriate genetic counselling, risk assessment and cancer surveillance to relatives from both sides of the family. Our analyses suggest that FA-D1 is indeed phenotypically distinctive among the Fanconi anaemia complementation groups, despite the known heterogeneity within Fanconi anaemia itself. Focusing on the specific types of mutations in BRCA2 may be instructive with regard to how patients with FA-D1 differ from patients with other types of Fanconi anaemia, and with regard to why biallelic mutations in a breast/ovarian cancer susceptibility gene give rise to a totally different syndrome from that seen in heterozygotes.
To tackle this question more definitively, it is important to note that two of the patients classified as FANCD1/BRCA2 actually had only one deleterious BRCA2 allele, with a benign polymorphism in the trans allele; they should be included in defining a new disorder with caution. In patient HSC230, the K3326X allele removes the last 92 amino acids at the C-terminus, does not change the function of the gene product, is a polymorphism found in >1% of normal individuals, and is commonly found in linkage disequilibrium with a known deleterious mutation.28,31 In addition, this patient clearly belongs to the FA-B complementation group, with a defined deleterious X-linked mutation in FANCB.15 The other patient with benign polymorphism, 900/1, has N372H, which also lacks known functional consequence, and is not associated with increased cancer risk.30,31 The report of very early-onset breast cancer in the grandmother on this side of the family, absent direct information that she carries this allele, is provocative but not informative. Unless a truly deleterious mutation is found to be in linkage disequilibrium with the N372H allele, it is possible that patient 900/1 belongs to a different complementation group than FA-D1, or even has another disorder. However, it must be emphasised that the usual methods for sequencing BRCA2 might miss large deletions or rearrangements, which might be in cis to the benign mutations reported in these two patients.32 Both patients carrying “benign” alleles had physical features consistent with Fanconi anaemia, as well as increased chromosome breakage, but 900/1 had T cell acute lymphoblastic leukaemia (not the typical AML of Fanconi anaemia) at age 5 years, whereas HSC230 had no cancer at age 2 years.
Six patients from five families with one deleterious and one missense BRCA2 mutation, the latter of unknown clinical significance with regard to familial breast cancer, are potentially very informative. Family histories of cancer were provided for only two of the five families, and neither was enriched with BRCA2-related cancers. However, it is striking that the five Fanconi anaemia-associated missense mutations clustered tightly between amino acid positions 2336 and 2729, whereas the 20 truncation/frameshift mutations were uniformly distributed across the BRCA2 gene. This “FA cluster” is located in the most highly conserved BRCA2 region in an interspecies comparison of human/mouse/chicken BRCA2, suggesting that these residues may be functionally constrained (data not shown). BRCA2 regions which are not implicated in breast/ovarian cancer pathogenesis in heterozygotic adults, nonetheless, may be critical interaction regions for a component of the Fanconi anaemia pathway, such as downstream of ubiquitination of FANCD2.7,33 This hypothesis provides a rationale for further evaluation of interactions between the BRCA2 protein and other proteins in the Fanconi anaemia DNA repair pathway. For example, these missense mutations (hypomorphs) may reduce the amount of functional BRCA2 protein.
Alternatively, at least some of the five families with an allele of unknown significance may be truly heterozygotic for a single biologically active BRCA2 mutation; their Fanconi anaemia could be due to unrecognised mutations in a different Fanconi anaemia gene, as we have shown with HSC230, who was reported to be an example of biallelic mutations in BRCA2 but who was clearly a patient with FA-B.
In 19 of the 27 patients, and in 13 of the 20 families, both alleles were interpreted to be deleterious or probably deleterious; patient 4 was a compound heterozygote for two different null mutations. No patient was homozygotic for the same null allele, and thus the data on live-born children with biallelic mutations in BRCA2 are insufficient to deal with the possibility that true homozygosity for mutations in BRCA2 is incompatible with life. Survival of homozygotic or compound heterozygotic mice with BRCA2 mutations seems dependent on mutation type and mouse strain.34,35 It would be of interest to determine whether mothers of patients with FA-D1 experience excessive fetal loss.
Are there genuine genotype/phenotype associations among patients who do seem to have bona fide deleterious mutations of both BRCA2 alleles? Short stature, microcephaly, abnormal thumbs, hyperpigmentation or café-au-lait spots and renal and gastrointestinal (GI) anomalies were reported frequently, and short stature, microcephaly and GI anomalies were more common in the FA-D1 group than in other patients with Fanconi anaemia.1 In the general population, the VATER association is >50 times more common than Fanconi anaemia (160 per 1 000 000 v 2.6/1 000 000 live births, respectively). It has been suggested that 5% of children in the VATER category may have Fanconi anaemia,36 and that 5% of patients with Fanconi anaemia have features consistent with the VATER association.24 The identification of five cases with VATER features among 27 patients (19%) in this series suggests that there may be a higher proportion of VATER association among patients with FA-D1 than among those with Fanconi anaemia overall (p = 0.01). However, the numbers are very small, and a VATER phenotype has also been reported in FA-A, C, E, F and G. There may be specific BRCA2 mutations that are linked with VATER association in the absence of a clinical diagnosis of Fanconi anaemia.
The risk of early onset of specific cancers is clear and dramatic in the FA-D1 group, in which the cumulative probability of leukaemia was 79% by age 10 years, that of a solid tumour was 83% by age 6.7 years, that of a brain tumour was 85% by age 9 years, that of a Wilms’ tumour was 63% by age 6.7 years (data not shown) and that of any malignancy was 97% by age 5.2 years. These probabilities are much higher than that we observed in genetically unclassified patients with Fanconi anaemia, and in our literature review,26,37 in which the cumulative probability of leukaemia reached a plateau of approximately 30% by age 30 years, and that of a solid tumour 25% by age 30 years and 75% by age 45 years. Thus, patients with FA-D1 have a distinctively higher risk of specific cancers than patients in other Fanconi anaemia complementation groups. In addition, the correlations between IVS7 with AML and both 886delGT and 6174delT with brain tumours suggest the possibility of specific genotype/cancer associations, although the numbers of cases and families reported to date are too small to draw firm conclusions.
The recent observation that all Fanconi anaemia groups, including FA-J, but excluding FA-D1, develop nuclear Rad51 foci after DNA damage29 underscores FA-D1’s distinctiveness. The aetiological role of BRCA2 mutations vis-à-vis the typical Fanconi anaemia birth defects or the specific cancers reported in these patients remains to be elucidated, although the associations with AML and brain tumours described above may be mutation-specific. Greater attention must be paid to the biological consequences of the specific BRCA2 mutations identified in this context, particularly as, by conventional criteria, several seem to be without known functional consequences. How do alleles, the importance of which would be dismissed in hereditary breast/ovarian cancer (HBOC), act to cause the unique phenotype observed in patients with FA-D1? Is there a protective interaction between a BRCA2 protein with a missense mutation and the normal BRCA2 protein, which is lost in patients with a truncated protein due to a deleterious mutation? Or might the recognition that some patients with FA-D1 have one missense mutation imply that such mutations, heretofore of “unknown” significance, are, in fact, biologically important?
A larger, more systematically ascertained series of patients with FA-D1 must be analysed to determine whether the severe clinical phenotype observed among the limited number of patients reported to date is truly characteristic of this Fanconi anaemia subgroup, or whether this represents biased ascertainment or reporting of patients with more severe disease. Most important, we must determine whether single copies of the presumably deleterious BRCA2 mutations detected in patients with FA-D1 also predispose to the HBOC spectrum of cancer in heterozygotic family members. Counselling those individuals is particularly difficult at present, as they are ascertained as members of families with Fanconi anaemia, rather than as members of HBOC kindreds. In addition, some of them have altered BRCA2 alleles that are currently of unknown clinical significance. The converse clinical presentation is also important: should women of child-bearing age from BRCA2-positive HBOC families be counselled regarding the theoretical risk of Fanconi anaemia in their offspring if their partners carried unrecognised truncating or even missense BRCA2 mutations? Such events are within the realm of possibility, particularly in specific population groups that have a higher prevalence of BRCA2 mutations (eg, 1% of unselected individuals of Ashkenazi Jewish descent carry the 6174delT founder mutation). The rarity of FA-D1 (∼3% of all Fanconi anaemia) suggests that a registry targeting patients with FA-D1 may be the most efficient means to deal with the many unanswered questions created by the unexpected nexus between the Fanconi anaemia and BRCA pathways.
Acknowledgments
We thank the astute geneticist, Markus Grompe, MD, for the diagnosis of Fanconi anaemia in our case, and assignment to group FANCD1, and Mark H Greene, MD for critical evaluation of the manuscript. We also thank the family of NCI 1 for their help and encouragement.
REFERENCES
Footnotes
-
Published Online First 6 July 2006
-
Funding: This research was supported in part by the Intramural Research Program of the National Cancer Institute of the National Institutes of Health.
-
Competing interests: None declared.