
Abstract
Hypoxic-ischemic injuries can evolve over several days, and recent studies suggest that further neuronal death may occur 6 to 72 h later. Because cerebral temperature is an important determinant of outcome during the primary injury, we investigated the effect of temperature, on outcome, during the later phases of injury. Hypoxic-ischemic injury was induced in 21-d-old rats by unilateral ligation of the right carotid artery followed by exposure to 15 min of hypoxia of 8% O2 at 34°C. Cerebral temperature changes were induced by modifying environmental temperature. The rats were divided into four treatment groups: group 1 (n = 15) remained at 34°C for 72 h; group 2 (n = 14) were kept at 34°C for 6 h and then at 22°C for the remaining 66 h; group 3 (n = 17) remained at 22°C for 6 h and 34°C for the next 66 h; group 4 (n = 16) remained at 22°C for 72 h. Rats kept at 22 or 34°C had cortical temperatures of 35.5 ± 0.1°C and 37.9 ± 0.2°C, respectively. Histologic outcome was assessed 72 h after hypoxia. The area of cortical infarction was reduced in group 4 compared with groups 1-3(p ≤ 0.05). Striatal damage was reduced in group 4 (p = 0.05). Hippocampal neuronal loss was not significantly altered. In a subsequent study the area of cortical infarction was 12.1 ± 3 mm2 in group 1 (n = 11) compared with 3.4 ± 1.5 mm2 group 4 treated rats (n = 10) 21 d after the injury(p < 0.01). Thus hypothermia spanning both the first 6 h and from 6 to 72 h after injury was needed to improve outcome. Conversely exposure to the thermoneutral environment exacerbated the injury. These observations suggest that prolonged moderate cerebral hypothermia can be used to suppress the cytotoxic processes that occur after hypoxic-ischemic injury.
Similar content being viewed by others

Main
The progressive events after HI injury to the developing brain are complex and dependent upon the nature and conditions of the insult. Both pathophysiologic and histopathologic data suggest that further neuronal death may occur many hours and even days after the primary injury(1, 2). Studies of the time course of high energy phosphate metabolites and of loss of membrane function suggest that there are two phases of injury. The first phase of injury is characterized by energy failure and occurs during the hypoxia-ischemia and is followed 12 to 48 h later by the second phase of energy failure in neonates(3) and piglets(4). Similarly there are primary and secondary phases of loss of membrane function in the parasagittal cortex of fetal sheep with the secondary phase developing from about 7 h after severe HI injury(5). Molecular evidence of delayed death may also be inferred from the time course of neuronal DNA degradation in the infant rat brain after HI injury(6). After severe HI injury in the rat, the DNA is intact at 5 h but is degraded in the striatum, cortex and in the hippocampus by 24 h. After moderate injuries, these processes are slowed and occur over a period of about 24 to 72 h(6).
The importance of cerebral temperature during and immediately after the injury in determining outcome has been highlighted(7–11). Hypothermia during the period of the insult has been shown to prevent or retard HI brain injury(10, 12–14). Conversely, mild hyperthermia during or immediately after an insult can exacerbate an injury(15). Hypothermia is thought to protect the brain by reducing metabolic demands and suppressing cytotoxic processes, such as excitatory amino acid accumulation(16) and free radical activity(17). However, it is uncertain as to whether the protective effect of hypothermia is restricted to the insult period or whether it extends into the period of delayed cell death. If the latter was the case, then this may be of therapeutic advantage. Therefore, we designed an experiment to examine the effects of prolonged environmental temperature modifications after a moderate HI injury during the period when delayed damage may occur.
MATERIALS
HI injury preparation. The experimental approach is based on the Levine(18) preparation and involves inducing a transient unilateral HI injury in the brain of an infant rat. This approach has been previously described(6, 19) and offers the advantage of allowing chronic instrumentation for monitoring cerebral temperature(19). The experimental protocol followed guidelines approved by the Animal Ethics Committee of the University of Auckland. Weaned 21-d-old Wistar rats of both sexes weighing between 40 and 49 g were used for the study. The rats were maintained on a 12-h cycle of light and dark with free access to food and water throughout the study. Rats were anesthetized with halothane, and the right common carotid artery was exposed through a midventral neck incision. The carotid artery was double ligated using 4 “0” silk, and the neck incision was closed. A randomized complete block design was used, and studies were performed in batches of litter mates of 8 to 12 infant rats. After surgery the rats were held for 1 h in a prewarmed infant incubator maintained at 34°C with >80% relative humidity, then exposed to 8% O2 for 15 min. After hypoxia the rats were kept caged in groups of four to eight until they were killed.
Study 1. Effect of temperature on histopathologic outcome. The effect of environmental temperature on histologic outcome was investigated in 62 rats. After the hypoxia, the rats were divided into four temperature treatment groups.
-
Group 1. Group 1 was held in an environment of 34°C for 72 h after the hypoxia (n = 15).
-
Group 2. Group 2 was held at 34°C for 6 h and then at 22°C for a further 66 h (n = 14).
-
Group 3. Group 3 was held at 22°C for 6 h and then at 34°C for 66 h (n = 17).
-
Group 4. Group 4 was held at 22°C for 72 h after the hypoxia(n = 16).
Study 2. Effect of temperature regime on long term histopathologic outcome. At the end of the hypoxia, the rats were divided into two groups corresponding to group 1 (34°C for 72 h, n = 11) and group 4(22°C for 72 h, n = 10) of study 1. Subsequently both groups of rats were maintained at 22°C for the next 19 d.
Study 3. Effect of environment on cortical temperature. Cortical temperature was recorded in a separate group of rats (n = 13) using telemetric temperature probes (XM-FH-BP, Mini Mitter Co., Inc., Sunriver, OR). For the cortical temperature measurements the thermistors were positioned on the dura 2 mm anterior to the ear bars(20) and 3 mm lateral to the midline and fixed in place with dental cement. The thermistors were calibrated in the 22°C environment at 34 and 40°C in a precision water bath. The measurements made in the 34°C environment were corrected by -0.4°C to compensate for the systematic error due to the effect of the 34°C environmental temperature on the probe body. Temperature was continuously recorded during the experiments and averages were calculated and stored to disk at 1-min intervals(21). Histologic outcome was not determined in these rats. Cortical temperature measurements were made over 72 h after hypoxia in ligated rats for each of the temperature regimes (see Table 1 and Fig. 2, a-d). Cortical temperature measurements were also made in a group of sham controls(n = 3) that were not ligated but exposed to hypoxia then kept at 22°C (see Table 1 and Fig. 2e).

The effect of environmental temperature upon cortical temperature. (a) Rats maintained at 34°C for 72h (n = 2). (b) Rats maintained at 34°C for 6 h then 22°C for 66 h(n = 3). (c) Rats maintained at 22°C for 6 h then 34°C for 66 h (n = 3). (d) Rats maintained at 22°C for 72 h (n = 2). (e) Sham-ligated rats maintained at 22°C for 72 h (n = 3).
Histologic preparation. The brains of the rats that died after the hypoxia were processed for histology only if they were collected within 6 h of death. Otherwise all rats were euthanized with sodium pentobarbitone 72 h or 21 d after hypoxia for studies 1 and 2, respectively. The brains were fixedin situ by transcardial perfusion with a 0.9% saline solution followed by 10% neutral buffered formalin. The fixed brains were sliced into 2-mm thick coronal sections using a rat brain matrix (RBM-2000C, Activational Systems, Inc., Warren, MI), dehydrated through graded alcohols, and embedded in paraffin. Serial 4-μm sections were cut and stained with thionine-acid fuchsin as described previously(19).
Histologic analysis. The quantification of histologic damage was carried out by a researcher blinded to the treatment protocols. The live and dead neurons were discriminated by the respective absence or presence of acidophilia in the cytoplasm as previously described(22–25). Light microscopy on these sections was carried out at 40×, 100×, and 400×. Only the extent of infarction was analyzed after study 2.
Cortex. The cortical damage was evaluated by measurement of the area of pannecrosis using an image analyzer (Java, Jandel Scientific, San Rafael, CA). The coronal brain slices analyzed were taken from between 2.9 and 3.5 mm anterior to the ear bars. The indirect technique was used(26), and outcome was calculated as follows:Equation where RI = infarct area of right ligated hemisphere, LT = total cortex area of nonligated (left) hemisphere, and RN = total area of viable cortex of the ligated (right) hemisphere in the same slide. These infarct measurements were taken from three slices in each brain. The measurement with the largest area was used for subsequent statistical analysis(26).
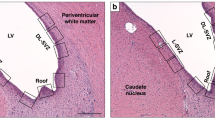
Hippocampus. The damage was measured by counting the neurons surviving in the CA1-CA3 regions of the dorsal hippocampus of both hemispheres (Fig. 1). These counts were done in a single slide of a coronal section taken from between 2.9 and 3.5 mm anterior to the ear bars. The percentage of surviving hippocampal neurons was calculated by using the following formula:

Schematic diagram of areas analyzed for histopathologic changes.
Striatum. Damage within the striatum was determined in coronal sections taken from between 6.2 and 7 mm anterior to the ear bars. The severity of striatal neuronal loss was assessed using a four-point scale as reported elsewhere(27). Grade 0, no neuronal loss; grade 1, between 1 and 5% neuronal loss; grade 2, between 6 and 50% neuronal loss; and grade 3, between 51 and 100% neuronal loss.
Statistical analysis. All data are presented as mean ± SEM. The histologic data in study 1 were compared nonparametrically by applying ANOVA on the rank-transformed data followed by Student-Newman-Keulspost hoc tests. The incidence of cortical infarction was compared using the χ2 test with the Bonferroni correction for multiple comparisons. The area of cortical infarction in study 2 was compared by Mann Whitney U test. Cortical temperature measurements were compared byt test (Sigmastat, Jandel Scientific, San Rafael, CA).
RESULTS
Temperature. The results of the temperature recording data are described in Table 1 and Figure 2. During the period 0-6 h after the HI, all rats (n = 5) kept at 22°C had a lower cortical temperature of 35.5 ± 0.1°C compared with 37.9 ± 0.2°C for all those (n = 5) kept at 34°C(p < 0.01). A similar trend was recorded during the period from 6-72 h and those kept at 22 or 34°C had cortical temperatures of 35.4± 0.2°C and 37.9 ± 0.4°C, respectively (p < 0.01).
Study 1. Histologic outcome. Three group 3 rats were excluded from analysis as their brains could not be collected within 6 h of death. These rats died between 24 and 48 h after hypoxia.
Cortex. The rats exposed to the 34°C environment for either the first 6 h or subsequently (groups 1-3) had an increased incidence and area of cortical infarction compared with group 4 rats that were kept at 22°C for the period 0-72 h (p < 0.05) (Table 2, Fig. 3, a and b). The extent of cortical infarction was 6.5 ± 1.7 mm2 in group 1, 4.5 ± 1.8 mm2 in group 2, 5.1 ± 1.5 mm2 in group 3, and 0.3 ± 0.3 mm2 in group 4. In the group 1 rats, the coronal sections showed shrinkage of cortex within the ligated hemisphere (Fig. 4a). The neuronal damage was apparent as a loss of viable neurons and the presence of acidophilic and pyknotic neurons. Most rats had laminar necrosis with loss of cortical neurons in the more superficial layers (layers 1-3) of the cortex. This neuronal loss extended through all the layers of the cortex in the more severely affected rats (Fig. 4a). The extracellular matrix was vacuolated and uneven, and there were reactive glia and an abundance of dilated capillaries. These findings are consistent with pannecrosis or infarction (Fig. 4b). The histologic outcome in groups 2 and 3 rats were similar to that of group 1 rats but with a lesser degree of severity. In contrast the coronal sections from the ligated hemisphere of group 4 rats did not show these gross morphologic changes (Fig. 5a). The histopathologic changes in the cortex generally consisted of selective neuronal loss predominantly occurring in cortical layers 4 and 5 within the middle cerebral artery territory of the lateral cortex. There were no gross histologic changes to the extracellular matrix (Fig. 5b).

The effect of environmental temperature on histologic outcome after the HI injury. (a) Incidence of cortical infarction(group 4 p < 0.05). (b) Area of cortical infarction(group 4 p < 0.05). (c) Number of surviving CA1-CA3 hippocampal neurons. (d) Striatal loss (group 4 p = 0.05).(e) Area of cortical infarction from rats kept 21 d (p< 0.01).

Photomicrographs of a representative brain from a rat kept at 34°C from 0 to 72 h after hypoxia (group 1). (a) Coronal section showing loss of definition between layer 1 (molecular layer) and layer 2 of the cortex (solid arrow) due to shrinkage resulting from infarction and presence of dilated capillaries (arrowheads). Bar = 1 mm. (b) High power photomicrograph of the cortex (boxed area) taken from (a). Examples of pyknotic neurons(arrows). The extracellular matrix is vacuolated and shows increased vascularization. An increased glial reaction (solid arrows) is evident around some of the pyknotic neurons. Thionine-acid fuchsin(400×). Bar = 100 μm.

Photomicrographs of a representative brain from a rat kept at 22°C from 0 to 72 h after the hypoxia (group 4). (a) Coronal section showing loss of hippocampal Nissl staining confined to the ligated hemisphere but no other gross changes to the cortical morphology. Bar= 1 mm. (b) High powered photomicrograph of the injured cortex(boxed area from a) showing several pyknotic neurons(arrows) scattered between a population of viable cortical neurons. The extracellular matrix appears normal. Thionine-acid fuchsin (400×). Bar = 100 μm.
Hippocampus. Environmental temperature did not significantly alter neuronal loss within CA1-3 region of the hippocampus, although there was a trend to reduced loss in group 4 (Fig. 3c). At low magnifications, the hippocampal damage in whole brain sections appeared as a unilateral loss of neuronal Nissl staining. The damaged neurons within the hippocampus were acidophilic and pyknotic. Compared with the group 4 rats, group 1 rats had increased gliosis and vacuolation in areas adjacent to CA1 region.
Striatum. The temperature treatments modified the severity of damage within the striatum and the group 4 rats maintained at 22°C had less damage (p = 0.05) (Fig. 3d). Injured striatal neurons were pyknotic or acidophilic, and the severity of injury in groups 1, 2, and 3 appeared to be similar.
Study 2. Three group 1 rats were excluded from analysis because their brains were not collected within 6 h of death. The rats exposed to the 34°C environment for 72 h after the hypoxia had an increased area of cortical infarction (12.1 ± 3 mm2) compared with those kept at 22°C (3.4 ± 1.5 mm2) for the period 0-72 h (p < 0.01) when assessed 21 d after the injury (Fig. 3e). In the former group, the coronal sections showed shrinkage of cortex within the ligated hemisphere. Rats in group 1 were lighter at 1 but not 3 wk postinjury(p < 0.01) (Table 3).
DISCUSSION
The results of this study suggest that moderate reductions in cerebral temperature of about 2°C throughout the period 0-72 h after HI injury can markedly reduce the severity of damage. However, reducing cerebral temperature for either the first 6 h or from 6 to 72 h did not significantly improve outcome, although there was a trend for cooling during the early and late neuronal injury phases to reduce cortical neuronal loss. These data suggest that prolonged reductions in cerebral temperature initiated after the insult itself are necessary to improve outcome.
Moderate hypothermia of about 2-3°C during hypoxic-ischemic injury is clearly neuroprotective in the mature and developing brain(28, 29). A recent study in adult gerbils showed that the efficacy of postischemic hypothermia is dependent on the severity of the primary insult. Twelve hours of hypothermia increased the number of surviving hippocampal neurons with more neurons surviving after 3 compared with 5 min of ischemia. Furthermore, there was a lesser percentage reduction in neuronal loss after the longer period of ischemia. It was further observed that increasing the duration of the hypothermia from 12 to 24 h produced much greater and long-term protection of hippocampal neurons after the 5 min of ischemia(30). A similar trend toward reduced loss of hippocampal neurons was observed when the duration of hypothermia was raised from 30 min to 5 h, starting 2 h after 10 min of ischemia in adult rats(31). In contrast a shorter duration (3 h) of hypothermia after 10 min of ischemia in adult rats(10) was not protective. Similarly 3 h of hypothermia after a prolonged (3 h) HI injury in 7-d-old rats(29) did not provide long-term protection. In the present study, 3 d of moderate hypothermia markedly reduced the extent of cerebral damage after a brief injury. Our results are compatible with observations in the mature brain that suggest prolonged hypothermia after HI injury provides a greater neuroprotective effect.
The cortex and striatum, but not the hippocampus, showed less damage when the rats were maintained at the lower temperature (Fig. 3,b-e). A similar regional neuroprotective effect occurs when moderate hypothermia is applied during ischemia in piglets(28). Given the superficial location of the cortex, it will have cooled to a greater degree due to conduction of heat through the skull and scalp(32). In contrast, the temperature of deeper brain structures is primarily determined by core temperature, particularly in species larger than rats(32). Work by others(28, 29) indicates that core temperature falls to a lesser degree than the cortex; however, the magnitude of this difference is likely to be influenced by the size of the species(32). Presumably the deeper structures are influenced to a lesser degree by the environmental temperature changes. This difference may partially explain the lack of significant protection in the hippocampus compared with the cortex, although it still does not clearly account for the more significant response in the striatum. Possibly the hippocampus was more severely injured or that different mechanisms were involved in the hippocampal neuronal death. A greater reduction in core temperature of at least 3-4°C might be necessary to achieve significant protection in deeper structures such as the hippocampus(31).
Infarction apparent as laminar necrosis of the cortex was seen in rats maintained in the warm environment (Fig. 4). This pattern of damage is similar to that seen in late gestation fetal sheep after an ischemic injury in utero(33). These cortical infarcts are associated with the development of seizures(33, 34). Thus it is possible that the increased cerebral temperature that occurs in the fetal environment exacerbates encephalopathies resulting from HI injuries occurring in utero.
The mechanisms causing delayed damage have not been clearly identified. The cascade of processes triggered by the primary injury can lead to secondary energy failure(35) and further neuronal death(6) in the developing brain. Apoptosis(6), excitoxicity(33), seizure activity(33), free radicals(36), and microglial reactions(6) may be involved in this cascade. Hypothermia is likely to interfere with several of these cytotoxic processes and can, at least around the period of the primary injury, reduce accumulation of excitatory amino acids(16), phospholipid degradation(37), the actions of free radicals(17), and cerebral metabolism. Clearly a difference of about 2°C can strongly modulate the cytotoxic mechanisms leading to cortical infarction. This effect may have important implications for some pharmacologic neuronal rescue studies because it is possible that some therapeutic agents may alter outcome by inducing moderate hypothermia. Surprisingly moderate hypothermia during either the first 6 h or from 6 to 72 h, when much of the neuronal death occurs in this preparation(6), did not significantly alter outcome. Only prolonged hypothermia was effective, suggesting that there is a critical period preceding and during the period of delayed neuronal death when cytotoxic processes need to be suppressed. In contrast to our results, Coimbra and Wieloch(31) observed that a delayed period (5 h) of cooling initiated at 6 and 12 h after 10 min ischemia in adult rats reduced the loss of hippocampal but not striatal neurons. This difference may be due to the use of lower temperatures (33°C) in the study by Coimbra and Wieloch(31). Alternatively regional differences in the rate of cell death(6, 31) or differences in neurologic maturation and the nature of the injury could account for this disparity.
The observations in this study may have important ramifications. Clinical studies suggest that sick premature newborn infants maintained in a warmer environment display a lower mortality(38–40). Thus current practice aims at maintaining thermoneutrality in all babies. However, this study suggests that exposure to a thermoneutral environment in the hours after HI injury can markedly worsen neurologic outcome (Fig. 3). In contrast, prolonged and mild cerebral hypothermia of about 2°C was protective. In terms of neurologic development, the rat preparation used in these studies is more relevant to the term or more mature infant(19). These findings raise questions regarding the routine management of temperature in asphyxiated term newborn infants. Further investigations are needed to evaluate the relevance of these observations.


Abbreviations
- HI:
-
hypoxic ischemic
References
Pulsinelli WA, Brierley JB, Plum F 1982 Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol 11: 491–498.
Kirino T 1982 Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239: 57–69.
Azzopardi D, Wyatt JS, Cady EB, Delpy DT, Baudin J, Stewart AL, Hope PL, Hamilton PA, Reynolds EO 1989 Prognosis of newborn infants with hypoxic-ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr Res 25: 445–451.
Lorek A, Takei Y, Cady EB, Wyatt JS, Penrice J, Edwards AD, Peebles D, Wylezinska M, Owenreece H, Kirkbride V, Cooper CE, Aldridge RF, Roth SC, Brown G, Delpy DT, Reynolds EOR 1994 Delayed(secondary) cerebral energy failure after acute hypoxia-ischemia in the newborn piglet: continuous 48-hour studies by phosphorus magnetic resonance spectroscopy. Pediatr Res 36: 699–706.
Williams CE, Gunn AJ, Gluckman PD 1991 The time course of intracellular edema and epileptiform activity following prenatal cerebral ischemia in sheep. Stroke 22: 516–521.
Beilharz EJ, Williams CE, Dragunow M, Sirimanne E, Gluckman PD 1995 Mechanisms of delayed cell death following hypoxic-ischemic injury in the immature rat: evidence for apoptosis during selective neuronal loss. Mol Brain Res 29: 1–14.
Busto R, Dietrich WD, Globus MY, Ginsberg MD 1989 The importance of brain temperature in cerebral ischemic injury. Stroke 20: 1113–1114.
Busto R, Dietrich WD, Globus MY, Ginsberg MD 1989 Postischemic moderate hypothermia inhibits CA1 hippocampal ischemic neuronal injury. Neurosci Lett 101: 299–304.
Buchan A, Pulsinelli WA 1990 Hypothermia but not theN-methyl-D-aspartate antagonist, MK-801, attenuates neuronal damage in gerbils subjected to transient global ischemia. J Neurosci 10: 311–316.
Dietrich WD, Busto R, Alonso O, Globus MY, Ginsberg MD 1993 Intraischemic but not postischemic brain hypothermia protects chronically following global forebrain ischemia in rats. J Cereb Blood Flow Metab 13: 541–549.
Boris-Moller F, Smith M-L, Siesjo BK 1989 Effects of hypothermia on ischemic brain damage: a comparison between preischemic and postischemic cooling. Neurosci Res Commun 5: 87–94.
Busto R, Dietrich WD, Globus MY, Valdes I, Scheinberg P, Ginsberg MS 1987 Small differences in intra ischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab 7: 729–738.
Dietrich WD, Busto R, Valdes I, Loor Y 1990 Effects of normothermic versus mild hyperthermic forebrain ischemia in rats. Stroke 21: 1318–1325.
Green EJ, Dietrich WD, Van Dijk F, Busto R, Markgraf CG, McCabe PM, Ginsberg MD, Schneiderman N 1992 Protective effects of brain hypothermia on behavior and histopathology following global cerebral ischemia in rats. Brain Res 580: 197–204.
Ginsberg MD, Sternau LL, Globus MY, Dietrich WD, Busto R 1992 Therapeutic modulation of brain temperature: relevance to ischemic brain injury. Cerebrovasc Brain Metab Rev 4: 189–225.
Illievich UM, Zornow MH, Choi KT, Scheller MS, Strnat MAP 1994 Effects of hypothermic metabolic suppression on hippocampal glutamate concentrations after transient global cerebral ischemia. Anesth Analg 78: 905–911.
Karibe H, Chen SF, Zarow GJ, Gafni J, Graham SH, Chan PH, Weinstein PR 1994 Mild intraischemic hypothermia suppresses consumption of endogenous antioxidants after temporary focal ischemia in rats. Brain Res 649: 12–18.
Levine S 1960 Anoxic encephalopathy in rats. Am J Pathol 36: 1–17.
Sirimanne ES, Guan J, Williams CE, Gluckman PD 1994 Two models for determining the mechanisms of damage and repair after hypoxic-ischemic injury in the developing rat brain. J Neurosci Methods 55: 7–14.
Sherwood Nancy M, Timiras Poala S 1970 A Stereotaxic Atlas of the Developing Rat Brain. University of California Press, Berkeley, Los Angeles, pp 5–12.
Dale PS, Ducsay CA, Gilbert RD, Koos BJ, Longo LD, Power GG 1989 A microcomputer program for real time data acquisition in the perinatal physiology laboratory. J Dev Physiol 11: 185–188.
Smith ML, Auer RN, Siesjo BK 1984 The density and distribution of ischemic brain injury in the rat following 2-10 min of forebrain ischemia. Acta Neuropathol Berl 64: 319–332.
Auer RN, Kalimo H, Olsson Y, Siesjo BK 1985 The temporal evolution of hypoglycemic brain damage. I. Light- and electron-microscopic findings in the rat cerebral cortex. Acta Neuropathol 67: 13–24.
Auer RN, Kalimo H, Olsson Y, Siesjo BK 1985 The temporal evolution of hypoglycemic brain damage. II. Light-and electron-microscopic findings in the hippocampal gyrus and subiculum of the rat. Acta Neuropathol 67: 25–36.
Guan J, Williams CE, Gunning M, Mallard EC, Gluckman PD 1993 The effects of IGF-1 treatment after hypoxic-ischemic brain injury in adult rats. J Cereb Blood Flow Metab 13: 609–616.
Lin T-N, He TT, Wu G, Khan M, Hsu CY 1993 Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke 24: 117–121.
Lundgren J, Smith ML, Siesjo BK 1992 Effects of dimethylthiourea on ischemic brain damage in hyperglycemic rats. J Neurol Sci 113: 187–197.
Laptook AR, Corbett RJT, Sterett R, Burns DK, Tollefsbol G, Garcia D 1994 Modest hypothermia provides partial neuroprotection for ischemic neonatal brain. Pediatr Res 35: 436–442.
Yager J, Towfighi J, Vannucci RC 1993 Influence of mild hypothermia on hypoxic-ischemic brain damage in the immature rat. Pediatr Res 34: 525–529.
Colbourne F, Corbett D 1994 Delayed and prolonged post-ischemic hypothermia is neuroprotective in the gerbil. Brain Res 654: 265–272.
Coimbra C, Wieloch T 1994 Moderate hypothermia mitigates neuronal damage in the rat brain when initiated several hours following transient cerebral ischemia. Acta Neuropathol 87: 325–331.
Dexter F, Hindman BJ 1994 Computer simulation of brain cooling during cardiopulmonary bypass. Ann Thorac Surg 57: 1171–1178.
Tan WKM, Williams CE, Gunn AJ, Mallard CE, Gluckman PD 1992 Suppression of postischemic epileptiform activity with MK-801 improves neural outcome in fetal sheep. Ann Neurol 32: 677–682.
Williams CE, Gunn AJ, Gluckman PD, Synek B 1990 Delayed seizures occurring with hypoxic-ischemic encephalopathy in the fetal sheep. Pediatr Res 27: 561–565.
Mehmet H, Yue X, Squier MV, Lorek A, Cady E, Penrice J, Sarraf C, Wylezinska M, Kirkbride V, Cooper C, Brown GC, Wyatt JS, Reynolds EOR, Edwards AD 1994 Increased apoptosis in the cingulate sulcus of newborn piglets following transient hypoxia-ischaemia is related to the degree of high energy phosphate depletion during the insult. Neurosci Lett 181: 121–125.
Palmer C, Towfighi J, Roberts RL, Heitjan DF 1993 Allopurinol administered after inducing hypoxia-ischemia reduces brain injury in 7-day-old rats. Pediatr Res 33: 405–411.
Haun SE, Trapp VL, Horrocks LA 1993 Hypothermia decreases astroglial injury and arachidonate release during combined glucose-oxygen deprivation. Brain Res 631: 352–356.
Silverman WA, Fertig JW, Berger PA 1958 The influence of the thermal environment upon the survival of newly born premature infants. Pediatrics 31: 876–885.
Buetow KC, Klein SW 1964 Effect of maintenance of normal skin temperature on survival of infants of low birthweight. Pediatrics 34: 163–170.
Day RL, Caliguiri L, Kamenski C, Ehrlich F 1964 Body temperature and survival of premature infants. Pediatrics 34: 171–181.
Acknowledgements
The authors thank Carina Mallard for her help and guidance in the preparation of the manuscript.
Author information
Authors and Affiliations
Additional information
Supported by grants provided by the New Zealand Neurologic Foundation and the New Zealand Health Research Council.
Rights and permissions
About this article
Cite this article
Sirimanne, E., Blumberg, R., Bossano, D. et al. The Effect of Prolonged Modification of Cerebral Temperature on Outcome after Hypoxic-Ischemic Brain Injury in the Infant Rat. Pediatr Res 39, 591–597 (1996). https://doi.org/10.1203/00006450-199604000-00005
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199604000-00005
This article is cited by
-
Does sex materially modulate responses to therapeutic hypothermia?
Pediatric Research (2023)
-
Future perspectives of cell therapy for neonatal hypoxic–ischemic encephalopathy
Pediatric Research (2018)
-
Therapeutic hypothermia translates from ancient history in to practice
Pediatric Research (2017)
-
Comparison of Three Hypothermic Target Temperatures for the Treatment of Hypoxic Ischemia: mRNA Level Responses of Eight Genes in the Piglet Brain
Translational Stroke Research (2013)
-
Safety and efficacy of topiramate in neonates with hypoxic ischemic encephalopathy treated with hypothermia (NeoNATI)
BMC Pediatrics (2012)
