
Abstract
In a previous study we showed that pneumococcal adherence to epithelial cells was enhanced by a preceding respiratory syncytial virus (RSV) infection. RSV-glycoproteins, expressed on the infected cell surface, may play a role in this enhanced pneumococcal binding, by acting as bacterial receptors. In the current study, it was attempted to analyze the capacity of pneumococci to interact directly with RSV virions. By flow-cytometry, a direct interaction between RSV and pneumococci could be detected. Heparin, an inhibitor of RSV infectivity that interacts with RSV protein-G, blocked RSV-pneumococcal binding, indicating that the latter interaction is indeed mediated by protein-G. RSV-pneumococcal complexes showed enhanced adherence to uninfected human epithelial cells, compared with pneumococcal adherence without bound RSV, and this enhancement was also blocked by heparin. In addition, the significance of these findings in vitro was explored in vivo in a murine model. Both mice that were pretreated with RSV at day 4 before pneumococcal challenge and mice infected with both agents simultaneously showed significantly higher levels of bacteraemia than controls. Simultaneous infection with both agents enhanced the development of pneumococcal bacteraemia most strongly. It was hypothesized that direct viral binding is another mechanism by which RSV can induce enhanced pneumococcal binding to epithelial cells, a phenomenon that is translated in vivo by a higher invasiveness of pneumococci when administered simultaneously with RSV to mice. Apparently, RSV acts in this process as a direct coupling particle between bacteria and uninfected epithelial cells, thereby increasing colonization by and enhancing invasiveness of pneumococci.
Similar content being viewed by others

Main
Clinical and epidemiologic data suggest that respiratory syncytial virus (RSV) infections in humans can be complicated by bacterial superinfection e.g., with Streptococcus pneumoniae, leading to increased morbidity (1–5). Mechanisms underlying bacterial superinfection include virus-induced local destruction of the epithelium, compromising the host's physiologic barrier, and virus-induced modulation of the immune response (6,7).
In addition, enhanced bacterial adherence to virus-infected cells is considered an important factor increasing the risk of bacterial superinfection (8–11). In a previous study in vitro, we obtained evidence for such a mechanism. A preceding RSV infection of human respiratory tract epithelium led to significantly enhanced adherence of S. pneumoniae (12).
The basis of RSV-enhanced pneumococcal adherence is not known. RSV infection both leads to expression of viral glycoproteins and up-regulation of cellular molecules on the host-cell membrane. Both could possibly serve as bacterial receptors, as has been described for Neisseria meningitidis: viral glycoprotein G as well as cellular molecules, like CD 14 and CD18, are involved in enhanced binding of N. meningitidis to RSV-infected cells (13,14).
The current study was designed to analyse the possibility that RSV glycoproteins directly interact with pneumococci. Therefore, we tested the binding of RSV to pneumococci and characterized this binding by studying the involvement of several bacterial components. We show here that RSV is capable of direct binding to pneumococci and provide evidence for the involvement of RSV glycoprotein G in this interaction. Finally, we explored the demonstrated synergism between RSV and S. pneumoniae in relation to epithelial cell binding as found in vitro also in vivo in a murine model. To this purpose, we studied the effect of a preceding or concurrent nasal infection with RSV on the kinetics of pneumococcal bacteraemia in mice after respiratory challenge with pneumococci.
MATERIALS AND METHODS
Bacteria.
Clinical pneumococcal isolates, serotypes 3, 9, 14, 18, 19, and 23, were a kind gift of dr. C. Neeleman, Intensive Care Department of the University Hospital of Nijmegen, The Netherlands. In addition, for the in vivo experiments a clinical isolate serotype 1 was kindly provided by dr Eirikur Saeland, Department of Molecular Cell Biology, Free University Medical Center, Amsterdam, the Netherlands. The bacteria were stored in micro-banks at −70°C (Pro-Lab Diagnostics, Austin, Texas). Wild-type S. pneumoniae serotype 3 strain WU2 and its pneumococcal surface protein A (PspA) deleted mutant JY 1123 (15) were kindly provided by Dr. L. S. McDaniel (Birmingham, Alabama). Unencapsulated mutant S. pneumoniae strain DW3.8, generated by conjugative transfer of transposon Tn916 from donor strain Enterococcus faecalis CG110 to the genome of WU2, was a gift by Dr. D. Watson and Dr. D. Musher (Veterans Affairs Medical Center, Houston, TX, USA) (16). The strains used are listed in Table 1.
Before testing, an aliquot of stored bacteria was transferred from a micro-bank bead to blood-agar plates (trypticase soy agar with 5% sheep blood, Becton Dickinson Company, Heidelberg, Germany) and was incubated overnight at 37°C in a CO2 incubator. The next day, the bacteria were transferred to Todd-Hewitt broth (Difco, Detroit, MI, USA), supplemented with 0.5% yeast extract (THY) and grown by static culture to logarithmic phase (t = 6.5 h) at 37°C in a CO2 incubator. This culture was spun at 3,270xg (Centrifuge GP8R, International Equipment Company (IEC) Milford, USA) for 15 min. Pelleted bacteria were washed twice in PBS (PBS) alternated by centrifugation at 9,300×g during 5 min (Eppendorf centrifuge micromax, International Equipment Company, Milford, USA).
For all in vitro experiments heat-killed (30 min, 56°C) bacteria were used. For cytometric adherence experiments, pelleted, heat-killed bacteria were labelled with saturated FITC (FITC) solution, 0.5 mg/mL in PBS (Sigma Chemical Co., St. Louis, MO, USA), and incubated during one hour at 4°C, whereafter they were washed thrice with PBS. In fluorescence activated cell sorter (FACS) analysis experiments unlabelled, heat-killed pneumococci were used. Finally, all bacterial samples were stored at −20°C until use. Before testing, the bacteria were suspended in PBS2+/BSA (BSA) 2.5% (Ca2+ 0.15 mM, Mg2+ 0.5 mM enriched with 2.5% BSA (albumin bovine fraction V, pH 7.0 Serva, Instruchemie, Delfzijl, The Netherlands) to avoid nonspecific binding. The bacteria were further adjusted in a spectrophotometer to a concentration of 108 bacteria/mL (??? OD (OD) 660 nm = 0.1).
For the preparation of viable log phase S. pneumoniae stocks, for the use in the in vivo infection experiments, a microbank bead was transferred to blood-agar plates (trypticase soy agar with 5% sheep blood, Becton Dickinson Company, Heidelberg, Germany) incubated overnight at 37°C in a CO2 incubator. The next day, the bacteria were inoculated in THY and grown by static culture to logarithmic phase for 6,5 h at 37°C in a CO2 incubator. The culture was centrifuged at 3270×g (Centrifuge GP8R, International Equipmant Company, Milford, USA) during 15 min and pelleted bacteria were resuspended and concentrated by adding a small volume of THY containing 10% glycerol. After resuspension, the bacteria were aliquoted for unique use in 150 μL vials, by freezing them instantaneously in liquid nitrogen after which they were stored at −70°C until use. Inoculation densities were determined by making serial dilutions in sterile saline, that were plated on blood agar plates to express colony-forming units (CFU) after overnight incubation at 37°C in a CO2 incubator.
Respiratory syncytial virus.
RSV serotype A (American Type Culture Collection, ATCC VR1302, Manassas, VA, USA) was kindly provided by dr. A. Brandenburg (Department of Clinical Virology, University Medical Center Erasmus, Rotterdam, The Netherlands). An RSV dilution of 1:1000 in 70 mL Iscove's Modified Dulbecco's Medium (IMDM, GIBCO, Paisley, UK) supplemented with hepes (25 mM), 5% fetal calf Serum (HyClone, Utah, USA) and gentamicin 0.01 mg/mL (GibcoBRL, Life Technologies, Paisley, UK) was added to monolayers of HEp-2 cells. Regularly, at day 4 postRSV infection cytopathologic effects (CPE) were detected by microscopic examination and supernatant was replaced by fresh IMDM. After 3 h of incubation at 37°C, supernatant containing RSV was harvested and filtered through a 0.22 μm filter. Plaque-forming units (PFU) were counted by the PFU assay according to Dulbecco (17). Stock aliquots were stored at −180°C in liquid N2, containing about 2.4 × 107 PFU/mL.
Cell culture.
HEp-2 human nasopharyngeal carcinoma cells (ATCC: CCL-23, Manassas, VA, USA) and A549 human pneumocyte type II carcinoma cells (ATCC: CCL-185) were used throughout the experiments. Stocks of cellular suspensions were stored at −180°C in liquid nitrogen.
Tissue culture flasks (Costar, Cambridge, MA, USA), confluently grown with HEp-2 or A549, were washed with PBS. A film of trypsin (GIBCO BRL, Paisley, UK) was added and the mixtures were incubated approximately 10 min at 37°C to detach the cells. Subsequently, IMDM was added. The cell suspension was diluted to approximately 2 × 105/mL and 100 μL was seeded in all wells of 96-well plates. The microtiter plates were incubated at 37°C to let them form confluent monolayers in two days.
Preincubation of pneumococci with RSV.
One ml of a suspension of heat-killed, FITC-labeled or unlabeled pneumococci (1 × 108/mL = OD 0.1) was spun at 9,300 × g for 3 min. Supernatant was removed and 400 μL undiluted RSV, heat-inactivated RSV (30 min at 56°C), or PBS/BSA 0.5% (controls) was added. These suspensions were incubated for 30 min at 37°C. Subsequently, suspensions were alternately centrifuged and washed with PBS2+ twice. Finally, RSV-pretreated or PBS/BSA-pretreated pneumococci were resuspended in 1 mL PBS2+/BSA 2.5%.
Cytometric fluorescence adherence assay.
The assay was adapted from a cytofluor-assay described by Tamura et al. (18). Medium was removed from monolayers of HEp-2 or A549 cells in 96-tissue culture plates. Depending on the experiment, 50 μL of FITC- labeled bacteria or 50 μL RSV-pretreated pneumococci were added. To study the effect of heparin, a solution of increasing concentration of sodium heparin (0.1, 1, 10, 100 IE/mL) was added to the monolayers before adding the bacteria. Monolayers were incubated for 30 min at 37°C in a CO2 incubator. Next the medium was removed and subsequently bacteria were added as described above.
The plates were spun at 160×g for 10 min in a plate spinner (IEC centra- 3C, International Equipment Company, Milford, USA) to allow the pneumococci to approximate the monolayers. Next, the plates were incubated at 21°C for 30 min. Total fluorescence was measured by using a fluorescence multi-well plate reader (CytoFluor II ™, PerSeptive Biosystems Inc., Framingham, MA, USA) with an excitation wave length of 485 nm and an emission filter of 530 nm, gain setting 54, readings per well: 3. Subsequently, the wells were washed four times with PBS2+ to remove unbound bacteria. After the addition of 50 μL PBS2+, final fluorescence was measured. Adherence percentages were calculated by: final/total fluorescence × 100.
Analysis of pneumococcal-RSV interaction by flow-cytometry.
During 5 min, a bacterial suspension of unlabeled, heat-killed pneumococci was spun at 9,300 × g. Next, undiluted RSV stock suspension or PBS/BSA 0.5% as a control, were added to the pelleted bacteria in a ratio of 1:2.5. These mixtures were incubated during 30 min at 37°C in a CO2 incubator. Subsequently, 1.5 mL PBS2+/BSA 0.5% was added to the suspensions that were alternately centrifuged at 9,300 × g and washed twice. Following the last wash, 25 μL of conjugate-containing MAb directed against RSV-glycoprotein F with FITC label (Imagen kit for RSV, Dakocytomation, Glostrup, Denmark) was incubated with pneumococci during 30 min at 4°C. Suspensions were twice washed again, to remove unbound antibodies, whereafter pellets were resuspended in PBS. Fluorescence per cell was analyzed in a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). Per teste sample ten thousand bacterial cells were analyzed. The fluorescence of test samples was compared with the fluorescence of control samples by setting a marker at the end of the auto-fluorescence peak of the controls. Fluorescence beyond the marker was attributed to RSV particles attached to the pneumococci. In the figures, percentages of positive pneumococci are depicted.
Mice.
Female BALB/c mice of 8–12 wk old were obtained from Harlan (Horst, The Netherlands). Groups of mice were housed in micro-isolator cages and received commercial food and water ad libitum.
Mouse pneumococcal challenge.
Mice were anesthetized by a mixture of halothane and oxygen inhalation and exposed to 50 μL S. pneumoniae serotype 1 stock inoculum, by intranasal inoculation of the bacterial suspension applied dropwise to the nares. For the experiments in combination with the RSV preand co-infection, concentrations of 3 × 105 and 1 × 106 CFU/mouse were administered.
Mice inoculation with RSV.
Mice were anesthetized as described above, and exposed to 50 μL of the RSV stock suspension by intranasal inoculation of the RSV suspension applied dropwise to the nares. The control mice received 50 μL sterile saline (i.n.), as described before. RSV treatment was done 4 d before the pneumococcal challenge. In addition, a simultaneous infection of RSV and pneumococci was performed.
Bacteremia evaluation.
For detection of bacteremia, blood samples (10 μL) were obtained by puncture from the tail vein at time points at 12, 18 and 24 h (h) after pneumococcal challenge. Blood samples were serially diluted 101- to 106 fold in sterile saline. Subsequently, 5 μL of undiluted blood and 5 μL of every dilution were plated onto blood agar plates for life counting of pneumococci (CFU/mL blood).
Statistics.
Interaction of RSV with S. pneumoniae by flow cytometry was tested in duplicate samples. Cytometric fluorescence adherence experiments were performed in duplicate or triplicate with each individual experiment performed in 7-fold. The significance of different values was calculated by t test. Differences with p values < 0.05 were considered significant.
The incidence of bacteraemia in animal experiments was compared between groups by the Fisher exact test. Comparison of quantitative blood bacterial counts between groups was done with the Mann-Whitney U-test. All reported p-values are two-sided. For statistical calculations, the Statistical Product and Service Solutions (SPSS) for Windows was used (version 11.0, SPSS Inc., Chigaco, IL, USA).
RESULTS
Interaction of RSV with S. pneumoniae.
RSV particles bound to all pneumococcal serotypes tested, although to various extents (Fig. 1A). RSV-pneumococcal complex formation (tested with serotype 19) occurred in a dose-dependent manner, since the percentage of RSV-positive pneumococci increased with the number of RSV incubations with pneumococci (Fig. 1B).

Detection of pneumococci positive for RSV glycoprotein-F antigen as detected by FACS immunofluorescence. A: Interaction of RSV with S. pneumoniae serotypes 3, 9, 14, 18, 19 and 23. X-axis: pneumococcal serotypes, y axis: percentage of RSV-positive pneumococci. B: Interaction of RSV with S. pneumoniae serotype 19: influence of RSV incubation frequency with pneumococci. X-axis: pneumococci incubated for 1×, 2× or 5× during 30 min with RSV particles, y axis: percentage of enhancement (100% = 1× incubation with RSV). Figure represents means with standard errors of the mean (SEM) (n = 4).
Pneumococcal cell wall but not capsule is required for binding to RSV.
Fig. 2 shows that RSV bound in higher amounts to the unencapsulated mutant DW 3.8 than to the wild-type WU2, indicating that the presence of pneumococcal capsular polysaccharides is not a prerequisite for RSV binding to pneumococci but on the contrary, appears to interfere with RSV-pneumococcal complex formation. In addition, we found that a PspA-negative mutant, JY1123, bound RSV in comparable amounts to its surface as the wild type WU2 did, suggesting that PspA is not likely to be involved in the binding to RSV.

Interaction of RSV with S. pneumoniae wild type WU2 and mutants JY1123 (PspA-negative mutant) and DW 3.8 (unencapsulated mutant) as measured by FACS immunofluorescence. X-axis: pneumococcal mutants, y axis: percentage of RSV-positive pneumococci. Figure represents means ± SD of one representative experiment performed in triplicate.
RSV binding to pneumococci is inhibited by heparin.
Several recent studies have pointed out that heparin can inhibit the infectivity of RSV, by interacting with RSV-glycoprotein G (19,20). We tested whether protein G was involved in RSV binding to pneumococci by incubating RSV particles with heparin before adding pneumococci (serotype 14). Indeed, RSV-pneumococcal complex formation was inhibited in a dose-dependent manner by heparin, suggesting that protein-G plays a role in RSV-pneumococcal binding (Fig. 3). Adherence of pneumococci to epithelial cells in the absence of RSV was not affected by heparin (data not shown).

Interaction of RSV with S. pneumoniae serotype 14: Influence of incubating RSV with heparin. X-axis: black bar: interaction of S. pneumoniae serotype 14 with RSV, open bars: interaction of pneumococci with RSV, pretreated with heparin 100, 10, 1 and 0.1 IE. Y-axis: percentage of RSV-positive pneumococci. Figure represents mean ± SEM (n = 4): * = p < 0.05.
Adherence of RSV-pretreated pneumococci to uninfected monolayers.
Whether RSV-pneumococcal complex formation contributed to pneumococcal adherence to epithelial cells, was investigated by preincubating pneumococcal serotypes 3, 9, 14, 18, 19, and 23 with RSV. Adherence to HEp-2 and A549 monolayers was compared between RSV-pretreated and buffer-treated pneumococci (Fig. 4A and 4B). Remarkably, pneumococcal adherence was strongly enhanced when pneumococci were pretreated with RSV. These differences were highly statistically significant for all serotypes, independent of the cell-line used. This RSV-enhanced pneumococcal adherence was also found when pneumococci were preincubated with heat-inactivated RSV (data not shown).

Adherence of RSV-pretreated vs PBS-treated pneumococci to HEp-2 (A) and A549 (B) monolayers. X-axis: pneumococcal serotypes, y axis: percentage of pneumococcal adherence. Black bars = pneumococci, treated with PBS (controls), open bars = RSV-pneumococcal complexes. Both figures represent means ± SEM (n = 21): * = p < 0.001.
Heparin inhibits RSV-enhanced pneumococcal binding to respiratory epithelial cells.
RSV attachment to host cells is mediated by protein G, which can be blocked by heparin. To investigate whether binding of RSV-pneumococcal complexes to HEp-2/A549 cells was mediated by RSV glycoproteins, HEp-2/A549 monolayers were pretreated with heparin, before adding RSV-pneumococcal complexes to the cells. A dose-dependent inhibition of the adherence of RSV-pneumococcal complexes to heparin treated- HEp-2/A549 cells was detected (Fig. 5A and 5B).

Adherence of S. pneumoniae serotype 18 to monolayers: influence of preincubating Hep-2 (A) /A549 (B) monolayers with heparin. X-axis: black bar = adherence of PBS-treated pneumococci to untreated monolayer, open bars = adherence of RSV-pneumococcal complexes to PBS (−) and heparin-treated (100, 10, 1 and 0.1 IE) monolayer. Y-axis: pneumococcal adherence percentage. Figures represent means ± SEM (n = 14): * = p < 0.01, ** = p < 0.001.
Influence of RSV infection on development of pneumococcal bacteraemia.
To determine whether nasal infection with RSV influenced the development of pneumococcal bacteraemia, mice were co-infected with RSV and pneumococci (serotype 1). To this purpose, groups of seven mice were either infected with RSV at day -4 before pneumococcal challenge (day 0) or inoculated with RSV and pneumococci simultaneously.
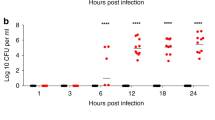
Mice that were pretreated with RSV 4 d before pneumococcal challenge, showed levels of bacteraemia similar to control animals at 12 and 18 h. However, at 24 h, CFU counts were significantly higher (p = 0.025) (Fig. 6A).

Influence of RSV infection on kinetics of pneumococcal bacteraemia (S. pneumoniae serotype 1). X-axis: time points at which blood samples were taken (following pneumococcal challenge). Y-axis log CFU levels measured in blood samples. A: RSV infection administered 4 d before pneumococcal challenge. B: Simultaneous inoculation of RSV and pneumococci (day 0). Figures represent results in groups of seven mice and show the CFU blood levels in individual mice in addition to the mean CFU blood levels. The dotted line is the detection level of blood CFU; minimal CFU/mL that could be determinded with this assay was 200 CFU/mL. * p < 0.025, ** p < 0.002.
Infection with both agents simultaneously enhanced the development of pneumococcal bacteraemia most strongly. Within 18 h after inoculation, a significantly higher frequency of bacteraemia (70%) could be detected in the RSV-treated group compared with controls (14%, p = 0.029). In addition, there was a strong increase in blood CFU counts of RSV-treated animals at 18 and 24 h postinfection (p = 0.002 and, p = 0.002, respectively) (Fig. 6B).
DISCUSSION
In a previous study we have demonstrated that a preceding RSV infection of human respiratory epithelial cells strongly and significantly enhanced pneumococcal adherence (12).
In the current study, we provided evidence that RSV virions can bind directly to pneumococci. In addition, we showed that the binding of RSV particles to pneumococci could be inhibited by preincubation of RSV with heparin. Heparin decreases RSV infectivity by interacting with the (attachment-) G-protein (21).
Therefore, it is likely that the enhanced binding of pneumococci to RSV-infected epithelial cells is mediated by the G-protein of RSV.
Our data show that RSV binding to pneumococci is to the cell wall rather than to the polysaccharide capsule. The cell wall consists of teichoic acids and peptidoglycan, the former acting as a binding site for several choline-binding proteins (22), including pneumococcal surface protein A (PspA). However, our results show that PspA is not likely to be involved in RSV binding to pneumococci, since JY1123, a PspA-negative mutant binds RSV in comparable amounts as the wild-type parent strain WU2. Further study is needed to detect which pneumococcal cell wall-related constituents are involved in binding RSV virions. Interestingly, in a recent paper Okamoto et al. show a similar direct interaction between influenza A virus and group A streptococci (23). In contrast to our findings they found that the group A streptococcal capsule was a prerequistite for binding.
The adherence of pneumococci to uninfected epithelial cells increased dramatically following complex formation with RSV. For all in vitro assays performed in this study, heat-killed pneumococci were used, to standardize bacterial growth conditions and to prevent autolysis to occur during the experiments. Therefore, it is unlikely that the enhanced pneumococcal adherence is the result of molecular alterations of the pneumococcal surface induced by the interaction with RSV.
In addition, it was found that heat-inactivated RSV particles could equally well enhance pneumococcal adherence to epithelial cells. This indicates that RSV infection of a cell is not an essential step in the enhancement of pneumococcal adherence, but that RSV virions by coupling with pneumococci already strongly enhance pneumococcal attachment. RSV-enhanced pneumococcal adherence to uninfected respiratory epithelial cells could be inhibited by heparin. Since heparin has been shown to inhibit RSV attachment to epithelial cells by interacting with the G-protein (19–21), we hypothesized that binding of the RSV-pneumococcal complex to the uninfected epithelial cell surface is mediated by RSV protein-G. The importance of these findings in vitro for the pathogenesis of pneumococcal infection was explored in vivo in a murine model.
Our findings in this model clearly demonstrate that mice infected with RSV four days before pneumococcal challenge as well as mice infected with both agents simultaneously not only showed an increased incidence, but also significantly increased levels of bacteremia. A striking finding of the present study was that simultaneous inoculation of RSV and pneumococci enhanced pneumococcal bacteraemia most strongly. It is unlikely that either the RSV-induced cytopathic effect on the epithelium or expression of viral glycoproteins or up-regulation of host proteins on the cellular surface contribute to the RSV-enhanced pneumococcal bacteremia when RSV and pneumococci are inoculated simultaneously. Okamoto et al. found that the direct interaction between influenza A virus and group A streptococci induced a lethal respiratory infection in a murine model (23,24). Similar findings with respect to a lethal synergism between influenza virus and pneumococci were obtained by Mc. Cullers et al. Specifically, these latter authors report that a preceding influenza infection resulted in a mortality rate of 100% compared with mortality rates of 15% and 35% for pneumococci and influenza virus alone, respectively. The authors speculate that influenza infection triggers inflammatory cytokines and may indirectly up-regulate adhesion molecules on epithelial cells, providing a receptor for bacterial adhesion and invasion. However, the authors also found that simultaneous inoculation with influenza virus and pneumococci produced a increased mortality of 60% and concluded that this was just an additive effect without speculating on the underlying mechanism any further (25). Together the findings in the present study with RSV and those by Okamoto et al. and McCullers et al. with influenza virus show that bacterial infection may run a more severe and even lethal course after simultaneous inoculation with a respiratory virus. A possible mechanism is suggested by our findings and those of Okamoto et al. in vitro, in which it was shown that viruses can form complexes with bacteria, leading to enhanced bacterial adherence and invasiveness. Apparently, by direct binding to bacteria respiratory viruses are able to act as a coupling agent between bacteria and respiratory epithelial cells. This phenomenon leads to enhanced bacterial adherence without the need for either expression of viral glycoproteins or up-regulation of host cell proteins.
In summary, we showed that S. pneumoniae is able to bind RSV directly. Therefore, it is likely that expression of viral glycoproteins on RSV-infected epithelial cells can serve as attachment molecules for pneumococci. We hypothesize that direct viral-bacterial binding is another, additional mechanism by which RSV can induce enhanced pneumococcal binding, not only to RSV-infected, but also to uninfected epithelial cells.
The findings in vitro were supported by experiments in vivo in a murine model that showed a synergism between RSV and pneumococci resulting in an increased invasiveness of the latter.
We propose that the direct interaction between viruses and bacteria inducing a more severe infection is probably a more general phenomenon in the pathogenesis of respiratory infections than has previously been appreciated.
Abbreviations
- CFU:
-
colony-forming units
- FACS:
-
fluorescence activated cell sorter
- PFU:
-
plaque-forming units
- PspA:
-
pneumococcal surface protein A
- RSV:
-
respiratory syncytial virus
References
Andrade MA, Hoberman A, Glustein J, Paradise JL, Wald ER 1998 Acute otitis media in children with bronchiolitis. Pediatrics 101: 617–619
Eskola J, Hovi T 1999 Respiratory viruses in acute otitis media. N Engl J Med 340: 312–314
Greenberg DP, Hoberman A 2001 Vaccine prevention of acute otitis media. Curr Allergy Asthma Rep 1: 358–363
Heiskanen-Kosma T, Korppi M, Jokinen C, Kurki S, Heiskanen L, Juvonen H, Kallinen S, Sten M, Tarkiainen A, Ronnberg PR, Kleemola M, Makela PH, Leinonen M 1998 Etiology of childhood pneumonia: serologic results of a prospective, population-based study. Pediatr Infect Dis J 17: 986–991
Kim PE, Musher DM, Glezen WP, Rodriguez-Barradas MC, Nahm WK, Wright CE 1996 Association of invasive pneumococcal disease with season, atmospheric conditions, air pollution, and the isolation of respiratory viruses. Clin Infect Dis 22: 100–106
Hament JM, Kimpen JL, Fleer A, Wolfs TF 1999 Respiratory viral infection predisposing for bacterial disease: a concise review. FEMS Immunol Med Microbiol 26: 189–195
Heikkinen T 2000 Role of viruses in the pathogenesis of acute otitis media. Pediatr Infect Dis J 19: S17–S22
Fainstein V, Musher DM, Cate TR 1980 Bacterial adherence to pharyngeal cells during viral infection. J Infect Dis 141: 172–176
Hakansson A, Kidd A, Wadell G, Sabharwal H, Svanborg C 1994 Adenovirus infection enhances in vitro adherence of Streptococcus pneumoniae. Infect Immun 62: 2707–2714
El Ahmer OR, Raza MW, Ogilvie MM, Weir DM, Blackwell CC 1999 Binding of bacteria to HEp-2 cells infected with influenza A virus. FEMS Immunol Med Microbiol 23: 331–341
Patel J, Faden H, Sharma S, Ogra PL 1992 Effect of respiratory syncytial virus on adherence, colonization and immunity of non-typable Haemophilus influenzae: implications for otitis media. Int J Pediatr Otorhinolaryngol 23: 15–23
Hament JM, Aerts PC, Fleer A, van Dijk H, Harmsen T, Kimpen JL, Wolfs TF 2004 Enhanced adherence of Streptococcus pneumoniae to human epithelial cells infected with respiratory syncytial virus. Pediatr Res 55: 972–978
Elahmer OR, Raza MW, Ogilvie MM, Blackwell CC, Weir DM, Elton RA 1996 The effect of respiratory virus infection on expression of cell surface antigens associated with binding of potentially pathogenic bacteria. Adv Exp Med Biol 408: 169–177
Raza MW, El Ahmer OR, Ogilvie MM, Blackwell CC, Saadi AT, Elton RA, Weir DM 1999 Infection with respiratory syncytial virus enhances expression of native receptors for non-piliate Neisseria meningitidis on HEp-2 cells. FEMS Immunol Med Microbiol 23: 115–124
Crain MJ, Waltman WD 2nd, Turner JS, Yother J, Talkington DF, McDaniel LS, Gray BM, Briles DE 1990 Pneumococcal surface protein A (PspA) is serologically highly variable and is expressed by all clinically important capsular serotypes of Streptococcus pneumoniae.. Infect Immun 58: 3293–3299
Watson DA, Musher DM 1990 Interruption of capsule production in Streptococcus pneumonia serotype 3 by insertion of transposon Tn916. Infect Immun 58: 3135–3138
Dulbecco R 1952 Production of plaques in monolayer tissue cultures by single particles of an animal virus. Proc Natl Acad Sci USA 38: 747–752
Tamura GS, Kuypers JM, Smith S, Raff H, Rubens CE 1994 Adherence of group B streptococci to cultured epithelial cells: roles of environmental factors and bacterial surface components. Infect Immun 62: 2450–2458
Hosoya M, Balzarini J, Shigeta S, De Clercq E 1991 Differential inhibitory effects of sulfated polysaccharides and polymers on the replication of various myxoviruses and retroviruses, depending on the composition of the target amino acid sequences of the viral envelope glycoproteins. Antimicrob Agents Chemother 35: 2515–2520
Krusat T, Streckert HJ 1997 Heparin-dependent attachment of respiratory syncytial virus (RSV) to host cells. Arch Virol 142: 1247–1254
Bourgeois C, Bour JB, Lidholt K, Gauthray C, Pothier P 1998 Heparin-like structures on respiratory syncytial virus are involved in its infectivity in vitro. J Virol 72: 7221–7227
Rosenow C, Ryan P, Weiser JN, Johnson S, Fontan P, Ortqvist A, Masure HR 1997 Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae.. Mol Microbiol 25: 819–829
Okamoto S, Kawabata S, Terao Y, Fujitaka H, Okuno Y, Hamada S 2004 The Streptococcus pyogenes capsule is required for adhesion of bacteria to virus-infected alveolar epithelial cells and lethal bacterial-viral superinfection. Infect Immun 72: 6068–6075
Okamoto S, Kawabata S, Nakagawa I, Okuno Y, Goto T, Sano K, Hamada S 2003 Influenza A virus-infected hosts boost an invasive type of Streptococcus pyogenes infection in mice. J Virol 77: 4104–4112
McCullers JA, Rehg JE 2002 Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis 186: 341–350
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hament, JM., Aerts, P., Fleer, A. et al. Direct Binding of Respiratory Syncytial Virus to Pneumococci: A Phenomenon That Enhances Both Pneumococcal Adherence to Human Epithelial Cells and Pneumococcal Invasiveness in a Murine Model. Pediatr Res 58, 1198–1203 (2005). https://doi.org/10.1203/01.pdr.0000188699.55279.1b
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.pdr.0000188699.55279.1b
This article is cited by
-
Airway response to respiratory syncytial virus has incidental antibacterial effects
Nature Communications (2019)
-
Direct interactions with influenza promote bacterial adherence during respiratory infections
Nature Microbiology (2019)
-
Burden of severe RSV disease among immunocompromised children and adults: a 10 year retrospective study
BMC Infectious Diseases (2018)
-
Impact of viral upper respiratory tract infection on the concentration of nasopharyngeal pneumococcal carriage among Kenyan children
Scientific Reports (2018)
-
Transcriptional adaptation of pneumococci and human pharyngeal cells in the presence of a virus infection
BMC Genomics (2013)
