
Abstract
Necrotizing enterocolitis (NEC) is the leading cause of death from gastrointestinal disease in the preterm infant. The dismal results of current treatment for NEC highlight the urgent need for greater understanding of the pathogenesis of this disease, and the importance of discovering novel, molecular-specific therapies for it. Current dogma indicates that NEC development reflects an abnormal response by the premature infant to the microbial flora that colonizes the gastrointestinal tract, although the mechanisms that mediate these abnormal bacterial-enterocyte interactions and the reasons for the particularly increased susceptibility of the premature infant to the development of NEC remain incompletely explained. Recent evidence has shed light on an emerging role for the Toll-like receptors (TLRs) of the innate immune system as central players in the pathways that signal in response to enteric bacteria resulting in the development of NEC. We now review recent advances in the field of NEC and identify several exciting potential avenues for novel treatments by focusing on abnormal TLR4 signaling in the premature intestine in the pathogenesis of NEC. In so doing, we seek to offer new hope to the patients and their families who are affected by this devastating disorder.
Similar content being viewed by others

BACTERIAL-ENTEROCYTE SIGNALING IN THE PATHOGENESIS OF NECROTIZING ENTEROCOLITIS
Necrotizing enterocolitis (NEC) is the leading cause of death from gastrointestinal disease in neonates and remains a major cause of morbidity in survivors (1,2). Despite many years of research into NEC pathogenesis and treatment, relatively little progress has been made toward improving the outcome of patients with NEC since its initial description in 1965 (3), and surgical survival remains ∼50% (4). Despite the marked improvements in overall neonatal care in general, the management approach to the infant with NEC has not changed in the past 30 y, and the outcome is generally as poor today as it was 3 decades ago (5). Based on these sobering statistics, it is clear that new therapeutic approaches to NEC are required and that such approaches will demand a greater understanding of the molecular mechanisms that contribute to the development of this disease. As was recently summarized by the 2006 NICHD workshop on NEC research, “NEC can be thought to arise from an uncontrolled exuberant inflammatory response to bacterial colonization that characterizes the intestine of premature infant” (6). Several investigators have now examined the mechanisms that mediate the signaling response of the newborn intestine to bacteria and have detailed the consequences of this signaling response to the pathogenesis of NEC. In particular, these studies have uncovered an essential role for a class of bacterial receptors named Toll-like receptors (TLRs) in the pathogenesis of NEC and have provided compelling evidence to suggest that blunting the ability of TLRs to signal within the intestinal epithelium of the newborn infant may either prevent or treat NEC. These findings place the spotlight on the molecular basis that underlies the interaction between the intestinal epithelium and the commensal microbial flora and have also identified that the ability of TLRs to respond to bacteria within the newborn intestinal epithelium may in part explain the particular susceptibility of the premature infant to the development of NEC. In this review, we will now describe the molecular events that regulate TLR signaling, highlight the evidence for TLR-bacterial signaling in the pathogenesis of NEC, and explain the basis for a novel therapeutic approach for NEC involving the selective silencing of TLRs within the newborn intestinal epithelium.
THE IMMUNE SYSTEM OF THE NEWBORN INTESTINE: MAINTAINING HOMEOSTASIS IN THE INFANT GUT
The recognition of microbial antigens by the newborn host may be accomplished through two intertwined arms of the immune system: the innate immune system, which consists of cells and their receptors that from a network of “first responders” that are programmed to respond rapidly to microbes; and the adaptive immune system, which requires prior exposure to antigenic stimuli and the release of antibodies by lymphocytes (7). Several authors have shown that the adaptive immune system is significantly underdeveloped in the newborn, which may in part contribute to the development of NEC (8). Recent work has also shed light on the important and exciting role of the innate immune receptors of the intestinal epithelium in the development of NEC. These receptors detect unique molecular sequences on bacteria and other potential pathogens, and because they share homology with the Toll protein of the fly innate immune system, they have been termed as the TLRs (9,10). Ten individual TLRs have been identified in humans—which are termed TLR1 through TLR10, respectively (11). TLR4 is known to be the receptor for lipopolysaccharide (LPS), which is the outer membrane component of Gram-negative bacteria (12). A role for LPS—and by extension for TLR4—in the pathogenesis of NEC is highlighted by the findings that LPS administration in association with systemic stress induces NEC in animals (13–15); serum levels of LPS are increased in patients with both NEC and inflammatory bowel disease, a disorder that shares histopathological features with NEC (16–19); and levels of LPS are significantly increased in the stools of infants and mice that develop NEC compared with healthy counterparts (20,21). Taken together, these findings indicate that LPS may play an important role in the pathogenesis of NEC and suggest that strategies that limit the responsiveness to LPS may provide a therapeutic approach to the management of this disease. On the basis of these lines of rationale, we will now consider the role of the TLR4 within the intestinal epithelium in the pathogenesis of NEC.
EVIDENCE TO SUPPORT AN ESSENTIAL ROLE FOR TLR4 IN THE PATHOGENESIS OF NEC
Based on the observation that NEC is known to develop after the intestine has been colonized with Gram-negative bacteria, several laboratories have sought to establish whether TLR4—the receptor for Gram-negative bacteria—may play a role in the pathogenesis of this disease. The Caplan laboratory showed in 2006 that mice with mutations in TLR4 are protected from the development of NEC (22), whereas the Hackam laboratory in 2007 confirmed that mice lacking TLR4 do not develop NEC and extended these observations by demonstrating that TLR4 signaling regulates the balance between injury and repair in the newborn intestine (21). Several laboratories have shown that TLR4 is increased in the intestinal mucosa of mice, rats, and humans with NEC compared with controls (21,23), and that activation of enterocyte TLR4 leads to an increase in death of the cells that line the intestine through the process of apoptosis (24). Along with the increase in epithelial death that accompanies TLR4 activation in the newborn small intestine, TLR4 activation within the intestine was also found to reduce the capacity of mucosal healing to occur because of a reduction in enterocyte proliferation (25) and reduced enterocyte migration (26). Taken together, these findings demonstrate that TLR4 activation in the newborn small intestine leads to the development of NEC through profound and deleterious effects on promoting intestinal injury and reducing the capacity for mucosal repair.
Other authors have shed light on additional roles played by TLR4 in the pathogenesis of NEC. Luk and coworkers (23) have shown that the activity of the TLR4 signaling pathway was up-regulated in intestinal tissues from premature neonates and rats with NEC compared with controls. Wolfs et al. (27) have shown that the absence of the key TLR4 regulatory molecule MD-2 in the immature infant bowel may lead to impaired bacterial sensing and thus predispose to NEC on microbial colonization of the premature intestine. This concept is supported by the work of Liu et al. (28) who reported that TLR4 expression and signaling is increased in the intestine of rats with NEC when compared with control rats, and that such increases precede histological evidence of mucosal injury in this disease. Further evidence of the physiological relevance of these findings is demonstrated by Lu et al. (29), who showed that while TLR4 expression is increased in the intestinal mucosa of rats with NEC, agents known to reduce NEC—namely polyunsaturated fatty acids—may protect from the development of NEC through a reduction in TLR4 expression.
HOW DOES TLR4 ACTIVATION IN THE NEWBORN SMALL INTESTINE INHIBIT MUCOSAL REPAIR?
As summarized earlier, defects in the newborn intestinal mucosa—as may be induced through the apoptosis of enterocytes—are rapidly repaired through the combined processes of restitution, a process that involves the migration of healthy enterocytes adjacent to the mucosal defect into the wound, and proliferation, which involves the generation of new enterocytes from intestinal stem cells that are present within the intestinal crypts. In seeking to explore the mechanisms by which TLR4 adversely affects mucosal repair, TLR4 activation was observed to increase the adhesiveness that enterocytes exert on their underlying matrix, profoundly restricting their ability to move along the basement membrane (30,31). TLR4 activation was also found to inhibit enterocyte proliferation through direct dysregulation of the wnt-β-catenin signaling pathway, which is the predominant signaling cascade that regulates cell division within the intestine (25). It is also important to emphasize that TLR4 activation in enterocytes—as opposed to other TLR4-expressing cells such as macrophages—was found to be important in the pathogenesis of experimental NEC, as the delivery of viral particles to the intestinal epithelium of mice that inhibited TLR4 in enterocytes reduced NEC severity (25). Taken in aggregate, these findings demonstrate that TLR4 activation within the intestinal epithelium exerts deleterious effects on the small intestinal mucosa by promoting intestinal injury and reducing mucosal repair and thus play a central role in the pathogenesis of NEC.
COULD TLR4 ACTIVATION EVER PROTECT AGAINST THE DEVELOPMENT OF NEC?
Although these studies suggest that TLR4 signaling on enterocytes is responsible for the development of NEC, they do not exclude a role for other TLRs on other cell types nor do they reject the possibility that TLR4 may serve a protective role within other parts of the intestine or in nonnewborn states. In support of this possibility, Zheng et al. (32) have recently shown that TLR4 signaling in response to the endogenous matrix protein hyaluronic acid led to enhanced mucosal repair via the production of prostaglandin E2 and protection from colonic inflammation. Moreover, in two separate publications from the same laboratory, Fukata et al. (33) have shown that TLR4 signaling is required for optimal proliferation and protection against apoptosis in the injured adult colon in experimental colitis, whereas adult mice that lack TLR4 demonstrate increased severity of colitis compared with WT counterparts (34). In a landmark article in Cell, Rakoff-Nahoum et al. (35) demonstrated that in adult mice under baseline conditions, the activation of TLRs by commensal microflora is required for protection against colonic injury. How can we therefore reconcile the data showing that TLR4 signaling is required for the development of NEC with studies showing that TLR4 exerts a protective role in colitis? The first possible explanation is that TLR4 activation may have different effects in different parts of the intestine. In support of this possibility, TLR4 activation with LPS was recently found to lead to mucosal injury because of enterocyte apoptosis in the terminal ileum of newborn mice but not adult mice and in the small intestine but not the newborn colon (24). These findings are not actually at odds with the work of Fukata et al. (33) or of Medzhitov and cworkers (35), who have elegantly shown that TLR4 activation is protective in the colon in mature (i.e. nonnewborn) mice. It should also be stated that the models used to induce NEC in the laboratory, which typically use a combination of systemic hypoxia and administration of formula by gavage, differ markedly from those used to induce colitis, which typically involve the administration of agents with local toxicity to the colon. Because TLR4 is expressed not only on enterocytes but also on macrophages present within the intestine, it is possible that the protective effects attributed to TLR4 signaling in the gut by Fukata et al. who use global TLR4 deficient mice may reflect in part the mitigating effects of TLR4 signaling on other cells. In support of this possibility, we note that Fukata et al. have recently shown in an elegant study using chimeric mice that TLR4 signaling in colonic epithelial cells worsened intestinal inflammation (i.e. was required for recruitment and activation of cox-2 expressing macrophages and determining the degree of histological injury) (36). Taken together, these findings strongly argue that the effect of TLR4 in the development of intestinal inflammation is demonstrably influenced by a variety of factors, including the effector cells involved (enterocyte versus immune cell), developmental factors (newborn versus adult), and region of the intestine (small bowel versus large bowel).
KEEPING TLR4 SIGNALING IN THE INTESTINAL EPITHELIUM IN CHECK: MECHANISMS THAT DAMPEN TLR4 SIGNALING
The fact that TLR4 signaling in the intestinal epithelium in response to commensal enteric flora can lead to NEC raises an important question: what prevents NEC from occurring spontaneously in every infant? It is apparent that the host has evolved several strategies that limit the extent of TLR4 signaling in enterocytes that occurs after LPS binding. Inhibition of the various downstream effector molecules that are required for intact TLR4 signaling can dampen the TLR4 response. As an example of this method by which TLR4 signaling can be reduced in enterocytes, Lotz et al. (37) have shown that the enterocytes of vaginally born—but not C-section delivered mice—are relatively resistant to TLR4 activation in vitro because of a posttranscriptional down-regulation of the IL-1 receptor-associated kinase 1, which has been shown to be essential for epithelial TLR4 signaling in vitro. Wang et al. (38) have shown that expression of MAPK phosphatase 1 leads to a reduction in the extent of signaling through TLR4—and other TLRs—in enterocytes, in what seems to be a negative feedback loop requiring nuclear factor kappa B (NFkB), which is the downstream effector of TLR4 activation. This same group also recently demonstrated that the ubiquitin-editing enzyme A20 can limit the extent of TLR4 signaling in enterocytes, as evidenced by the increased degree of intestinal inflammation occurring in A20-deficient mice, although the precise mechanisms involved remain incompletely understood (39). Recent evidence also suggests that cross-talk between two other innate immune receptors—namely TLR9, which is a homologue of TLR4 and a receptor for bacterial DNA, and nucleotide oligomerization domain 2 (NOD2), which is the receptor for the bacterial component muramyl-di-peptide (MDP)—can prevent exaggerated TLR4 signaling. TLR9 is the receptor in enterocytes for bacterial DNA, which is rich in CpG groups and significantly hypomethylated, in contrast to mammalian DNA. We demonstrated that activation of TLR9 with CpG-DNA in enterocytes both in vitro and in the newborn intestine led to reduced TLR4 signaling as manifest by reduced cytokine production and decreased apoptosis through a mechanism that involved the up-regulation of the cytoplasmic inhibitor IRAK-M, which interferes with the downstream signaling of TLR4 (40). The reciprocal expression of TLR9 and TLR4 was found to influence the extent of TLR4 signaling, and the development of NEC was accompanied by a relative increase in TLR4 with a concomitant reduction in the protective TLR9 (40). Indeed, these findings may explain the relative protective value observed from probiotic administration to infants with NEC (5), as these probiotic preparations are rich in bacterial DNA, through which activation of TLR9 on the host would be expected to limit TLR4 signaling and reduce NEC severity. In recent experiments, our laboratory has shown that the cytoplasmic bacterial sensor NOD2—which recognizes the bacterial motif MDP and which has risen to recent prominence because of the mutations in NOD2 are linked to the development of inflammatory bowel disease in humans (41)—limits TLR4 signaling through posttranslational effects on TLR4 and through the up-regulation of the proapoptosis protein SMAC-diablo (24). In seeking to identify the signaling pathways involved, we now uncover a novel pathway linking these innate immune receptors with the mitochondrial protein SMAC-diablo, which serves to regulate the extent by which NOD2 activation with MDP limits the extent of TLR4- induced intestinal injury and thus serve as a potential therapeutic agent for NEC. From a therapeutic point of view, activation of TLR9 by the administration of CpG-DNA or activation of NOD2 with administration of MDP resulted in a marked reduction in the severity of experimental NEC in mice. These findings not only highlight the mechanisms that maintain levels of TLR4 signaling under physiological conditions but also suggest novel therapeutic approaches to this devastating disease.
These studies raise the possibility that the administration of CpG-DNA may provide therapeutic benefit to infants with NEC, or alternatively, may serve as a preventive strategy when administered to premature infants who are at risk for the development of the disease in the first place. Although we are actively pursuing such possibilities, we readily acknowledge that the administration of CpG-DNA to infants may carry some risk, either from activation of immune effector cells and/or by inhibiting any protective effects otherwise conferred by TLR4. Carefully designed studies will be required to determine the beneficial versus adverse effects of strategies that use molecules like CpG-DNA or related isoforms in the prevention or treatment of NEC.
COULD TLR4 ACTIVATION BE LINKED WITH PLATELET-ACTIVATING FACTOR SIGNALING IN THE PATHOGENESIS OF NEC?
As described earlier, work from various laboratories has demonstrated that TLR4 signaling plays a pivotal role in the pathogenesis of NEC in large part through the regulation of mucosal injury and repair in the newborn gut. However, it is important to point out the effects of TLR4 on mucosal repair mechanisms do not occur in isolation, and indeed, are likely to act in concert with other proinflammatory signaling pathways that lead to the development of NEC. In this regard, platelet-activating factor (PAF) is a potent lipid mediator that is released by a variety of cells including peritoneal cells (42) and macrophages (43) and is capable of eliciting proinflammatory responses in several cell types (44). Caplan and coworkers and others (45–48) have elegantly demonstrated an important role for PAF in NEC pathogenesis. The neonatal intestine was found to have high PAF biosynthetic activity (45), and PAF levels were significantly increased in the stools of infants with NEC compared with healthy controls (46). Moreover, the PAF receptor antagonist WEB2170 was found to prevent NEC in rats (47) and recombinant PAF acetylhydrolase—which degrades PAF—was found to reduce the incidence of NEC in rats (48). These findings suggest the possibility of that a potential relationship may exist between PAF and TLR4. In support of this concept, Worthen et al. (49) have previously shown that LPS—the receptor for TLR4—enhances PAF release from neutrophils. Taken together, these lines of evidence argue for a link between TLR4 and PAF signaling in the premature infant and provide a conceptual bridge between two pivotal pathways required for the pathogenesis of NEC.
HOW TLR4 SIGNALING IN THE GUT MAY EXPLAIN WHY THE PREMATURE INFANT IS AT PARTICULAR RISK FOR THE DEVELOPMENT OF NEC
Prematurity is the greatest risk factor for the development of NEC, and 90% of cases of the disease occur in premature infants. Several authors have provided insights into the possible explanations for the unique susceptibility of the premature infant to the devastating and acute onset of necrosis that leads to NEC. Specifically, intestinal peristalsis, macrophage phagocytosis, gastric acidification, and even epithelial barrier integrity have all reportedly been shown to be impaired in premature infants compared with full-term counterparts (50). However, although these features may provide a satisfactory explanation as to why the premature infant is at particularly increased risk for sepsis in general, none of these features—either alone or in combination—is sufficient to explain how the premature infant intestine develops the massive gross and microscopic features of NEC. By contrast, a focus on the role of TLR4 signaling in the intestinal epithelium may offer additional insights into the unique susceptibility of the premature infant to the NEC development. We have recently demonstrated that the expression of TLR4 within the intestine rises during gestation in the mouse and then falls shortly before birth (40). The finding that TLR4 expression rises in the intestine of the fetus during development suggests the distinct possibility that TLR4 may signal in response to agonists within the microenvironment of the developing intestine other than LPS (31). The increased expression of TLR4 in the fetal intestine toward the end of development would expectedly be reflected in an increase in expression of TLR4 in the intestine of the premature infant, who is essentially under developed, assuming that similar mechanisms regulate murine and human TLR4 expression in the gut. In support of this possibility, we and others have determined that TLR4 expression is increased in the bowel of infants with NEC compared with control bowel (21). On the basis of these findings, we now believe that the premature infant has a selective predisposition to the development of NEC because of the persistently increased expression of TLR4 in its underdeveloped intestine.
PUTTING IT ALL TOGETHER—AN OVERRIDING HYPOTHESIS TO EXPLAIN HOW TLR4 SIGNALING LEADS TO THE DEVELOPMENT OF NEC
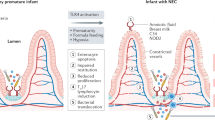
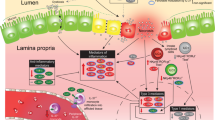
On the basis of the work performed in a variety of laboratories as described earlier, we now submit that NEC is a disease that is characterized by impaired signaling in response to the indigenous microflora of the newborn infant. On the basis of our recent discovery that prematurity, hypoxia, and endotoxemia—three clinical features that are characteristic of NEC—lead to persistent up-regulation of intestinal TLR4 (21), we now propose that part of the mechanism of NEC reflects the inability of the intestine to down-regulate TLR4 signaling to become tolerant to the luminal bacteria. This would be expected to lead to exaggerated TLR4 signaling on bacterial colonization, resulting in increased intestinal injury by apoptosis and reduced healing because of impaired enterocyte proliferation and migration, leading to persistent susceptibility to mucosal injury and the development of NEC. We further speculate that other factors that are known to lead to the development of NEC (such as PAF, various chemokines, and reactive oxygen species) may act at least in part through increased expression or signaling of TLR4 in the intestinal mucosa of the premature infant. These findings expand the current understanding of the role of TLR4 in intestinal homeostasis, while also providing novel insights into the molecular mechanisms that lead to NEC and are summarized in Figure 1.

The role of TLR4 signaling in the pathogenesis of necrotizing enterocolitis. As described in the text, hypoxia, infection, and prematurity together increase the expression of TLR4 in the intestinal mucosa, whose subsequent activation by enteric bacteria leads to increased barrier injury and reduced epithelial repair. Inhibition of TLR4, as may occur via activation of the cytoplasmic innate immune receptors NOD2 and TLR9 leads to an inhibition of TLR4, restoration of the intestinal epithelial barrier, and a reduction in the severity of NEC.
SUMMARY AND FUTURE DIRECTIONS
NEC still remains a major unsolved medical challenge, for which no specific therapy exists. We have highlighted an important role for TLR4 signaling within the intestinal epithelium in the development of NEC, a finding that may account in part for the increased susceptibility of the premature infant—whose levels of TLR4 remain persistently high—to develop this devastating disease. Placing the spotlight on TLR4 has allowed us to explore potential therapies for this disease, in particular, the compound CpG-DNA, which acts to inhibit TLR4 signaling via effects on TLR9, and MDP, which inhibits TLR4 signaling via activation on NOD2, thereby dramatically reducing the severity of NEC in mice. Future directions in the field to understand the roles of vascular networks, intestinal stem cells, genetic factors, and the intestinal microbiome are likely to provide tremendous insights into the development of this disease. Moreover, the possibility that the expression levels of TLR4 on the intestinal epithelium or in the stool may serve as a biomarker for infants at risk for the development of NEC is likely to be an area of active investigation now and in the future. Through these combined efforts, it is hoped that a new understanding of NEC will arise, thereby alleviating the effects of this devastating disorder.
Abbreviations
- NEC:
-
necrotizing enterocolitis
- PAF:
-
platelet-activating factor
- TLR4:
-
Toll-like receptor 4
References
Luig M, Lui K, NSW & ACT NICUS Group 2005 Epidemiology of necrotizing enterocolitis—part I: changing regional trends in extremely preterm infants over 14 years. J Paediatr Child Health 41: 169–173
Gagliardi L, Bellu R, Cardilli V, De Curtis M, Network Neonatale Lombardo 2008 Necrotising enterocolitis in very low birth weight infants in Italy: incidence and non-nutritional risk factors. J Pediatr Gastroenterol Nutr 47: 206–210
Mizrahi A, Barlow O, Berdon W, Blanc WA, Silverman WA 1965 Necrotizing enterocolitis in premature infants. J Pediatr 66: 697–705
Blakely ML, Lally KP, McDonald S, Brown RL, Barnhart DC, Ricketts RR, Thompson WR, Scherer LR, Klein MD, Letton RW, Chwals WJ, Touloukian RJ, Kurkchubasche AG, Skinner MA, Moss RL, Hilfiker ML; Network NECSotNNR 2005 Postoperative outcomes of extremely low birth-weight infants with necrotizing enterocolitis or isolated intestinal perforation: a prospective cohort study by the NICHD Neonatal Research Network. Ann Surg 241: 984–989; discussion 989–994.
Lin HC, Su BH, Chen AC, Lin TW, Tsai CH, Yeh TF, Oh W 2005 Oral probiotics reduce the incidence and severity of necrotizing enterocolitis in very low birth weight infants. Pediatrics 115: 1–4
Grave GD, Nelson SA, Walker WA, Moss RL, Dvorak B, Hamilton FA, Higgins R, Raju TN 2007 New therapies and preventive approaches for necrotizing enterocolitis: report of a research planning workshop. Pediatr Res 62: 510–514
Iwasaki A, Medzhitov R 2010 Regulation of adaptive immunity by the innate immune system. Science 327: 291–295
Wynn J, Cornell TT, Wong HR, Shanley TP, Wheeler DS 2010 The host response to sepsis and developmental impact. Pediatrics 125: 1031–1041
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr 1997 A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388: 394–397
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA 1996 The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86: 973–983
Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A 2005 The evoluation of vertebrate Toll-like receptors. Proc Natl Acad Sci U S A 102: 9577–9582
Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B 1998 Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282: 2085–2088
Hotta T, Yoshida N, Yoshikawa T, Sugino S, Kondo M 1986 Lipopolysaccharide-induced colitis in rabbits. Res Exp Med (Berl) 186: 61–69
Feng J, El-Assal ON, Besner GE 2005 Heparin-binding EGF-like growth factor (HB-EGF) and necrotizing enterocolitis. Semin Pediatr Surg 14: 167–174
Feng J, Besner GE 2007 Heparin-binding epidermal growth factor-like growth factor promotes enterocyte migration and proliferation in neonatal rats with necrotizing enterocolitis. J Pediatr Surg 42: 214–220
Kruis W, Schussler P, Weinzierl M, Galanos C, Eisenburg J 1984 Circulating lipid A antibodies despite absence of systemic endotoxemia in patients with Crohn's disease. Dig Dis Sci 29: 502–507
Caradonna L, Amati L, Lella P, Jirillo E, Caccavo D 2000 Phagocytosis, killing, lymphocyte-mediated antibacterial activity, serum autoantibodies, and plasma endotoxins in inflammatory bowel disease. Am J Gastroenterol 95: 1495–1502
Noerr B 2003 Current controversies in the understanding of necrotizing enterocolitis. Adv Neonatal Care 3: 107–120
Sharma R, Tepas JJ III, Hudak ML, Mollitt DL, Wludyka PS, Teng RJ, Premachandra BR 2007 Neonatal gut barrier and multiple organ failure: role of endotoxin and proinflammatory cytokines in sepsis and necrotizing enterocolitis. J Pediatr Surg 42: 454–461
Duffy LC, Zielezny MA, Carrion V, Griffiths E, Dryja D, Hilty M, Rook C, Morin F III 1997 Concordance of bacterial cultures with endotoxin and interleukin-6 in necrotizing enterocolitis. Dig Dis Sci 42: 359–365
Leaphart CL, Cavallo JC, Gribar SC, Cetin S, Li J, Branca MF, Dubowski TD, Sodhi CP, Hackam DJ 2007 A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol 179: 4808–4820
Jilling T, Simon D, Lu J, Meng FJ, Li D, Schy R, Thomson RB, Soliman A, Arditi M, Caplan MS 2006 The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol 177: 3273–3282
Chan KL, Wong KF, Luk JM 2009 Role of LPS/CD14/TLR4-mediated inflammation in necrotizing enterocolitis: pathogenesis and therapeutic implications. World J Gastroenterol 15: 4745–4752
Richardson WM, Sodhi CP, Russo A, Siggers RH, Afrazi A, Gribar SC, Neal MD, Dai S, Prindle TJ, Branca M, Ma C, Ozolek J, Hackam DJ 2010 Nucleotide-binding oligomerization domain-2 inhibits Toll like receptor-4 signaling in the intestinal epithelium. Gastroenterology 139: 904–917
Sodhi CP, Shi XH, Richardson WM, Grant ZS, Shapiro RA, Prindle TJ, Branca M, Russo A, Gribar SC, Ma C, Hackam DJ 2010 Toll-like receptor-4 inhibits enterocyte proliferation via impaired beta-catenin signaling in necrotizing enterocolitis. Gastroenterology 138: 185–196
Qureshi FG, Leaphart C, Cetin S, Li J, Grishin A, Watkins S, Ford HR, Hackam DJ 2005 Increased expression and function of integrins in enterocytes by endotoxin impairs epithelial restitution. Gastroenterology 128: 1012–1022
Wolfs TG, Derikx JP, Hodin CM, Vanderlocht J, Driessen A, de Bruïne AP, Bevins CL, Lasitschka F, Gassler N, van Gemert WG, Buurman WA 2010 Localization of the lipopolysaccharide recognition complex in the human healthy and inflamed premature and adult gut. Inflamm Bowel Dis 16: 68–75
Liu Y, Zhu L, Fatheree NY, Liu X, Pacheco SE, Tatevian N, Rhoads JM 2009 Changes in intestinal Toll-like receptors and cytokines precede histological injury in a rat model of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 297: G442–G450
Lu J, Jilling T, Li D, Caplan MS 2007 Polyunsaturated fatty acid supplementation alters proinflammatory gene expression and reduces the incidence of necrotizing enterocolitis in a neonatal rat model. Pediatr Res 61: 427–432
Cetin S, Ford HR, Sysko LR, Agarwal C, Wang J, Neal MD, Baty C, Apodaca G, Hackam DJ 2004 Endotoxin inhibits intestinal epithelial restitution through activation of Rho-GTPase and increased focal adhesions. J Biol Chem 279: 24592–24600
Dai S, Sodhi CP, Cetin S, Richardson W, Branca M, Neal MD, Prindle T, Ma C, Shapiro RA, Li B, Wang JH, Hackam DJ 2010 Extracellular high mobility group box1 (HMGB1) inhibits enterocyte migration via activation of Toll like receptor 4 and increased cell-matrix adhesiveness. J Biol Chem 285: 4995–5002
Zheng L, Riehl TE, Stenson WF 2009 Regulation of colonic epithelial repair in mice by Toll-like receptors and hyaluronic acid. Gastroenterology 137: 2041–2051
Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, Xu R, Inoue H, Arditi M, Dannenberg AJ, Abreu MT 2006 Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: role in proliferation and apoptosis in the intestine. Gastroenterology 131: 862–877
Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, Nast CC, Lechago J, Xu R, Naiki Y, Soliman A, Arditi M, Abreu MT 2005 Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol 288: G1055–G1065
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R 2004 Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell 118: 229–241
Fukata M, Hernandez Y, Conduah D, Cohen J, Chen A, Breglio K, Goo T, Hsu D, Xu R, Abreu MT 2009 Innate immune signaling by Toll-like receptor-4 (TLR4) shapes the inflammatory microenvironment in colitis-associated tumors. Inflamm Bowel Dis 15: 997–1006
Lotz M, Gutle D, Walther S, Menard S, Bogdan C, Hornef MW 2006 Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J Exp Med 203: 973–984
Wang J, Ford HR, Grishin AV 2010 NF-kappaB-mediated expression of MAPK phosphatase-1 is an early step in desensitization to TLR ligands in enterocytes. Mucosal Immunol 3: 523–534
Wang J, Ouyang Y, Guner Y, Ford HR, Grishin AV 2009 Ubiquitin-editing enzyme A20 promotes tolerance to lipopolysaccharide in enterocytes. J Immunol 183: 1384–1392
Gribar SC, Sodhi CP, Richardson WM, Anand RJ, Gittes GK, Branca MF, Jakub A, Shi XH, Shah S, Ozolek JA, Hackam DJ 2009 Reciprocal expression and signaling of TLR4 and TLR9 in the pathogenesis and treatment of necrotizing enterocolitis. J Immunol 182: 636–646
Borzutzky A, Fried A, Chou J, Bonilla FA, Kim S, Dedeoglu F 2010 NOD2-associated diseases: bridging innate immunity and autoinflammation. Clin Immunol 134: 251–261
Shindou H, Ishii N, Uozumi T, Shimizu T 2000 Roles of cytosolic phospholipase A2 and platelet-activating factor receptor in the Ca-induced biosynthesis of PAF. Biochem Biophys Res Commun 271: 812–817
Svetlov SI, Liu H, Chao W, Olson MS 1997 Regulation of platelet-activating factor (PAF) biosynthesis via coenzyme A-independent transacylase in the macrophage cell line IC-21 stimulated with lipopolysaccharide. Biochim Biophys Acta 1346: 120–130
Izumi T, Shimizu T 1995 Platelet-activating factor receptor: gene expression and signal transduction. Biochim Biophys Acta 1259: 317–333
Muguruma K, Gray PW, Tjoelker LW, Johnston JM 1997 The central role of PAF in necrotizing enterocolitis development. Adv Exp Med Biol 407: 379–382
Amer MD, Hedlund E, Rochester J, Caplan MS 2004 Platelet-activating factor concentration in the stool of human newborns: effects of enteral feeding and neonatal necrotizing enterocolitis. Biol Neonate 85: 159–166
Caplan MS, Hedlund E, Adler L, Lickerman M, Hsueh W 1997 The platelet activating factor receptor antagonist WEB 2170 prevents neonatal necrotizing enterocolitis in rats. J Pediatr Gastroenterol Nutr 24: 296–301
Caplan MS, Lickerman M, Adler L, Dietsch GN, Yu A 1997 The role of recombinant platelet activating factor acetylhydrolase in a neonatal rat model of necrotizing enterocolitis. Pediatr Res 42: 779–783
Worthen GS, Seccombe JF, Clay KL, Guthrie LA, Johnston RB Jr 1988 The priming of neutrophils by lipopolysaccharide for production of intracellular platelet-activating factor: potential role in mediation of enhanced superoxide secretion. J Immunol 140: 3553–3559
Milla PJ, Fenton TR 1983 Small intestinal motility patterns in the perinatal period. J Pediatr Gastroenterol Nutr 2: S141–S144
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by Grants DK083752 and GM078238 from the National Institutes of Health and The Hartwell Foundation, Memphis, TN.
Rights and permissions
About this article
Cite this article
Afrazi, A., Sodhi, C., Richardson, W. et al. New Insights Into the Pathogenesis and Treatment of Necrotizing Enterocolitis: Toll-Like Receptors and Beyond. Pediatr Res 69, 183–188 (2011). https://doi.org/10.1203/PDR.0b013e3182093280
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3182093280
This article is cited by
-
IL-17-related signature genes linked to human necrotizing enterocolitis
BMC Research Notes (2021)
-
Sialylated human milk oligosaccharides prevent intestinal inflammation by inhibiting toll like receptor 4/NLRP3 inflammasome pathway in necrotizing enterocolitis rats
Nutrition & Metabolism (2021)
-
Surfactant protein A reduces TLR4 and inflammatory cytokine mRNA levels in neonatal mouse ileum
Scientific Reports (2021)
-
Irf5 deficiency in myeloid cells prevents necrotizing enterocolitis by inhibiting M1 macrophage polarization
Mucosal Immunology (2019)
-
Single-Immunoglobulin Interleukin-1-Related Receptor regulates vulnerability to TLR4-mediated necrotizing enterocolitis in a mouse model
Pediatric Research (2018)
