




Abstract
Development of model systems that recapitulate the molecular heterogeneity observed among glioblastoma multiforme (GBM) tumors will expedite the testing of targeted molecular therapeutic strategies for GBM treatment. In this study, we profiled DNA copy number and mRNA expression in 21 independent GBM tumor lines maintained as subcutaneous xenografts (GBMX), and compared GBMX molecular signatures to those observed in GBM clinical specimens derived from the Cancer Genome Atlas (TCGA). The predominant copy number signature in both tumor groups was defined by chromosome-7 gain/chromosome-10 loss, a poor-prognosis genetic signature. We also observed, at frequencies similar to that detected in TCGA GBM tumors, genomic amplification and overexpression of known GBM oncogenes, such as EGFR, MDM2, CDK6, and MYCN, and novel genes, including NUP107, SLC35E3, MMP1, MMP13, and DDX1. The transcriptional signature of GBMX tumors, which was stable over multiple subcutaneous passages, was defined by overexpression of genes involved in M phase, DNA replication, and chromosome organization (MRC) and was highly similar to the poor-prognosis mitosis and cell-cycle module (MCM) in GBM. Assessment of gene expression in TCGA-derived GBMs revealed overexpression of MRC cancer genes AURKB, BIRC5, CCNB1, CCNB2, CDC2, CDK2, and FOXM1, which form a transcriptional network important for G2/M progression and/or checkpoint activation. Our study supports propagation of GBM tumors as subcutaneous xenografts as a useful approach for sustaining key molecular characteristics of patient tumors, and highlights therapeutic opportunities conferred by this GBMX tumor panel for testing targeted therapeutic strategies for GBM treatment.
Glioblastoma multiforme (GBM; WHO grade IV) is the most common type of CNS tumor, the prognosis for which remains dismal despite intervention with surgery, radiation, and chemotherapy.1 A large number of genetic and epigenetic alterations have been identified in GBMs,2,3 many of which enhance the ability of tumor cells to proliferate, invade surrounding brain tissue, and evade therapeutic treatments. Although the mRNA and protein products of these genes are attractive candidates for targeted therapeutics, realization of the potential of targeted therapeutics for improved treatment of GBM will require more extensive understanding of the molecular pathways that underlie tumorigenesis and preclinical models that closely recapitulate the human disease.
Ex vivo cell culture models of GBM have provided valuable insights into the mechanisms by which oncogene and tumor suppressor dysfunction promote GBM development. However, it is well known that GBM cell lines cultured ex vivo lack amplification and associated overexpression of epidermal growth factor receptor (EGFR), which occurs in 40%–50% of primary tumors. In addition, tumors that develop in rodents following intracranial implantation of cultured GBM cells often lack key phenotypes observed in patient tumors, such as angiogenesis and infiltrative growth. In contrast, GBM tumors maintained as subcutaneous xenografts in nude mice demonstrate maintenance of EGFR gene amplification through serial in vivo propagation,4 and additionally recapitulate the invasive growth pattern of patient tumors when transplanted intracranially in rodents.5 A GBM xenograft (GBMX) tumor panel has enabled studies aimed at directly assessing the effect of EGFR amplification on GBM radiation response,6 and correlating tumor PTEN (phosphatase and tensin homolog deleted on chromosome 10) and EGFR status with response to the EGFR kinase inhibitor erlotinib.7 While these studies suggest that subcutaneously propagated GBMX tumors more accurately model GBM molecular biology and therapeutic responses than do permanent cell lines, it remains unclear the extent to which this GBMX tumor panel represents the molecular subtypes of patient GBMs.
In this study, we assessed DNA copy number aberrations and mRNA transcript levels in a 21-member GBMX tumor panel and compared these molecular data sets with data sets derived from GBM clinical specimens. This comparative genomic approach enabled the identification of a number of aberrantly overexpressed transcripts in GBM and GBMX tumors for which targeted inhibition may prove efficacious for disease treatment.
Materials and Methods
Samples
Subcutaneous GBMX tumors were surgically removed in accordance with procedures approved by the Institutional Animal Care and Use Committee and snap frozen in liquid nitrogen. Snap-frozen nonneoplastic brain tissues were derived from the temporal lobes of epileptic patient surgeries and were composed primarily of cortex with mild to moderate reactive astrocytosis and neurons. Nonneoplastic controls were obtained from the Brain Tumor Research Center tissue core at the University of California–San Francisco in accordance with procedures approved by the Committee on Human Research. All samples were ground to a powder using a liquid-nitrogen–cooled pestle and mortar, and DNA and RNA were extracted from separate aliquots of ground tissue.
DNA Copy Number Analyses
DNA extractions8 and hybridizations to Affymetrix 50K Xba single nucleotide polymorphism (SNP) chip arrays9 (Affymetrix, Inc., Santa Clara, CA, USA) were performed as previously described. SNP array data were preprocessed as follows: perfect-match (PM) probe intensities of the 21 GBMX tumors were quantile normalized with those of 90 normal tissue controls (HapMap trios, Affymetrix). The total hybridization intensities, PM A + PM B (in logarithm base two), were median-summarized over the five to seven probe quartets for each SNP, followed by a fragment-length adjustment using cubic splines.10 We then calculated the log2 copy number ratio for each SNP by subtracting the mean SNP log2 intensity of all 90 HapMap reference samples from the per-SNP intensity within each GBMX tumor to remove SNP-specific effects. Copy number was segmented along each chromosome into regions of equal copy number changes with a circular binary segmentation algorithm11 implemented in the DNAcopy package of R/Bioconductor (version BioC 2.2; www.bioconductor.org,12 using the National Center for Biotechnology Information Build 36.1 annotation from Affymetrix [dated July 12, 2007]). Neighboring genomic segments were merged if their estimated copy numbers did not differ by more than one standard deviation. Frequencies of copy number gain or loss were calculated using segment mean thresholds of ±0.3. For analyses of GBM clinical specimens, segment mean thresholds of ±0.3 were used on data generated from SNP arrays,13 and thresholds of ±0.1 were used on data generated from bacterial artificial chromosome (BAC) arrays14,15 and Agilent 244K oligonucleotide arrays2 (Agilent Technologies, Santa Clara, CA, USA). All array data included in this manuscript are accessible through Gene Expression Omnibus Series accession no. GSE14806 (www.ncbi.nlm.nih.gov/geo).
mRNA Expression Analyses
Total RNA was extracted from GBMX tumors and nonneoplastic control brain using the mirVana RNA isolation system (Ambion, Inc., Austin, TX, USA), further purified using RNeasy columns (Qiagen, Inc., Valencia, CA, USA), and RNA integrity assessed using a bioanalyzer (Agilent). RNA from all samples was hybridized in parallel to Human U133A GeneChip arrays on the Affymetrix HTA system (HT_HG-U133A). CEL raw data files were read into R/Bioconductor using the Affy/affyPLM package,16 and RMA (robust multiarray average) intensity in log2 scale was generated for each probe set (gene). The 11 PM intensities per probe set were background-corrected, quantile-normalized (to make the distribution of intensities the same for all arrays), and summarized for each probe set using a robust fit of linear models as previously described.17
Unsupervised hierarchical clustering based on the most variably expressed genes, defined by the medium absolute deviation, was conducted using Pearson's correlation coefficient or Euclidean distance as the similarity metrics and Ward's linkage method or the complete linkage method as the between-cluster distance metrics. Separate analyses were conducted for the top n = 50,200,500 variably expressed genes among all samples (nonneoplastic control brain and GBMX tumors) and among GBMX tumors only. Analyses of the proneural (Pn), mesenchymal (Mes), and proliferative (Pr) gene expression signatures in GBMX tumors, GBM tumors from M.D. Anderson Cancer Center, and GBM tumors from the University of California, San Francisco, were conducted using 478 U133A genes from the set of 725 U133A and U133B survival-associated all-marker genes.15 Relative expression was determined by comparing the median expression of the Pn, Mes, and Pr signature genes in each tumor group relative to the median expression in respective nonneoplastic control tissues.
For comparisons of gene expression between GBMX tumors and nonneoplastic controls, paired t-tests were performed on the average log2 intensity of each probe set in GBMX tumors and nonneoplastic controls. p-Values were adjusted for multiple comparisons using Bonferroni-corrected p-values of the moderated t-statistics. GOstat analysis18 (GOstat is software made freely available at http://gostat.wehi.edu.au) was conducted using 607 genes that were significantly upregulated at least 2-fold in GBMX tumors. This gene list was searched against the AFFY_HG_U133A gene ontology (GO) gene association database; the maximal p-value in the GO output list was set to 1–10, and the minimal length of considered GO paths was set to 5. GOs were merged if the indicating gene lists were inclusions or differed by fewer than 10 genes.
Assessment of gene expression from Cancer Genome Atlas (TCGA)2 was conducted on GBM tumors (n = 201) and 100% nontumor controls (n = 5) analyzed with the Affymetrix exon array 1.0 platform. Raw data were preprocessed with RMA17 and aroma.affymetrix.19 The average and maximum fold changes in the GBMs were calculated relative to the median log2 expression value of the nontumor samples.
Results
Molecular Subclassification of GBMX
Prior global assessments of DNA copy number and mRNA expression suggest the presence of distinct molecular subsets of GBM.14,15,20–22 To assess molecular subclass representation among GBMX tumors, we examined DNA copy number aberrations (CNAs) and mRNA expression profiles in 21 distinct xenografts (Supplementary Table 1). For CNA assessment, we used the Affymetrix 50K Xba SNP chip, which enabled identification of the expected EGFR amplifications4 and CDKN2A (cyclin-dependent kinase inhibitor 2A) homozygous deletions (CD James, personal communication; Supplementary Fig. 1). We next compared the frequencies of genomic CNA between GBMX tumors and a series of de novo GBM specimens.2,13,14 The patterns of recurrent CNA were highly similar between both tumor groups (Fig. 1); the most frequently observed CNAs were whole chromosome 7 gains, whole chromosome 10 losses, CDKN2A homozygous deletions, and EGFR amplifications.
To assess transcriptional heterogeneity in the GBMX tumor panel, we next determined mRNA expression profiles of GBMX tumors and nonneoplastic control brain tissues using Human U133A GeneChip arrays on the Affymetrix HTA system (HT_HG-U133A). As expected, unsupervised hierarchical clustering of the most variably expressed genes among all samples segregated tumors and nonneoplastic controls into distinct classes (Fig. 2A). Also, GBMX tumors from within a tumor line, but from distinct tumor passages, showed a higher extent of identity than when compared to any other tumor line (Fig. 2A), suggesting that GBMX transcriptional signatures are stable in association with subcutaneous propagation. However, unlike expression profiling studies of patient tumors,15,21 GBMX tumors did not reliably segregate into two or three distinct subclasses.
We next performed a supervised classification of the GBMX tumors with respect to the proneural-mesenchymal-proliferative signature gene classification scheme of high-grade astrocytomas.15 This revealed that all GBMX tumors invariably contained a strong proliferative expression signature, whereas no GBMX tumors contained evidence of a proneural signature (Fig. 2B). Expression of the mesenchymal signature was variable across GBMX tumor lines (Fig. 2B), consistent with observations in primary tumors.15
Cell Cycle Gene Expression Networks in GBMX Tumors
To further investigate the proliferative signature in GBMX tumors, we compared the average expression of all HT_HG-U133A probe sets between GBMX tumors and nonneoplastic control brain samples. This analysis revealed 809 probe sets (607 unique genes) that were significantly (p < 0.01, adjusted for multiple comparisons) overexpressed at least 2-fold on average in GBMX tumors. To determine the biological processes associated with these 607 genes, we assessed their GO classifications using GOstat.18 This analysis revealed four main GO clusters that were significantly overrepresented in this gene list (Supplementary Table 2), comprising highly significant enrichment for genes associated with mitosis (p = 0), DNA replication (p = 1.3 × 10–51), RNA splicing (p = 7.9 × 10–28), and chromosome organization and biogenesis (p = 9.6 × 10–21). Interestingly, these GO biological processes closely resembled the mitosis and cell-cycle module (MCM) gene expression signature previously identified in GBM and breast cancer.23 Indeed, analyses of MCM hub genes (genes that show high intermolecular physical and/or functional interactions) revealed that 27 of 35 (77%) of the hub genes were among the most highly overexpressed genes (99th percentile) in GBMX tumors relative to nonneoplastic controls; each of the 27 hub genes were overexpressed >8-fold on average in GBMX tumors relative to nonneoplastic controls. Therefore, the predominant gene expression signatures observed in GBMX tumors significantly overlap with signatures observed in human GBM clinical specimens.
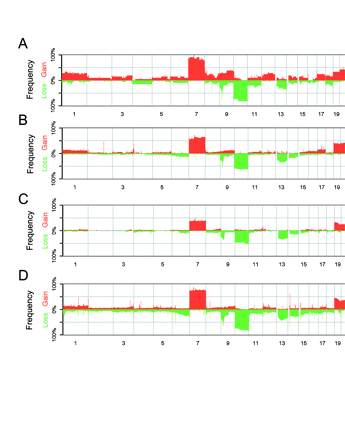
Frequencies of genomic copy number gains and losses in glioblastoma multiforme (GBM) xenograft (GBMX) tumor lines and GBM clinical samples. (A) GBMX tumors (n = 21) analyzed on the Affymetrix Xba 50K SNP array platform. (B) GBM tumors (n = 82) analyzed on the Affymetrix Xba 50K SNP array platform.13 (C) GBM tumors (n = 56) analyzed on the BAC array platform.14 (D) GBM tumors (n = 221) analyzed on the Agilent 244K oligonucleotide array platform.2
To determine which of the mitosis, DNA replication, and chromosome organization (MRC) genes overexpressed in GBMX tumors (n = 389) were also overexpressed in GBM clinical samples, we analyzed expression data from 201 GBM clinical specimens derived from the TCGA. This revealed that 41 of 389 (11%) of these genes were overexpressed at least 2-fold on average in GBM clinical specimens, as well as in GBMX tumors, relative to nontumor controls (Table 1). Characterization of these genes using ingenuity pathways analysis revealed two main cell-cycle expression networks: (1) cellular assembly and organization (Fig. 3A), and (2) DNA replication, recombination, and repair (Fig. 3B). Within the cellular assembly and organization network, aurora kinase B (AURKB), cyclin B1 (CCNB1), cyclin-dependent kinase 1 (CDC2), cyclin-dependent kinase 2 (CDK2), and forkhead box M1 (FOXM1) were the principal hub genes, showing the highest degree of intranetwork connectivity. These genes have been implicated in the development of multiple malignancies, including GBM, and play important roles in ensuring appropriate progression through mitosis. Within the DNA replication, recombination, and repair network, the main hub gene was the tumor suppressor TP53, suggesting that loss of TP53 function in GBM results in transcriptional upregulation of a gene expression network important for transition through S-phase of the cell cycle.
Mitosis, DNA replication, and chromosome (MRC) organization genes overexpressed in glioblastoma multiforme xenograft (GBMX) tumors and de novo GBM tumors
| Fold Change | ||||||||
|---|---|---|---|---|---|---|---|---|
| GBMX Tumors | GBM Tumors | |||||||
| Gene | Gene Symbol | Average | Maximum | Average | Maximum | |||
| 1 | ASPM | 71 | 140 | 13 | 28 | |||
| 2 | AURKB | 11 | 23 | 3 | 6 | |||
| 3 | BIRC5 | 42 | 109 | 3 | 6 | |||
| 4 | BUB1 | 22 | 45 | 5 | 9 | |||
| 5 | BUB1B | 26 | 53 | 6 | 13 | |||
| 6 | CCNB2 | 48 | 103 | 3 | 7 | |||
| 7 | CDC2 | 50 | 126 | 5 | 17 | |||
| 8 | CDC45L | 10 | 30 | 6 | 10 | |||
| 9 | CDCA8 | 9 | 22 | 3 | 7 | |||
| 10 | CDK2 | 16 | 31 | 3 | 5 | |||
| 11 | CENPE | 17 | 32 | 8 | 18 | |||
| 12 | CENPF | 33 | 67 | 16 | 22 | |||
| 13 | CHEK1 | 14 | 40 | 3 | 5 | |||
| 14 | DLG7 | 38 | 85 | 11 | 16 | |||
| 15 | DTL | 41 | 74 | 4 | 11 | |||
| 16 | E2F8 | 7 | 18 | 6 | 9 | |||
| 17 | FOXM1 | 18 | 32 | 3 | 6 | |||
| 18 | GINS2 | 11 | 20 | 3 | 6 | |||
| 19 | HELLS | 5 | 16 | 3 | 9 | |||
| 20 | IGF2BP3 | 64 | 167 | 2 | 3 | |||
| 21 | KIF14 | 13 | 33 | 8 | 14 | |||
| 22 | KIF18A | 9 | 18 | 6 | 17 | |||
| 23 | KIF20A | 32 | 70 | 5 | 9 | |||
| 24 | KIF23 | 21 | 42 | 6 | 11 | |||
| 25 | KIF2C | 33 | 81 | 3 | 6 | |||
| 26 | KIF4A | 24 | 50 | 7 | 11 | |||
| 27 | MLF1IP | 57 | 140 | 6 | 20 | |||
| 28 | MYBL2 | 5 | 13 | 3 | 7 | |||
| 29 | NCAPG | 22 | 37 | 6 | 13 | |||
| 30 | NDC80 | 30 | 73 | 8 | 11 | |||
| 31 | NEK2 | 16 | 31 | 2 | 4 | |||
| 32 | PBK | 106 | 252 | 7 | 14 | |||
| 33 | RAD51AP1 | 21 | 46 | 2 | 6 | |||
| 34 | RRM2 | 112 | 285 | 5 | 13 | |||
| 35 | SMC4 | 28 | 59 | 3 | 6 | |||
| 36 | SPC25 | 16 | 30 | 3 | 13 | |||
| 37 | TOP2A | 83 | 152 | 10 | 14 | |||
| 38 | TPX2 | 47 | 113 | 6 | 10 | |||
| 39 | TTK | 25 | 50 | 10 | 19 | |||
| 40 | UBE2C | 49 | 112 | 3 | 4 | |||
| 41 | WEE1 | 13 | 24 | 4 | 6 | |||
| Fold Change | ||||||||
|---|---|---|---|---|---|---|---|---|
| GBMX Tumors | GBM Tumors | |||||||
| Gene | Gene Symbol | Average | Maximum | Average | Maximum | |||
| 1 | ASPM | 71 | 140 | 13 | 28 | |||
| 2 | AURKB | 11 | 23 | 3 | 6 | |||
| 3 | BIRC5 | 42 | 109 | 3 | 6 | |||
| 4 | BUB1 | 22 | 45 | 5 | 9 | |||
| 5 | BUB1B | 26 | 53 | 6 | 13 | |||
| 6 | CCNB2 | 48 | 103 | 3 | 7 | |||
| 7 | CDC2 | 50 | 126 | 5 | 17 | |||
| 8 | CDC45L | 10 | 30 | 6 | 10 | |||
| 9 | CDCA8 | 9 | 22 | 3 | 7 | |||
| 10 | CDK2 | 16 | 31 | 3 | 5 | |||
| 11 | CENPE | 17 | 32 | 8 | 18 | |||
| 12 | CENPF | 33 | 67 | 16 | 22 | |||
| 13 | CHEK1 | 14 | 40 | 3 | 5 | |||
| 14 | DLG7 | 38 | 85 | 11 | 16 | |||
| 15 | DTL | 41 | 74 | 4 | 11 | |||
| 16 | E2F8 | 7 | 18 | 6 | 9 | |||
| 17 | FOXM1 | 18 | 32 | 3 | 6 | |||
| 18 | GINS2 | 11 | 20 | 3 | 6 | |||
| 19 | HELLS | 5 | 16 | 3 | 9 | |||
| 20 | IGF2BP3 | 64 | 167 | 2 | 3 | |||
| 21 | KIF14 | 13 | 33 | 8 | 14 | |||
| 22 | KIF18A | 9 | 18 | 6 | 17 | |||
| 23 | KIF20A | 32 | 70 | 5 | 9 | |||
| 24 | KIF23 | 21 | 42 | 6 | 11 | |||
| 25 | KIF2C | 33 | 81 | 3 | 6 | |||
| 26 | KIF4A | 24 | 50 | 7 | 11 | |||
| 27 | MLF1IP | 57 | 140 | 6 | 20 | |||
| 28 | MYBL2 | 5 | 13 | 3 | 7 | |||
| 29 | NCAPG | 22 | 37 | 6 | 13 | |||
| 30 | NDC80 | 30 | 73 | 8 | 11 | |||
| 31 | NEK2 | 16 | 31 | 2 | 4 | |||
| 32 | PBK | 106 | 252 | 7 | 14 | |||
| 33 | RAD51AP1 | 21 | 46 | 2 | 6 | |||
| 34 | RRM2 | 112 | 285 | 5 | 13 | |||
| 35 | SMC4 | 28 | 59 | 3 | 6 | |||
| 36 | SPC25 | 16 | 30 | 3 | 13 | |||
| 37 | TOP2A | 83 | 152 | 10 | 14 | |||
| 38 | TPX2 | 47 | 113 | 6 | 10 | |||
| 39 | TTK | 25 | 50 | 10 | 19 | |||
| 40 | UBE2C | 49 | 112 | 3 | 4 | |||
| 41 | WEE1 | 13 | 24 | 4 | 6 | |||
Mitosis, DNA replication, and chromosome (MRC) organization genes overexpressed in glioblastoma multiforme xenograft (GBMX) tumors and de novo GBM tumors
| Fold Change | ||||||||
|---|---|---|---|---|---|---|---|---|
| GBMX Tumors | GBM Tumors | |||||||
| Gene | Gene Symbol | Average | Maximum | Average | Maximum | |||
| 1 | ASPM | 71 | 140 | 13 | 28 | |||
| 2 | AURKB | 11 | 23 | 3 | 6 | |||
| 3 | BIRC5 | 42 | 109 | 3 | 6 | |||
| 4 | BUB1 | 22 | 45 | 5 | 9 | |||
| 5 | BUB1B | 26 | 53 | 6 | 13 | |||
| 6 | CCNB2 | 48 | 103 | 3 | 7 | |||
| 7 | CDC2 | 50 | 126 | 5 | 17 | |||
| 8 | CDC45L | 10 | 30 | 6 | 10 | |||
| 9 | CDCA8 | 9 | 22 | 3 | 7 | |||
| 10 | CDK2 | 16 | 31 | 3 | 5 | |||
| 11 | CENPE | 17 | 32 | 8 | 18 | |||
| 12 | CENPF | 33 | 67 | 16 | 22 | |||
| 13 | CHEK1 | 14 | 40 | 3 | 5 | |||
| 14 | DLG7 | 38 | 85 | 11 | 16 | |||
| 15 | DTL | 41 | 74 | 4 | 11 | |||
| 16 | E2F8 | 7 | 18 | 6 | 9 | |||
| 17 | FOXM1 | 18 | 32 | 3 | 6 | |||
| 18 | GINS2 | 11 | 20 | 3 | 6 | |||
| 19 | HELLS | 5 | 16 | 3 | 9 | |||
| 20 | IGF2BP3 | 64 | 167 | 2 | 3 | |||
| 21 | KIF14 | 13 | 33 | 8 | 14 | |||
| 22 | KIF18A | 9 | 18 | 6 | 17 | |||
| 23 | KIF20A | 32 | 70 | 5 | 9 | |||
| 24 | KIF23 | 21 | 42 | 6 | 11 | |||
| 25 | KIF2C | 33 | 81 | 3 | 6 | |||
| 26 | KIF4A | 24 | 50 | 7 | 11 | |||
| 27 | MLF1IP | 57 | 140 | 6 | 20 | |||
| 28 | MYBL2 | 5 | 13 | 3 | 7 | |||
| 29 | NCAPG | 22 | 37 | 6 | 13 | |||
| 30 | NDC80 | 30 | 73 | 8 | 11 | |||
| 31 | NEK2 | 16 | 31 | 2 | 4 | |||
| 32 | PBK | 106 | 252 | 7 | 14 | |||
| 33 | RAD51AP1 | 21 | 46 | 2 | 6 | |||
| 34 | RRM2 | 112 | 285 | 5 | 13 | |||
| 35 | SMC4 | 28 | 59 | 3 | 6 | |||
| 36 | SPC25 | 16 | 30 | 3 | 13 | |||
| 37 | TOP2A | 83 | 152 | 10 | 14 | |||
| 38 | TPX2 | 47 | 113 | 6 | 10 | |||
| 39 | TTK | 25 | 50 | 10 | 19 | |||
| 40 | UBE2C | 49 | 112 | 3 | 4 | |||
| 41 | WEE1 | 13 | 24 | 4 | 6 | |||
| Fold Change | ||||||||
|---|---|---|---|---|---|---|---|---|
| GBMX Tumors | GBM Tumors | |||||||
| Gene | Gene Symbol | Average | Maximum | Average | Maximum | |||
| 1 | ASPM | 71 | 140 | 13 | 28 | |||
| 2 | AURKB | 11 | 23 | 3 | 6 | |||
| 3 | BIRC5 | 42 | 109 | 3 | 6 | |||
| 4 | BUB1 | 22 | 45 | 5 | 9 | |||
| 5 | BUB1B | 26 | 53 | 6 | 13 | |||
| 6 | CCNB2 | 48 | 103 | 3 | 7 | |||
| 7 | CDC2 | 50 | 126 | 5 | 17 | |||
| 8 | CDC45L | 10 | 30 | 6 | 10 | |||
| 9 | CDCA8 | 9 | 22 | 3 | 7 | |||
| 10 | CDK2 | 16 | 31 | 3 | 5 | |||
| 11 | CENPE | 17 | 32 | 8 | 18 | |||
| 12 | CENPF | 33 | 67 | 16 | 22 | |||
| 13 | CHEK1 | 14 | 40 | 3 | 5 | |||
| 14 | DLG7 | 38 | 85 | 11 | 16 | |||
| 15 | DTL | 41 | 74 | 4 | 11 | |||
| 16 | E2F8 | 7 | 18 | 6 | 9 | |||
| 17 | FOXM1 | 18 | 32 | 3 | 6 | |||
| 18 | GINS2 | 11 | 20 | 3 | 6 | |||
| 19 | HELLS | 5 | 16 | 3 | 9 | |||
| 20 | IGF2BP3 | 64 | 167 | 2 | 3 | |||
| 21 | KIF14 | 13 | 33 | 8 | 14 | |||
| 22 | KIF18A | 9 | 18 | 6 | 17 | |||
| 23 | KIF20A | 32 | 70 | 5 | 9 | |||
| 24 | KIF23 | 21 | 42 | 6 | 11 | |||
| 25 | KIF2C | 33 | 81 | 3 | 6 | |||
| 26 | KIF4A | 24 | 50 | 7 | 11 | |||
| 27 | MLF1IP | 57 | 140 | 6 | 20 | |||
| 28 | MYBL2 | 5 | 13 | 3 | 7 | |||
| 29 | NCAPG | 22 | 37 | 6 | 13 | |||
| 30 | NDC80 | 30 | 73 | 8 | 11 | |||
| 31 | NEK2 | 16 | 31 | 2 | 4 | |||
| 32 | PBK | 106 | 252 | 7 | 14 | |||
| 33 | RAD51AP1 | 21 | 46 | 2 | 6 | |||
| 34 | RRM2 | 112 | 285 | 5 | 13 | |||
| 35 | SMC4 | 28 | 59 | 3 | 6 | |||
| 36 | SPC25 | 16 | 30 | 3 | 13 | |||
| 37 | TOP2A | 83 | 152 | 10 | 14 | |||
| 38 | TPX2 | 47 | 113 | 6 | 10 | |||
| 39 | TTK | 25 | 50 | 10 | 19 | |||
| 40 | UBE2C | 49 | 112 | 3 | 4 | |||
| 41 | WEE1 | 13 | 24 | 4 | 6 | |||
Expression of Genomically Amplified Genes in GBMX Tumors
As expected from previous fluorescence in situ hybridization studies,4 our microarray analyses revealed high-level EGFR amplification (log2 ratio > 4; 32 copies) in a significant proportion (8 of 21) of GBMX tumors. This frequency is very consistent with frequencies reported in association with the analysis of large series of patient tumors,24–26 suggesting that there is no selection bias for establishing xenografts based on patient tumor EGFR amplification status. Among GBMX tumors, EGFR transcript levels were highly correlated with genomic amplification (Fig. 4A). Because of this, we identified all genomic loci for which the segment mean log2 copy number ratio was greater than 4 in at least one GBMX tumor line to identify additional amplification–copy number relationships. We identified 15 such amplicons (including the EGFR amplicon), which ranged in size from 215 kb to 3.0 Mb (Table 2). Within these amplicons, we identified a total of 58 National Center for Biotechnology Information Reference Sequence genes, 51 of which contained probe sets on the U133A gene expression arrays (Supplementary Table 3). Of these, 33 were selectively overexpressed >5-fold in the amplicon-bearing tumors relative to the average expression in non-amplified tumors (Supplementary Table 3).
High-level genomic amplicons (Log2 ratio > 4) in glioblastoma multiforme xenograft (GBMX) tumors
| Frequency of Focal Amplifications (TCGA GBM) | Focal CNA Region (TCGA GBM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GBMX Amplicon | Chromosome | Minimal Amplicon (NCBI Build 36.1) | Maximum Copy No. (Log2) | Width (kb) | GBMX Line | GISTIC | RAE | GTS | Amplified and Overexpressed Genes (GBMX) | |
| 1 | 2 | chr2:14372951–15050902 | 5.7 | 678 | 28 | 2.4 | — | — | ||
| 2 | 2 | chr2:15614969–16256395 | 6.2 | 641 | 28 | 2.4 | — | — | DDX1, MYCN | |
| 3 | 2 | chr2:120958377–121603516 | 6.3 | 645 | 28 | — | — | Y | GLI1 | |
| 4 | 7 | chr7:51987798–53893473 | 5.0 | 1906 | 26 | — | — | Y | ||
| 5 | 7 | chr7:54970126–55186653 | 6.2 | 215 | a | 43.2 | 44.0 | Y | EGFR | |
| 6 | 7 | chr7:90939401–91161022 | 6.5 | 222 | 34 | — | — | — | ||
| 7 | 7 | chr7:91236115–94061316 | 5.0 | 2825 | 34 | — | 3.4 | 3.7 | BET1, CASD1, CDK6, DKFZ564O0523, KRIT1, MTERF, PEX1, SAMD9 | |
| 8 | 7 | chr7:150881861–150906739 | 5.7 | 347 | 12, 34 | — | — | — | ||
| 9 | 8 | chr8:36998497–37472939 | 4.2 | 474 | 22 | — | — | — | ||
| 10 | 8 | chr8:129006828–131970985 | 4.8 | 2964 | 5 | — | — | — | DDEF1, FAM49B | |
| 11 | 8 | chr8:137526870–137892295 | 4.7 | 365 | 22 | — | — | — | ||
| 12 | 11 | chr11:101575571–102626961 | 5.1 | 1051 | 22 | — | — | — | BIRC2, BIRC3, MMP1, MMP10, MMP12, MMP13, MMP27, MMP3, MMP8, TMEM123, YAP1 | |
| 13 | 12 | chr12:66409069–67695739 | 4.9 | 1287 | 5 | 10.7 | 10.2 | Y | CPM, IFNG, IL26, MDM1, MDM2, NUP107, SLC35E3 | |
| 14 | 13 | chr13:23598616–24008514 | 5.3 | 410 | 6 | — | — | — | PARP4 | |
| 15 | 13 | chr13:66424482–67844382 | 4.5 | 1420 | 6 | — | — | — | — | |
| Frequency of Focal Amplifications (TCGA GBM) | Focal CNA Region (TCGA GBM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GBMX Amplicon | Chromosome | Minimal Amplicon (NCBI Build 36.1) | Maximum Copy No. (Log2) | Width (kb) | GBMX Line | GISTIC | RAE | GTS | Amplified and Overexpressed Genes (GBMX) | |
| 1 | 2 | chr2:14372951–15050902 | 5.7 | 678 | 28 | 2.4 | — | — | ||
| 2 | 2 | chr2:15614969–16256395 | 6.2 | 641 | 28 | 2.4 | — | — | DDX1, MYCN | |
| 3 | 2 | chr2:120958377–121603516 | 6.3 | 645 | 28 | — | — | Y | GLI1 | |
| 4 | 7 | chr7:51987798–53893473 | 5.0 | 1906 | 26 | — | — | Y | ||
| 5 | 7 | chr7:54970126–55186653 | 6.2 | 215 | a | 43.2 | 44.0 | Y | EGFR | |
| 6 | 7 | chr7:90939401–91161022 | 6.5 | 222 | 34 | — | — | — | ||
| 7 | 7 | chr7:91236115–94061316 | 5.0 | 2825 | 34 | — | 3.4 | 3.7 | BET1, CASD1, CDK6, DKFZ564O0523, KRIT1, MTERF, PEX1, SAMD9 | |
| 8 | 7 | chr7:150881861–150906739 | 5.7 | 347 | 12, 34 | — | — | — | ||
| 9 | 8 | chr8:36998497–37472939 | 4.2 | 474 | 22 | — | — | — | ||
| 10 | 8 | chr8:129006828–131970985 | 4.8 | 2964 | 5 | — | — | — | DDEF1, FAM49B | |
| 11 | 8 | chr8:137526870–137892295 | 4.7 | 365 | 22 | — | — | — | ||
| 12 | 11 | chr11:101575571–102626961 | 5.1 | 1051 | 22 | — | — | — | BIRC2, BIRC3, MMP1, MMP10, MMP12, MMP13, MMP27, MMP3, MMP8, TMEM123, YAP1 | |
| 13 | 12 | chr12:66409069–67695739 | 4.9 | 1287 | 5 | 10.7 | 10.2 | Y | CPM, IFNG, IL26, MDM1, MDM2, NUP107, SLC35E3 | |
| 14 | 13 | chr13:23598616–24008514 | 5.3 | 410 | 6 | — | — | — | PARP4 | |
| 15 | 13 | chr13:66424482–67844382 | 4.5 | 1420 | 6 | — | — | — | — | |
Abbreviations: NCBI, National Center for Biotechnology Information; TCGA, The Cancer Genome Atlas; CNA, Copy Number Aberration; GISTIC, Genomic Identification of Significant Targets in Cancer; GTS, Genome Topography Scan. Amplicons shown in boldface are those present in both GBMX tumors (this study) and TCGA-derived GBM tumors
EGFR (epidermal growth factor receptor) amplification (log2 > 4) was observed in 8 of 21 independent GBMX lines: 6, 8, 12, 15, 26, 34, 38, and 39
High-level genomic amplicons (Log2 ratio > 4) in glioblastoma multiforme xenograft (GBMX) tumors
| Frequency of Focal Amplifications (TCGA GBM) | Focal CNA Region (TCGA GBM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GBMX Amplicon | Chromosome | Minimal Amplicon (NCBI Build 36.1) | Maximum Copy No. (Log2) | Width (kb) | GBMX Line | GISTIC | RAE | GTS | Amplified and Overexpressed Genes (GBMX) | |
| 1 | 2 | chr2:14372951–15050902 | 5.7 | 678 | 28 | 2.4 | — | — | ||
| 2 | 2 | chr2:15614969–16256395 | 6.2 | 641 | 28 | 2.4 | — | — | DDX1, MYCN | |
| 3 | 2 | chr2:120958377–121603516 | 6.3 | 645 | 28 | — | — | Y | GLI1 | |
| 4 | 7 | chr7:51987798–53893473 | 5.0 | 1906 | 26 | — | — | Y | ||
| 5 | 7 | chr7:54970126–55186653 | 6.2 | 215 | a | 43.2 | 44.0 | Y | EGFR | |
| 6 | 7 | chr7:90939401–91161022 | 6.5 | 222 | 34 | — | — | — | ||
| 7 | 7 | chr7:91236115–94061316 | 5.0 | 2825 | 34 | — | 3.4 | 3.7 | BET1, CASD1, CDK6, DKFZ564O0523, KRIT1, MTERF, PEX1, SAMD9 | |
| 8 | 7 | chr7:150881861–150906739 | 5.7 | 347 | 12, 34 | — | — | — | ||
| 9 | 8 | chr8:36998497–37472939 | 4.2 | 474 | 22 | — | — | — | ||
| 10 | 8 | chr8:129006828–131970985 | 4.8 | 2964 | 5 | — | — | — | DDEF1, FAM49B | |
| 11 | 8 | chr8:137526870–137892295 | 4.7 | 365 | 22 | — | — | — | ||
| 12 | 11 | chr11:101575571–102626961 | 5.1 | 1051 | 22 | — | — | — | BIRC2, BIRC3, MMP1, MMP10, MMP12, MMP13, MMP27, MMP3, MMP8, TMEM123, YAP1 | |
| 13 | 12 | chr12:66409069–67695739 | 4.9 | 1287 | 5 | 10.7 | 10.2 | Y | CPM, IFNG, IL26, MDM1, MDM2, NUP107, SLC35E3 | |
| 14 | 13 | chr13:23598616–24008514 | 5.3 | 410 | 6 | — | — | — | PARP4 | |
| 15 | 13 | chr13:66424482–67844382 | 4.5 | 1420 | 6 | — | — | — | — | |
| Frequency of Focal Amplifications (TCGA GBM) | Focal CNA Region (TCGA GBM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GBMX Amplicon | Chromosome | Minimal Amplicon (NCBI Build 36.1) | Maximum Copy No. (Log2) | Width (kb) | GBMX Line | GISTIC | RAE | GTS | Amplified and Overexpressed Genes (GBMX) | |
| 1 | 2 | chr2:14372951–15050902 | 5.7 | 678 | 28 | 2.4 | — | — | ||
| 2 | 2 | chr2:15614969–16256395 | 6.2 | 641 | 28 | 2.4 | — | — | DDX1, MYCN | |
| 3 | 2 | chr2:120958377–121603516 | 6.3 | 645 | 28 | — | — | Y | GLI1 | |
| 4 | 7 | chr7:51987798–53893473 | 5.0 | 1906 | 26 | — | — | Y | ||
| 5 | 7 | chr7:54970126–55186653 | 6.2 | 215 | a | 43.2 | 44.0 | Y | EGFR | |
| 6 | 7 | chr7:90939401–91161022 | 6.5 | 222 | 34 | — | — | — | ||
| 7 | 7 | chr7:91236115–94061316 | 5.0 | 2825 | 34 | — | 3.4 | 3.7 | BET1, CASD1, CDK6, DKFZ564O0523, KRIT1, MTERF, PEX1, SAMD9 | |
| 8 | 7 | chr7:150881861–150906739 | 5.7 | 347 | 12, 34 | — | — | — | ||
| 9 | 8 | chr8:36998497–37472939 | 4.2 | 474 | 22 | — | — | — | ||
| 10 | 8 | chr8:129006828–131970985 | 4.8 | 2964 | 5 | — | — | — | DDEF1, FAM49B | |
| 11 | 8 | chr8:137526870–137892295 | 4.7 | 365 | 22 | — | — | — | ||
| 12 | 11 | chr11:101575571–102626961 | 5.1 | 1051 | 22 | — | — | — | BIRC2, BIRC3, MMP1, MMP10, MMP12, MMP13, MMP27, MMP3, MMP8, TMEM123, YAP1 | |
| 13 | 12 | chr12:66409069–67695739 | 4.9 | 1287 | 5 | 10.7 | 10.2 | Y | CPM, IFNG, IL26, MDM1, MDM2, NUP107, SLC35E3 | |
| 14 | 13 | chr13:23598616–24008514 | 5.3 | 410 | 6 | — | — | — | PARP4 | |
| 15 | 13 | chr13:66424482–67844382 | 4.5 | 1420 | 6 | — | — | — | — | |
Abbreviations: NCBI, National Center for Biotechnology Information; TCGA, The Cancer Genome Atlas; CNA, Copy Number Aberration; GISTIC, Genomic Identification of Significant Targets in Cancer; GTS, Genome Topography Scan. Amplicons shown in boldface are those present in both GBMX tumors (this study) and TCGA-derived GBM tumors
EGFR (epidermal growth factor receptor) amplification (log2 > 4) was observed in 8 of 21 independent GBMX lines: 6, 8, 12, 15, 26, 34, 38, and 39
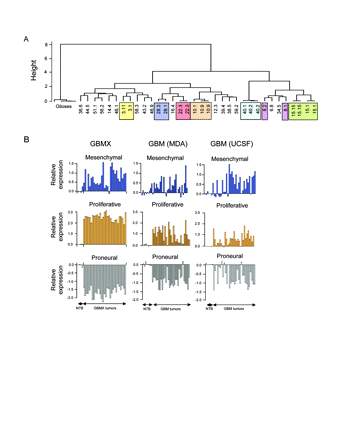
(A) Unsupervised hierarchical clustering dendrogram (Pearson-Ward) of glioblastoma multiforme xenograft (GBMX) tumors and nonneoplastic controls based on the 100 most variably expressed genes among the samples. Colored boxes represent tumors from the same line but from distinct tumor passages (the number after the period represents the generation number). (B) Expression of proneural, mesenchymal, and proliferative genes in GBMX tumors and GBM clinical samples (MDA, M.D. Anderson Cancer Center; UCSF, University of California, San Francisco).15
We next assessed whether DNA copy number and mRNA expression were increased for the 33 amplified and overexpressed GBMX genes in 228 human GBMs from TCGA.2 We first compared the amplicons observed in GBMX tumors with the amplicons detected in TCGA GBM copy number data sets; a locus was considered amplified in GBMs if it was observed on at least one of three microarray platforms and with at least one of three analytical algorithms.2 This revealed that 7 of the 15 GBMX amplicons were focally amplified in TCGA GBMs (Table 2). Further, a majority of the amplified and overexpressed GBMX genes (19 of 33) showed evidence for high overexpression (4- to 321-fold) in a subset of TCGA GBMs (Supplementary Table 3). Of particular interest, in the 12p13 amplicon, expression of MDM2 (Mdm2 p53 binding protein homolog), SLC35E3 (solute carrier family 35, member E3; UniProt ID Q7Z769), and NUP107 (nucleoporin 107kDa) clearly separated GBMs into two distinct expression groups (Fig. 4B). In the 11q22 amplicon, the collagenase genes matrix metallopeptidase 1 (MMP1) and MMP13 were transcriptionally overexpressed at least 10-fold in approximately 5% of GBMs relative to the median expression in all tumors (Fig. 4C); MMP13 was overexpressed more than 100-fold in multiple GBMs. Finally, in the 2p24 amplicon, both DDX1 (DEAD [Asp-Glu-Ala-Asp] box polypeptide 1) and MYCN (v-myc myelocytomatosis viral related oncogene, neuroblastoma derived) were clearly overexpressed in a small proportion (1%, 2 of 228) of GBMs (Fig. 4D).
Discussion
Molecular Subclassification of GBMX Tumors
Results from the microarray analyses conducted in this study revealed that GBMX tumors recapitulate many of the key molecular features described in GBM clinical samples. For example, DNA copy number aberrations in GBMX tumors showed significant similarity with results from previously published studies of patient tumors, with chromosome 7 gains, chromosome 10 losses, EGFR amplifications, and CDKN2A homozygous deletions representing the most frequent alterations in both tumor groups (Fig. 1). With respect to mRNA expression, the strongest expression signature in GBMX tumors was defined by genes that promote transition through S phase and mitosis during the cell cycle, and was highly similar to the MCM previously described in patient GBMs.23 However, our study also revealed differences between GBMX tumors and sets of patient GBMs. The most prominent example is the evident lack of the proneural GBM expression signature and overrepresentation of the proliferative expression signature15 (Fig. 2). This discrepancy suggests a selection bias in xenograft establishment (i.e., preferential successful engraftment of patient tumors with proliferative signatures) or that GBMs that successfully engraft in nude mice adopt a proliferative gene expression signature, irrespective of the classification signature of the original patient tumor. With regard to this latter possibility, it is important to note that three stable expression subclasses of high-grade astrocytomas could be established only if the gene list for clustering was weighted to include fewer proliferative markers.15 This gene weighting requirement argues that most, if not all, GBMs harbor a strong proliferative component that must be computationally masked to permit the unveiling of additional expression signatures (e.g., Mesenchymal, Proneural). Extending this line of reasoning, it is reasonable to hypothesize that it is this proliferative component/signature of GBM that emerges during subcutaneous xenograft growth. The consequence of this type of selection, regarding the ability of xenograft panels to recapitulate the variability of patient tumor therapeutic response, has yet to be extensively investigated, although results from preliminary reports indicate significant differences are evident between xenografts regarding their inherent radiation sensitivity and their response to the EGFR inhibitor erlotinib.6,7
The MRC Expression Signature in GBMX Tumors
Previous molecular profiling analyses of GBM tumors have defined expression signatures composed of genes that promote G1/S and G2/M cell cycle progression.15,23 We observed significant overlap of these expression signatures with the predominant expression signatures observed in GBMX tumors, defined by genes that drive MRC. Analysis of TCGA exon array expression data revealed that a subset (11%) of MRC signature genes were overexpressed more than 2-fold on average in GBMs compared to nonneoplastic controls (Table 1). Network analysis of the 41 MRC signature genes revealed two principal cell cycle networks: cellular assembly and organization (Fig. 3A) and DNA replication, recombination, and repair (Fig. 3B). The cellular assembly and organization network was primarily composed of genes that promote mitotic progression, many of which have been implicated in cancer etiology including AURKB, BIRC5 (survivin), CCNB1 (cyclin B1), CCNB2 (cyclin B2), CDC2 (CDK1), CDK2, and FOXM1. Activation of these genes induces tumorigenic phenotypes in a number of cancers, whereas their inhibition, such as has been shown for FOXM1, abrogates tumor growth and invasion27–30 and induces genomic instability.31,32 Further, many of G2/M genes identified in GBMX tumors play a role in mediating the DNA damage response in cancer cells. For example, inhibition or loss of BIRC5 sensitizes GBM cells33 and pancreatic cancer cells34,35 to ionizing radiation, and CHK2 (CHK2 checkpoint homolog) mediates stabilization of FOXM1 to stimulate expression of DNA repair genes.36 Therefore, the GBMX tumor panel should enable investigations of therapeutics that specifically inhibit genes that promote G2/M cell cycle progression in the face of DNA damage (genomic instability, ionizing radiation, temozolomide) and other cellular stresses.
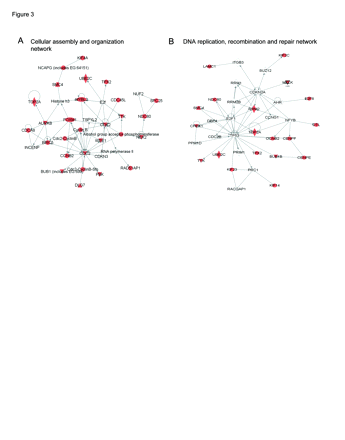
Ingenuity networks identified from M phase, DNA replication, and chromosome organization (MRC) gene list. (A) Cellular assembly and organization network. (B) DNA replication, recombination, and repair network. Genes overexpressed in glioblastoma multiforme xenograft (GBMX) and GBM tumors (Table 1) are shown by red symbols.
DNA Amplifications in GBMX Tumors and Primary GBMs
Our studies revealed a number of genomically amplified, highly overexpressed genes in GBMX tumors that are similarly amplified and/or overexpressed in GBM clinical specimens (Table 2, Fig. 4A, Supplementary Table 3). We identified well-established GBM oncogenes such as EGFR, MDM2, and CDK6 and identified additional amplified and/or overexpressed genes of potential biological or therapeutic interest in GBM. For example, NUP107 and SLC35E3 were coamplified and overexpressed with MDM2 in the 12p13 amplicon in GBMX tumor line 5, and the expression patterns of each gene clearly separated TCGA GBM clinical specimens into two distinct groups (Fig. 4B). NUP107 is a nuclear pore protein essential for kinetocore function and spindle assembly during mitosis.37,38SLC35E3 is a predicted multipass membrane protein39 that may enable targeted delivery of therapeutic agents to SLC35E3/MDM2-amplified GBMs. In the second example, we identified a cluster of MMP genes on chromosome 11q22 that were upregulated in an 11q22-amplified GBMX tumor line (GBMX line 22). Within this amplicon, the collagenase genes MMP1 and MMP13 were transcriptionally upregulated 10- to 100-fold in approximately 5% of GBM clinical specimens (Fig. 4C); both collagenases are known to promote growth and invasion of cancer cells.40–43 In the final example, we identified coamplification and overexpression of NMYC and DDX1 at chromosome locus 2p34 in GBMX line 28, as well as in a small proportion (1%) of TCGA GBMs (Fig. 4D). The MYCN transcription factor is a well-known oncogene in neuroblastoma,44 and MYCN amplifications have been previously observed in GBM.45,46 DDX1 is a member of the DEAD box protein family of RNA helicases that play important roles in RNA metabolism through modulation of inter- or intramolecular RNA structures or dissociation of RNA–protein secondary structures.47 Recent data suggest that through interaction with ATM (ataxia telangiectasia mutated), DDX1 plays an RNA clearance role at ionizing-radiation–induced DNA double-strand break sites, thereby facilitating template-guided repair of transcriptionally active regions of the genome.48
Collectively, the GBMX tumor panel provides a valuable resource with which to dissect the biology of the amplicons described herein, as it is becoming clearer that multiple genes encoded within amplicons play important roles in driving tumor biology.49,50 This in turn may lead to the development of novel therapeutic agents and strategies for disease treatment.
Targeted Molecular Therapeutics and Personalized Medicine
We have identified a number of candidate therapeutic targets in GBM, composed of genes that are genomically amplified and/or overexpressed in clinical specimens and xenografted tumors. The GBMX tumor panel provides an important resource with which to develop and test the efficacy of targeted molecular therapeutics such as novel small molecule inhibitors and RNA interference (RNAi) therapeutics51,52 as monotherapies or in combination with DNA-damaging agents such as temozolomide and ionizing radiation. RNAi utilizes small double-stranded RNA-based molecules such as small interfering RNAs (siRNAs) and microRNAs to inhibit gene expression in a nucleic-acid-sequence–specific manner. The principal advantage of siRNAs over small-molecule inhibitors and antibodies is that all genes are potential targets for inhibition; drug targeting is not limited to kinases and cell surface proteins. This dramatically expands the repertoire of candidate therapeutic targets in GBM, to include so-called “undrugable” targets such as transcription factors and oncogenes that have not been amenable to direct inhibition with small-molecule inhibitors. Examples of such genes identified in this study include FOXM1, MYCN, and BIRC5.
![Expression of amplified genes in glioblastoma multiforme (GBM) and GBM xenograft (GBMX) tumors. (A) Correlation of epidermal growth factor receptor (EGFR) expression versus EGFR copy number in GBMX tumors. (B) Expression of amplified genes NUP107 (nucleoporin 107kDa), SLC35E3 (solute carrier family 35, member E3), and MDM2 (Mdm2 p53 binding protein homolog) at 12q15 in GBMX tumors and the Cancer Genome Atlas (TCGA) GBMs. (C) Expression of amplified genes MMP1 (matrix metallopeptidase 1) and MMP13 at 11q22 in GBMX tumors and TCGA GBMs. (D) Expression of amplified genes DDX1 (DEAD [Asp-Glu-Ala-Asp] box polypeptide 1) and MYCN (v-myc myelocytomatosis viral related oncogene, neuroblastoma derived) at 2p24 in GBMX and TCGA GBMs. For B–D, open circles represent expression in the respective amplified GBMX line; expression for nonneoplastic controls and GBMX tumors has been normalized to the median expression of nonneoplastic controls; and expression for TCGA GBMs has been normalized to the median expression of all tumors.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/neuro-oncology/11/5/10.1215/15228517-2008-113/2/m_08-00284_HodgsonFIG4.gif?Expires=1716371571&Signature=RU23NRRXRFLRZUhqthGHRqm3a2cCuK9JwA3d1zviQhabIv1BvaKpa8galLLrZO53YIIo9Z8wzclP-6UMneYVy0W05KIFdSb25QjIKNi2wMVp3TNfdUuzr9pGLhM8sDYa48px9IDXgaXoaA5ATqvfcTmrH49G2~1AD55ysoKURLEHuOt9UjDIIaMwfg32-2OOSy6ODD93aW08ReMjVjJvaaQUeJmi7sBEMqER~IZ-5ltVl6v8lenzDLCCJcv6~x2MmR4vWE0F--nU1Sshaa2VyiIDqrVhTNrJ3eFXDXsuasxKEfBqwchXyL2zNYhGHeJox1CbQYGlbrNpPPsHXH5PEw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Expression of amplified genes in glioblastoma multiforme (GBM) and GBM xenograft (GBMX) tumors. (A) Correlation of epidermal growth factor receptor (EGFR) expression versus EGFR copy number in GBMX tumors. (B) Expression of amplified genes NUP107 (nucleoporin 107kDa), SLC35E3 (solute carrier family 35, member E3), and MDM2 (Mdm2 p53 binding protein homolog) at 12q15 in GBMX tumors and the Cancer Genome Atlas (TCGA) GBMs. (C) Expression of amplified genes MMP1 (matrix metallopeptidase 1) and MMP13 at 11q22 in GBMX tumors and TCGA GBMs. (D) Expression of amplified genes DDX1 (DEAD [Asp-Glu-Ala-Asp] box polypeptide 1) and MYCN (v-myc myelocytomatosis viral related oncogene, neuroblastoma derived) at 2p24 in GBMX and TCGA GBMs. For B–D, open circles represent expression in the respective amplified GBMX line; expression for nonneoplastic controls and GBMX tumors has been normalized to the median expression of nonneoplastic controls; and expression for TCGA GBMs has been normalized to the median expression of all tumors.
While delivery of therapeutics to the CNS is particularly challenging because of the blood–brain barrier, a number of promising strategies have recently been developed that may circumvent this problem. These include intranasal delivery of oligonucleotides,53 lipid encapsulation and targeted delivery of nucleic acids,54,55 and direct administration of therapeutic agents to brain tumor tissues by convection enhanced delivery.56,57 Because the GBMX tumor lines described in this study form invasive GBMs when implanted intracranially in rodents, they enable development and testing of novel strategies for targeted delivery of therapeutics to intracranial GBMX in a preclinical setting.
Development of panels of tumor lines that closely model the molecular heterogeneity and biology of patient tumors will be invaluable for developing and testing personalized molecular therapeutic strategies. The GBMX tumor panel described in this study and tumor panels described in other cancers58 constitute an important component of realizing the long-term goal of personalized medicine in cancer, wherein molecular diagnostics is closely coupled to therapeutic intervention. Future preclinical efficacy studies in the GBMX tumor panel will enable the development of predictive markers of response to a variety of inhibitory therapeutics and may also provide insights into the mechanisms of acquired resistance to these agents.
J.G.H. and R.-F.Y. contributed equally to this work.
This work was supported by the NIH grants CA101777 (J.G.H.), CA097257 (S.R.V., M.S.B., C.D.J.), and NSO49720 (C.D.J.); by the Director, Office of Science, Office of Basic Energy Sciences of the U.S. Department of Energy, under contract DE-AC02-05CH11231 (A.R., J.W.G., P.T.S.); by NIH grant U54 CA 112970 (A.R., J.W.G., P.T.S.).
References
Butowski NA, Sneed PK, Chang SM. Diagnosis and treatment of recurrent high-grade astrocytoma.
McLendon R, Friedman A, Bigner D, et al., for the Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways.
Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme.
Pandita A, Aldape KD, Zadeh G, Guha A, James CD. Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR.
Giannini C, Sarkaria JN, Saito A, et al. Patient tumor EGFR and PDG-FRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme.
Sarkaria JN, Carlson BL, Schroeder MA, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response.
Sarkaria JN, Yang L, Grogan PT, et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel.
Hodgson G, Hager JH, Volik S, et al. Genome scanning with array CGH delineates regional alterations in mouse islet carcinomas.
Zhao X, Weir BA, LaFramboise T, et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis.
Ishikawa S, Komura D, Tsuji S, et al. Allelic dosage analysis with genotyping microarrays.
Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data.
Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics.
Kotliarov Y, Steed ME, Christopher N, et al. High-resolution global genomic survey of 178 gliomas reveals novel regions of copy number alteration and allelic imbalances.
Nigro JM, Misra A, Zhang L, et al. Integrated array-comparative genomic hybridization and expression array profiles identify clinically relevant molecular subtypes of glioblastoma.
Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis.
Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias.
Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data.
Beissbarth T, Speed TP. GOstat: find statistically overrepresented gene ontologies within a group of genes.
Bengtsson H, Irizarry R, Carvalho B, Speed TP. estimation and assessment of raw copy numbers at the single locus level.
Misra A, Pellarin M, Nigro J, et al. Array comparative genomic hybridization identifies genetic subgroups in grade 4 human astrocytoma.
Mischel PS, Shai R, Shi T, et al. Identification of molecular subtypes of glioblastoma by gene expression profiling.
Maher EA, Brennan C, Wen PY, et al. Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities.
Horvath S, Zhang B, Carlson M, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target.
Ekstrand AJ, James CD, Cavenee WK, et al. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo.
Libermann TA, Nusbaum HR, Razon N, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin.
Wong AJ, Bigner SH, Bigner DD, et al. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification.
Liu M, Dai B, Kang SH, et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells.
Chan D, Yu SY, Chiu PM, et al. Over-expression of FOXM1 transcription factor is associated with cervical cancer progression and pathogenesis.
Dai B, Kang SH, Gong W, et al. Aberrant FoxM1B expression increases matrix metalloproteinase-2 transcription and enhances the invasion of glioma cells.
Wang Z, Banerjee S, Kong D, Li Y, Sarkar FH. Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells.
Laoukili J, Kooistra MR, Brás A, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability.
Schüller U, Zhao Q, Godinho SA, et al. Forkhead transcription factor FoxM1 regulates mitotic entry and prevents spindle defects in cerebellar granule neuron precursors.
Chakravarti A, Zhai GG, Zhang M, et al. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms.
Guan HT, Xue XH, Dai ZJ, Wang XJ, Li A, Qin ZY. Down-regulation of survivin expression by small interfering RNA induces pancreatic cancer cell apoptosis and enhances its radiosensitivity.
Kami K, Doi R, Koizumi M, et al. Downregulation of survivin by siRNA diminishes radioresistance of pancreatic cancer cells.
Tan Y, Raychaudhuri P, Costa RH. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes.
Orjalo AV, Arnaoutov A, Shen Z, et al. The Nup107-160 nucleoporin complex is required for correct bipolar spindle assembly.
Zuccolo M, Alves A, Galy V, et al. The human Nup107-160 nuclear pore subcomplex contributes to proper kinetochore functions.
Clark HF, Gurney AL, Abaya E, et al. The secreted protein discovery initiative (SPDI), a large-scale effort to identify novel human secreted and transmembrane proteins: a bioinformatics assessment.
Pulukuri SM, Rao JS. Matrix metalloproteinase-1 promotes prostate tumor growth and metastasis.
Stojic J, Hagemann C, Haas S, et al. Expression of matrix metalloproteinases MMP-1, MMP-11 and MMP-19 is correlated with the WHO-grading of human malignant gliomas.
Chu CY, Cha ST, Chang CC, et al. Involvement of matrix metalloproteinase-13 in stromal-cell-derived factor 1 alpha-directed invasion of human basal cell carcinoma cells.
Ala-aho R, Ahonen M, George SJ, et al. Targeted inhibition of human collagenase-3 (MMP-13) expression inhibits squamous cell carcinoma growth in vivo.
Grimmer MR, Weiss WA. Childhood tumors of the nervous system as disorders of normal development.
Hui AB, Lo KW, Yin XL, Poon WS, Ng HK. Detection of multiple gene amplifications in glioblastoma multiforme using array-based comparative genomic hybridization.
Rocak, S. Linder, P. DEAD-box proteins: the driving forces behind RNA metabolism.
Li L, Monckton EA, Godbout R. A role for DEAD box 1 at DNA double-strand breaks.
Guan Y, Kuo WL, Stilwell JL, et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer.
Holtkamp N, Ziegenhagen N, Malzer E, Hartmann C, Giese A, von Deimling A. Characterization of the amplicon on chromosomal segment 4q12 in glioblastoma multiforme.
Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges.
Mathupala SP, Guthikond M, Sloan AE. RNAi based approaches to the treatment of malignant glioma.
Hashizume R, Ozawa T, Gryaznov SM, et al. New therapeutic approach for brain tumors: intranasal delivery of telomerase inhibitor GRN163.
Hayes ME, Drummond DC, Hong K, et al. Increased target specificity of anti-HER2 genospheres by modification of surface charge and degree of PEGylation.
Hayes ME, Drummond DC, Kirpotin DB, et al. Genospheres: self-assembling nucleic acid-lipid nanoparticles suitable for targeted gene delivery.
Saito R, Krauze MT, Noble CO, et al. Convection-enhanced delivery of Ls-TPT enables an effective, continuous, low-dose chemotherapy against malignant glioma xenograft model.
Yamashita Y, Krauze MT, Kawaguchi T, et al. Convection-enhanced delivery of a topoisomerase I inhibitor (nanoliposomal topotecan) and a topoisomerase II inhibitor (pegylated liposomal doxorubicin) in intracranial brain tumor xenografts.



{kind=link}
{kind=link}
{kind=link}
{kind=link}