

Abstract
We investigated the mechanisms that contribute to spontaneous regular bursting in adult Purkinje neurons in acutely prepared cerebellar slices. Bursts consisted of 3–20 spikes and showed a stereotypic waveform. Each burst developed with an increase in firing rate and was terminated by a more rapid increase in firing rate and a decrease in spike height. Whole-cell current-clamp recordings showed that each burst ended with a rapid depolarization followed by a hyperpolarization. Dual dendritic and somatic extracellular recordings revealed that each burst was terminated by a dendritic calcium spike. The contributions of T- and P/Q-type calcium current, large (BK) and small (SK) conductance calcium-activated potassium currents, and hyperpolarization-activated (IH) current to bursting were investigated with specific channel blockers. None of the currents, except for P/Q, were required to sustain spontaneous bursting or the stereotypic burst waveform. T-type calcium, BK, and SK channels contributed to interspike and interburst intervals. The effect of T-type calcium channel block was more pronounced after BK channel block and vice versa, indicating that these two currents interact to regulate burst firing. Block of IH current had no effect on bursting. Partial block of P/Q-type calcium channels concurrently eliminated dendritic calcium spikes and caused a switch from regular bursting to tonic firing or irregular bursting. Dendritic calcium spikes persisted in the presence of tetrodotoxin, indicating that their initiation did not require somatic sodium spikes. Our results demonstrate an important role for dendritic conductances in burst firing in intact Purkinje neurons.
Introduction
Cerebellar Purkinje neurons are the sole output neurons of the cerebellar cortex. Their activity is crucial for motor coordination. The rate and pattern of firing of Purkinje neurons are controlled both by synaptic input and by intrinsic ion channels that allow the neurons to fire spontaneously in the absence of synaptic input. A striking feature of spontaneously active Purkinje neurons is their tendency to fire in bursts. In dissociated cells (Swensen and Bean, 2003), in cerebellar slices (Cingolani et al., 2002; Womack and Khodakhah, 2002a), and in vivo (Jaeger and Bower, 1994), Purkinje neurons exhibit trains of spontaneous bursts that often arise from their intrinsic membrane properties (Womack and Khodakhah, 2002a; Swensen and Bean, 2003).
The mechanism of burst firing in acutely dissociated Purkinje neurons has been described recently (Swensen and Bean, 2003). In these cells, T-type calcium channels and small (SK) and large (BK) conductance calcium-activated potassium currents control bursting. T-type calcium channels provide a depolarizing current between spikes, as large in magnitude as the resurgent sodium current, to push the membrane toward threshold for the next sodium spike. The inward calcium and sodium currents are opposed by calcium-activated potassium currents that are dominated by BK current. A cumulative inactivation of sodium channels and a cumulative increase in SK current terminate bursts. The finding that T-type calcium, SK, and BK channels control burst firing in Purkinje cells is consistent with their function in controlling burst firing in many other types of neurons (Jahnsen and Llinás, 1984; Pape, 1996; Golding et al., 1999; Grillner et al., 2001; Del Negro et al., 2002). The bursts in dissociated Purkinje neurons, however, differ significantly from those seen in Purkinje neurons in acutely prepared cerebellar slices (Womack and Khodakhah, 2002a). One reason for this difference may be that dissociated Purkinje neurons lack dendrites. The dendrites of Purkinje neurons have a high density of voltage-gated calcium channels (Usowicz et al., 1992) and exhibit prominent dendritic calcium spikes (Llinás and Sugimori, 1980). When evoked, regenerative dendritic potentials can cause bursting in Purkinje cells (Hounsgaard and Yamamoto, 1979; Llinás and Sugimori, 1980; Lev-Ram et al., 1992) as they do in hippocampal and neocortical pyramidal neurons (Wong and Prince, 1978; Schiller et al., 1997; Golding et al., 1999; Larkum et al., 1999). Because the dendrites make a significant contribution to the spontaneous firing of Purkinje neurons (Womack and Khodakhah, 2002a), it is also plausible that they contribute to spontaneous bursting.
We have characterized spontaneous bursting in intact Purkinje neurons in cerebellar slices. As found in dissociated Purkinje neurons in slices, T-type calcium, SK, and BK channels contribute to bursting; however, in Purkinje neurons in slices, all bursts are terminated by dendritic calcium spikes. Intriguingly, with the exception of P/Q-type calcium channels, which are required for dendritic calcium spikes, none of the other channels tested were necessary to sustain regular spontaneous bursting.
Materials and Methods
Preparation of slices. CD1 mice or Wistar rats at 15–30 d old were anesthetized with halothane and killed by decapitation. Sagittal slices (300 μm thick) were prepared from the vermis of the cerebellum using a Vibratome (Oxford Vibratome series 1000). Slices were maintained at room temperature in the recording solution until use (1–8 hr).
Recording and analysis. Slices were mounted in a chamber on the stage of an upright Zeiss microscope and visualized using a 40× water immersion objective with infrared optics. Slices were superfused continuously at a rate of 1.5 ml/min with recording solution containing (in mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 10 glucose 10, pH 7.4, when gassed with 5% CO2/95% O2. To block rapid synaptic transmission, the recording solution also contained 5 mm kynurenic acid, a broad-spectrum ionotropic glutamate receptor antagonist (Stone, 1993), and 100 μm picrotoxin, a GABAA receptor antagonist (Yoon et al., 1993). The slice temperature was maintained at 35°C(±0.5°C) by adjusting the temperature of the bathing solution. The volume of the chamber was 2 ml, requiring several minutes for complete wash-in of the antagonists or blockers. Extracellular recordings were made from individual Purkinje neurons using a homemade differential amplifier with glass pipette electrodes (tip size 0.3–1 μm) filled with the recording solution. The pipette tip was positioned just above, or lightly touching, the cell body near the axon hillock where the largest potential changes were usually recorded. Action potentials appeared as fast negative deflections of 50–1000 μV. Larger pipettes (10–20 μm) were used for dendritic recordings, and the pipette tip was positioned in the molecular layer ∼30 μm from the soma. Whole-cell recordings were made with borosilicate glass pipettes (3–6MΩ) filled with (in mm): 140 K methyl sulfate, 10 KCl, 5 NaCl, 2 MgATP, 0.01 EGTA, 10 HEPES, pH 7.2. Whole-cell data were recorded using an Optopatch amplifier (Cairn Research, Faversham, UK). Data were sampled at 10 kHz for extracellular recordings and at 20 kHz for whole-cell recordings using a National Instruments (Austin, TX) analog-to-digital card (MIO-16XE-10) and an IBM-compatible computer. Data acquisition and analyses were done with software written in-house using LabView (National Instruments). To analyze firing rate, a threshold level for spike detection was set by eye during the experiment. To analyze bursts, several cycles of the trimodal pattern of activity were selected under both control and experimental conditions. Trains of action potentials <500 msec were analyzed as bursts. Interspike intervals were calculated as the time from the peak of one action potential to the peak of the subsequent spike. An interspike interval was flagged as an interburst interval if it was larger than the preceding two intervals by a factor of 1.2–3.5. This factor was adjusted for each cell by visual inspection to ensure that the analysis routine accurately identified all bursts. A special subroutine also identified bursts that consisted of two spikes. On the basis of independent visual inspection, the program identified bursts with >95% accuracy. Once bursts were identified, burst duration, interburst and interspike intervals, and peak amplitude for the first and last spikes were determined. Kynurenic acid, picrotoxin, and apamin were obtained from Sigma (St. Louis). Iberiotoxin was from Tocris (Ellisville, MO), and ω-conotoxin MVIIC was from Bachem (Torrance, CA). All other chemicals were obtained from Sigma or Fisher Scientific (Fairlawn, NJ). Mibefradil was a generous gift of Hoffman-LaRoche (Basel, Switzerland).
Results
Characterization of spontaneous bursts in Purkinje neurons
In the absence of fast synaptic input, Purkinje neurons in cerebellar slices from mature animals show periods of burst firing as part of a trimodal pattern of activity (Womack and Khodakhah, 2002a). Using extracellular recording to avoid any alterations in the activity of the cells, a total of 19,349 bursts recorded from 27 Purkinje neurons were analyzed under control conditions. Bursts occurred in trains consisting of 16–630 bursts (mean = 271 ± 34; SEM; n = 27). The total number of spikes per burst varied from 2 to 250; most bursts had fewer than 10 spikes. Figure 1A shows a typical recording of bursts from a Purkinje neuron. The number of spikes per burst varied as indicated by the cumulative probability plot shown to the right of the raw data trace. The median number of spikes per burst varied from 3 to 20 among all Purkinje neurons tested (mean = 8.3 ± 0.7; SEM; n = 27) (Fig. 1B). Burst duration, interburst interval, and average firing rate during the burst also varied widely within each cell and among cells. Cumulative probability histograms of these parameters are also shown in Figure 1A. In all cases the cumulative probability plots were obtained from analysis of several cycles of the trimodal pattern of activity. Among neurons, the median burst duration ranged from 25 to 180 msec (mean = 60.1 ± 7.3; SEM; n = 27), and the median interburst interval ranged from 5 to 100 msec (mean = 35.9 ± 4.3; n = 27) (Fig. 1B). The average firing rate ranged from 30 to 280 spikes per second (mean = 150 ± 12; SEM; n = 27) (Fig. 1B). Within each cell there was a good correlation between the number of spikes per burst and burst duration (Fig. 1A); there was no correlation between any of the other parameters measured.

Characterization of spontaneous bursts in Purkinje neurons. A, Extracellular recording from a Purkinje neuron during spontaneous bursting. By analyzing bursts from several cycles of the trimodal pattern of activity, cumulative probability plots of spikes per burst (S/B), burst duration (BD), interburst interval (IBI), and average firing rate (FR AVG) were generated. Also shown is a plot of BD versus S/B. B, The median value of each parameter measured is shown for each cell (open circles). The mean of all the medians for each parameter is also shown (filled squares). Error bars represent ±SEM (n = 27).
Typically the firing rate increased as each burst progressed, increasing more rapidly between the last two to three spikes (Fig. 1A). To quantify the increase in firing rate, the instantaneous firing rate between the first two (FR BEG) and the last two (FR END) spikes in each burst was determined (Fig. 2A), and the ratio of FR BEG to FR END was calculated (FR B/E). Cumulative probability plots of FR BEG, FR END, and FR B/E for the cell described in Figure 1A show that >90% of bursts end with an increase in firing rate (FR B/E <1) (Fig. 2B). Among all neurons tested, median values of FR B/E ranged from 0.15 to 0.9 (mean = 0.45 ± 0.03; SEM; n = 27) (Fig. 2C).

Spontaneous bursts end with an increase in firing rate and a decrease in spike amplitude. Each burst was analyzed for changes in firing rate and spike amplitude. A, The diagram shows burst parameters that were measured. ISI BEG, First interspike interval; ISI END, last interspike interval; AMP BEG, first spike amplitude; AMP END, last spike amplitude. The instantaneous firing rate at the beginning (FR BEG) and end (FR END) of each burst was calculated by taking the reciprocal of the corresponding interspike interval. B, Cumulative probability plots of FR BEG (thin trace), FR END (thick trace), FR B/E, and AMP B/E are shown for the cell described in Figure 1 A. C, Median values of FR AVG, FR B/E, and AMP B/E for each Purkinje neuron tested (open circles). Median values of FR B/E were <1, whereas most median values of AMP B/E were >1. Average values of all the medians are indicated by the squares. Error bars represent ±SEM, (n = 27). D, FR B/E versus burst duration for the cell described in Figure 1 A. E, AMP B/E versus burst duration for the cell described in Figure 1 A. No correlation between FR B/E and burst duration or AMP B/E and burst duration was observed for any of the neurons tested.
An increase in the firing rate during each burst indicates that as each burst progresses the membrane depolarizes. It might be expected, therefore, that the firing rate increases to a greater extent with longer bursts; however, no correlation was found between burst duration and FR B/E ratio (Fig. 2D). This observation, combined with the finding that the firing rate of the last few spikes increases more rapidly, suggests that there is a relatively large, abrupt depolarization at the end of each burst.
Single-spike extracellular recordings measure potential changes at the tip of the electrode that correspond to the net current flow at that location. Assuming that the current recorded is flowing solely into or out of the cell under study, the kinetics of the recorded signal corresponds to the derivative of the change in the membrane potential. Thus, the peak of the negative spike recorded corresponds to the upstroke of the action potential, the time at which the rate of change of the membrane potential is the greatest. The dominant current during the upstroke of the action potential in Purkinje cells is the inward sodium current, because potassium channels have much slower kinetics of activation. The negative spike amplitude can therefore be used as an estimate of the sodium current.
Within each burst, even for very long bursts, spike amplitude is fairly constant until the last few spikes, during which it decreases abruptly (Figs. 1A, 2A). To quantify the change, the amplitude of the first (AMP BEG) and last (AMP END) spike in each burst was measured (Fig. 2A), and the ratio of AMP BEG to AMP END was calculated (AMP B/E). The cumulative probability plot of AMP B/E for the cell discussed in Figure 1A shows that for most bursts AMP B/E is >1, indicating that the bursts end with a decrease in spike amplitude (Fig. 2B). The decrease in spike amplitude is most likely caused by cumulative inactivation of sodium channels. Among all neurons tested, median values of AMP B/E ranged from 0.92 to 2.2 (mean = 1.4 ± 0.08; SEM; n = 27) (Fig. 2C), with only three cells showing a median AMP B/E <1. These three cells had a low (three to four) median number of spikes per burst. No correlation was found between burst duration and AMP B/E (Fig. 2E).
The findings that spike amplitude is constant until the final spikes in each burst and that burst duration is not correlated with AMP B/E suggest that significant cumulative inactivation of sodium current occurs only during these final few spikes. In bursts from dissociated Purkinje neurons, however, significant cumulative sodium channel inactivation occurs with each spike and is thought to terminate short bursts and significantly contribute to termination of longer bursts (Swensen and Bean, 2003). Such a mechanism is unlikely to be the cause of burst termination in intact Purkinje neurons, because no progressive decrease in spike amplitude or decrease in firing frequency is observed. Furthermore, there are three- to fourfold more spikes per burst in intact Purkinje neurons than in dissociated neurons.
Bursts are terminated by calcium spikes
The data presented in the previous section suggest that at the end of each burst the membrane depolarizes rapidly. This hypothesis was tested using whole-cell current-clamp recording. Bursts in Purkinje neurons occur at regular intervals as part of a trimodal pattern of activity (Fig. 3A). Whole-cell recordings were obtained from nine Purkinje neurons that exhibited the trimodal pattern. A recording of one cycle of the pattern is shown in the top trace of Figure 3B. The cell was hyperpolarized during the silent period and more depolarized during the tonic and bursting modes. The bottom two traces of Figure 3B show recordings from the same cell during the bursting period. The records show that the membrane depolarizes as each burst progresses. Each burst ends with a rapid depolarization followed by a rapid hyperpolarization that persists through the interburst interval. The kinetics of the depolarizations that end the bursts strongly resembles those of regenerative calcium spikes described in Purkinje neurons by Llinás and Sugimori (1980).

Bursts end with a rapid depolarization. Whole-cell current-clamp recordings were obtained from spontaneously firing Purkinje neurons. A, Average firing rate (calculated every 500 msec) for one Purkinje neuron. Two cycles of the trimodal pattern of activity are shown. Tonic (T), bursting (B), and silent (S) periods are indicated. B, Whole-cell current-clamp recording from the same cell. Top trace, A single cycle of the trimodal pattern. Middle and bottom traces, Records obtained during the bursting period are shown on expanded time scales.
Bursts are terminated by dendritic spikes
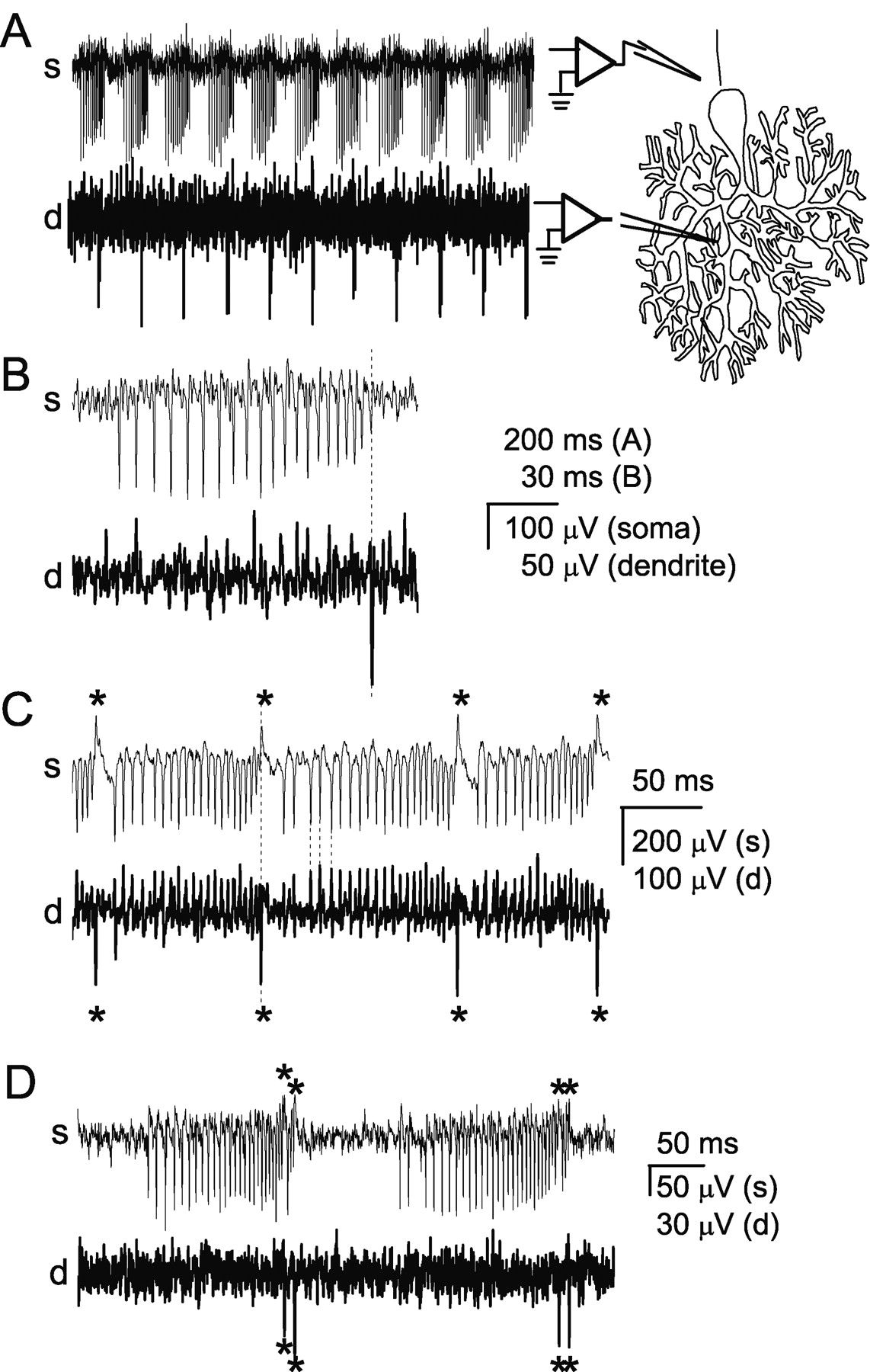
In Purkinje neurons, regenerative calcium spikes are generated in the dendrites and invade the soma (Llinás and Sugimori, 1980). To confirm that bursts are terminated by dendritic calcium spikes, simultaneous extracellular recordings were made from the soma and dendrites of Purkinje neurons. Dendritic recordings from bursting Purkinje neurons are shown in Figure 4A–D (bottom traces). By carefully positioning the dendritic recording pipette, it was possible to locate regions within the molecular layer in which trains of dendritic spikes occurred at regular intervals (Fig. 4A–D, bottom traces). Sodium-dependent action potentials were simultaneously recorded from the soma of the same neurons (Fig. 4A–D, top traces). Simultaneous recordings showed the presence of regular dendritic spikes during the bursting, but not tonic and silent modes of the trimodal pattern of activity. Furthermore, a calcium spike was recorded at the end of each sodium spike burst (Fig. 4A). The peak of the dendritic spike occurred after, or coincident with, the final sodium spike of each burst (Fig. 4B). Similar dual recordings were obtained from a total of 27 Purkinje neurons. In each of these neurons a dendritic spike occurred only during the bursting period of the trimodal pattern of activity. The calcium spikes were always associated with the end of bursts.

Bursts are terminated by dendritic spikes. Simultaneous extracellular recordings were made from the soma, near the axon hillock, and from the dendrites, at least 30 μm from the soma. A, Simultaneous somatic (s) and dendritic (d) recordings of spontaneous bursts in a Purkinje neuron. The diagram at the right shows the approximate location of the recording pipettes. At the end of each burst of somatic action potentials a rapid negative voltage change is seen in the dendrite. B, A single burst from the cell described in A. The dashed line shows that the peak of the dendritic spike occurs after the final somatic spike. C, Simultaneous somatic and dendritic recordings from another Purkinje neuron show that with each negative dendritic spike (denoted by asterisk), a corresponding positive potential change was recorded at the soma. Each sodium spike produced a negative potential change at the axon hillock and a corresponding positive potential change in the dendrites. The current source for the action potentials is confined to the soma, whereas the current source for the spikes that terminate the bursts is localized to the dendrites. D, Simultaneous recordings from a Purkinje neuron in which each burst of ∼25 spikes was followed immediately by a much shorter burst of 3 spikes. Both long and short bursts were terminated with a dendritic calcium spike (denoted by asterisk).
Extracellular electrodes record potential changes resulting from current flow through the cellular membrane near the electrode tip; a negative potential change indicates positive current flow into the cell. It was never possible to record the burst-terminating spikes as negative potential changes at the soma. In fact, to record the dendritic spikes it was usually necessary to move the recording pipette ≥30 μm into the molecular layer. These findings confirm the dendritic origin of the depolarizations that terminated the bursts. In some cases, somatic recordings showed brief positive potential changes that occurred at the end of each burst (Fig. 4C, top trace). Simultaneous dendritic recordings showed that the positive potential changes in somatic recordings coincided with the negative dendritic spikes (Fig. 4C). Similarly, dendritic recordings sometimes showed positive potential changes that corresponded to the negative somatic sodium spikes (Fig. 4C). These observations are consistent with those of Llinás and Sugimori (1980), who suggested that the two types of regenerative signals that can be recorded in Purkinje neurons are initiated in separate electrical compartments. Dendrites of Purkinje neurons have a low density of voltage-gated sodium channels and do not support sodium action potentials (Llinás and Sugimori, 1980; Stuart and Hausser, 1994). The dendritic spikes recorded here are therefore the regenerative calcium spikes shown to be present in these cells (Llinás and Sugimori, 1980).
In some neurons, each burst was followed immediately by a second short burst consisting of only a few spikes (Fig. 4D). In each case, both long and short bursts were terminated by a dendritic calcium spike. An intriguing possibility is that the two dendritic calcium spikes in such close succession may occur when calcium spikes are generated sequentially at two different locations in the dendrites.
Calcium spikes are required for regular spontaneous bursts in Purkinje neurons
The data presented earlier show that bursts are terminated by dendritic calcium spikes. We explored the consequences of preventing the generation of calcium spikes on the spontaneous bursts seen in Purkinje cells. P/Q-type calcium channels comprise ∼80% of the calcium current in Purkinje cell bodies (Regan, 1991) and are present at even higher densities in the dendrites (Usowicz et al., 1992). Because P/Q-type calcium channels are required to sustain spontaneous firing in Purkinje neurons (Womack and Khodakhah, 2002a), in the experiments reported here we blocked only a fraction of P/Q-type calcium channels to prevent regenerative calcium spikes but allow spontaneous firing. Because the bulk of calcium current is contributed by P/Q-type calcium channels, we used either low concentrations of cadmium (10 μm) or a selective calcium channel toxin to reduce the P/Q calcium channel density. In Purkinje neurons, SK channels are selectively coupled to P/Q-type calcium channels (M. D. Womack, C. Chevez, and K. Khodakhah, unpublished data). Partial block of P/Q channels significantly increases firing rate and bursting because of the reduced activation of SK channels. To isolate the direct effects of P/Q-type calcium channel block from those that result from decreased SK channel conductance, cadmium or ω-conotoxin MVIIC was applied after application of apamin (100 nm).
Regular spontaneous bursts disappeared in 8 of 10 cells after application of 10 μm cadmium and in 7 of 9 cells after application of 250–600 nm ω-conotoxin MVIIC. The immediate effects of cadmium or ω-conotoxin MVIIC were to abolish burst firing so that most cells only cycled through periods of tonic firing and silence (Fig. 5A, middle trace). With longer applications (5–10 min after wash-in of cadmium), the tonic firing mode was replaced by irregular bursting (Fig. 5A, bottom trace). The irregular bursts seen in the presence of cadmium or ω-conotoxin MVIIC were very different from those in control conditions, which showed a progressive increase in firing rate, and ended with a more rapid increase in firing rate and decrease in spike amplitude. In cadmium or ω-conotoxin MVIIC, the bursts were short (two to four spikes) and showed no change, or a decrease, in firing rate.

P/Q-type calcium channels are required for dendritic spikes and for bursting. A, Extracellular recordings from a Purkinje neuron under control conditions (top trace) and at 3 min (middle trace) and 10 min (bottom trace) after superfusion with cadmium (10 μm). B, Dual extracellular recordings from the soma (s) and dendrites (d) of a Purkinje neuron. Dendritic recordings show dendritic spikes as negative voltage deflections, whereas the somatic sodium spikes are positive. The polarity of the spikes is reversed in the somatic recordings. Under control conditions, each burst is terminated by a dendritic spike (denoted by asterisk). Cadmium (10 μm) inhibits dendritic but not somatic spikes (bottom traces).
To determine whether the loss of regular bursting was the result of loss of dendritic calcium spikes, dual somatic and dendritic recordings were made as calcium channel blockers were applied. Figure 5B shows simultaneous somatic and dendritic recordings made from a bursting Purkinje neuron in the presence of apamin. A dendritic spike is seen at the end of every burst. In the presence of cadmium, regular bursting ceased, and the cell began to fire with irregular bursts. The dendritic spikes disappeared concurrent with the change in spontaneous firing. In all four Purkinje neurons tested, dendritic spikes stopped coincident with the loss of regular spontaneous bursting. The irregular bursts seen in the presence of calcium channel antagonists are similar to the bursts seen in Purkinje cells when the electrical contribution of dendrites is removed by local perfusion with isotonic sucrose (Womack and Khodakhah, 2002a) or by mechanical dissociation (Swensen and Bean, 2003).
Dendrites generate calcium spikes spontaneously in the presence of tetrodotoxin
In Purkinje neurons, dendritic calcium spikes have been observed after direct injection of depolarizing current (Llinás and Sugimori, 1980; Lev-Ram et al., 1992) or intense synaptic stimulation provided by climbing fiber activation (Miyakawa et al., 1992). In several neurons, dendritic potentials can drive spontaneous bursting. In these instances, however, generation of the dendritic spikes is facilitated by rapid sodium-dependent action potentials that are generated at an axonal or somatic initiation site (Golding et al., 1999; Larkum et al., 1999). To determine whether spontaneous firing of calcium spikes in Purkinje neurons requires somatic action potentials, dendritic spikes were recorded in the presence of tetrodotoxin (TTX). Figure 6A shows simultaneous recordings of spontaneous activity from soma and dendrites of a Purkinje neuron. Somatic sodium spikes appear as negative potential changes in the somatic recording and positive potential changes in the dendritic recording, whereas the spikes generated in the dendrites are recorded as negative deflections in the dendrites but positive deflections at the soma. In the presence of TTX (1 μm), the somatic spikes cease but the dendritic spikes remain (Fig. 6A). Most adult Purkinje neurons exhibit a trimodal pattern of activity in which they cycle through regular periods of tonic firing, bursting, and silence. Several cycles of this pattern of activity are shown in the plot of average firing rate in Figure 6B (top trace). Dendritic spikes also occur in regular cycles that correspond to the bursting phases of the somatic spikes (Fig. 6B, bottom trace). When sodium spikes are blocked by TTX, the dendritic spikes continue to fire cyclically. Dendritic spikes persist for up to 30 min in the presence of tetrodotoxin, the longest time tested. Identical results were obtained in seven additional Purkinje neurons. These results show that Purkinje neurons do not require TTX-sensitive sodium channels to spontaneously generate dendritic calcium spikes. Block of sodium spikes, in most cases, prolonged the cycle period and, paradoxically, increased the rate of calcium spike generation (Fig. 6C). Thus, although calcium spikes remain in the absence of somatic sodium spikes, clearly the somatic sodium spikes interact with dendrites to regulate the generation of dendritic calcium spikes and cycle period.

Dendritic spikes occur spontaneously in the absence of sodium-dependent action potentials. A, Somatic (top trace) and dendritic (bottom trace) recordings before and after application of TTX (1 μm). Dendritic spikes produced positive potential changes at the soma. Each occurrence of a dendritic spike is indicated by an asterisk. B, The average firing rate (top trace) and average occurrence of dendritic calcium spikes (bottom trace) calculated every 500 msec are shown. TTX blocked firing of somatic but not dendritic spikes. Dendritic spikes continued to occur in bursts at regular intervals even in the presence of TTX. C, Average occurrence of dendritic spikes under control conditions and in TTX (left plot). The average cycle period of the bursts of calcium spikes for each cell under control conditions and in TTX is shown in the right plot. For each cell the ratio of the value measured in TTX to the value measured in control conditions was calculated for both the rate of calcium spike occurrence and cycle period. The averages of the ratios for all cells tested are shown in the histogram. Error bars represent ±SEM (n = 4, calcium spike rate; n = 8, cycle period).
Contribution of ion channels to bursting
The data presented in the earlier sections described the properties of spontaneous bursts in Purkinje cells in slices. As delineated, the firing rate within each burst increases and the burst is terminated coincident with the occurrence of a P/Q channel-dependant dendritic calcium spike. To explore further the mechanism of burst firing in Purkinje cells, we used selective blockers to evaluate whether specific voltage- or calcium-activated channels are required for spontaneous bursting. Specifically, we investigated the role of T-type calcium channels, hyperpolarization-activated channels, and SK and BK conductance calcium-activated potassium channels because these conductances have been shown to play a significant role in bursting in other cells (Huguenard, 1996; Luthi and McCormick, 1998; Del Negro et al., 1999; Grillner et al., 2001) or in dissociated Purkinje neurons (Swensen and Bean, 2003).
As discussed by Swensen and Bean (2003), a disadvantage of using pharmacological blockers to evaluate the exact contribution of ion channels in spontaneously active cells is that block of each channel type is likely to alter the voltage trajectory and thus affect activation, deactivation, or inactivation of other voltage-gated channels. A better approach would be to voltage clamp a cell and study the individual ionic currents that flow when a burst membrane potential waveform recorded from the same cell is applied as command voltage (Swensen and Bean, 2003). The later approach, however, is only applicable to cells in which adequate space clamp can be achieved, for example in the near-spherical soma of dissociated Purkinje cells that have lost their dendrites and axon. The extensive dendritic tree of intact Purkinje cells, on the other hand, is a poor preparation for voltage-clamp experiments. Furthermore, whole-cell recording alters the ionic composition of the cells and the extent of their endogenous calcium buffering, factors that are likely to play important roles in spontaneous firing. Thus, the approach taken here provides a good complement to the voltage-clamp experiments done in reduced preparations, although great caution should be taken in interpreting the results.
T-type calcium, SK, BK, and hyperpolarization-activated channels are not required to sustain regular spontaneous bursts in cerebellar Purkinje cells
Although a quantitative description of the effects of specific channel blockers on the various burst parameters is given in the subsequent sections, the most surprising finding of these experiments is that T-type calcium, SK, BK, and hyperpolarization-activated channels are not required to maintain the trimodal pattern of activity or for the generation of regular spontaneous bursts in cerebellar Purkinje cells. Extracellular recordings from Purkinje neurons in the presence of specific blockers for each of these channels are shown in Figure 7. The recordings demonstrate that Purkinje neurons continue to fire trains of regular bursts that are qualitatively similar to bursts observed in control conditions. Each of these bursts terminates with a rapid increase in its firing rate accompanied by a reduction in the amplitude of the last few spikes. Given the importance of T-type calcium, SK, and BK channels in the generation and support of burst firing in dissociated Purkinje cells (Swensen and Bean, 2003), these data suggest that the mechanism of burst firing in intact Purkinje cells differs from that in the dissociated cells.

Block of SK, BK, T-type calcium, or hyperpolarization-activated channels does not prevent spontaneous bursting. Recordings of spontaneous bursting in Purkinje neurons in the presence of specific blockers for the ion channels as indicated. SK channels were blocked with apamin (100 nm), BK channels with iberiotoxin (100 nm), and hyperpolarization-activated channels with cesium (1 mm). Mibefradil (1 μm) was used to block both T- and R-type voltage-gated calcium channels. Pharmacological block of these conductances abolished neither the trimodal pattern of activity nor the generation of regular spontaneous bursts.
SK channels contribute to interspike and interburst intervals
Calcium-activated potassium channels have been shown to contribute to bursting (Pape and Driesang, 1998; Del Negro et al., 1999; Grillner et al., 2001). Both SK and BK channels are present in Purkinje neurons (Jacquin and Gruol, 1999; Cingolani et al., 2002; Edgerton and Reinhart, 2003; Womack and Khodakhah, 2002b, 2003) and both contribute to burst firing in dissociated Purkinje cells (Swensen and Bean, 2003). BK channels also contribute to repolarization of dendritic calcium spikes (Edgerton and Reinhart, 2003). We therefore investigated the contribution of these channels to spontaneous bursting in intact Purkinje neurons.
To determine the role of SK channels, recordings were made in the presence of apamin, a specific blocker of the SK channels present in Purkinje cells (Cingolani et al., 2002). Bursts recorded from a Purkinje neuron under control conditions and in the presence of apamin are shown in Figure 8A. Comparison of the data records shows an increase in firing rate throughout each burst and a decrease in both burst duration and interburst interval. Figure 8B shows the average cumulative probability plots for each of the parameters measured in all six cells tested. The plots show a clear increase in the initial and average firing rates. The end firing rate also increased but to a lesser extent. The fact that there was a greater change in the firing rate at the beginning than at the end is reflected in an increase in FR B/E. The increase in the firing rate during the burst is consistent with previous results showing that block of SK channels increases spontaneous firing rate (Cingolani et al., 2002; Edgerton and Reinhart, 2003; Womack and Khodakhah, 2003). The increase in the firing rate is caused by elimination of an afterhyperpolarization because the membrane potential does not depolarize significantly in the presence of apamin (Cingolani et al., 2002; Edgerton and Reinhart, 2003; Womack and Khodakhah, 2003). The beginning firing rate increased to a greater extent than the end firing rate, possibly because at the end of the bursts, even under control conditions, the firing rate was already quite high and may have been close to the maximum for the cells. In dissociated Purkinje cells the amplitude of the SK current at the end of the bursts is larger than at the beginning (Swensen and Bean, 2003). If, in intact Purkinje neurons, the same accumulation of SK current occurs, then block of SK channels should increase the end firing rate to a greater extent than the beginning. The fact that this is not observed suggests that either the SK current does not accumulate during bursts in intact Purkinje cells or that other conductances contribute more prominently to the last interspike interval.

SK channels contribute to interspike and interburst intervals. A, Burst firing recorded from a Purkinje neuron under control conditions and in the presence of apamin (100 nm). B, Average cumulative probability distributions for each parameter under control conditions (open symbols) and in apamin (closed symbols) for all cells studied (mean ± SEM; n = 6). C, The ratio of the median value in apamin to the median value in control conditions was determined for each parameter in each cell tested. For each parameter, the mean of this ratio for all cells is shown. The horizontal bar indicates a ratio value of 1 corresponding to no effect of the blocker on the parameter measured. Asterisks indicate that the mean is significantly different from 1 (*p < 0.08; **p < 0.006; determined by one-way ANOVA). Error bars represent ±SEM (n = 6). The number of bursts analyzed was 2790 under control conditions and 5045 in apamin. For each cell, bursts from at least two cycles of the trimodal pattern of activity were analyzed under control conditions and in apamin.
Apamin also decreased the interburst interval and burst duration, but there was little change in the other parameters measured (Fig. 8B,C). The decrease of the interburst interval was statistically significant when compared among all neurons tested (p < 0.05 by one-way ANOVA). On average there was a general tendency toward shorter burst durations in apamin (Fig. 8B), but this difference was not statistically significant when the medians were compared (Fig. 8C). These observations are in contrast to the finding that block of SK channels increases the burst duration in dissociated neurons (Swensen and Bean, 2003).
The results presented in this section show that in intact neurons, SK channels make a significant contribution to controlling the firing rate during the bursts and to the interburst interval but make little contribution to burst termination.
BK channels modulate burst duration and interburst interval
In hippocampal pyramidal neurons, BK channels regulate burst duration by causing repolarization of dendritic calcium spikes that cause the bursts (Golding et al., 1999). The contribution of BK channels to bursting in Purkinje neurons was assessed by applying iberiotoxin, a specific BK channel blocker (Giangiacomo et al., 1992). Block of BK channels had subtle effects on burst parameters. Comparison of results from 11 cells shows that, on average, there was a slight increase in firing rate throughout the bursts, but this difference was not statistically significant (Fig. 9A). There was a small but significant increase in FR B/E (p < 0.01) and small but significant decreases in interburst interval (p < 0.05), burst duration (p < 0.01), and the number of spikes per burst (p < 0.05). In dissociated Purkinje neurons, BK channels are the dominant outward current during the interspike intervals (Swensen and Bean, 2003). Block of BK channels in dissociated Purkinje neurons decreases the first interspike interval but makes little contribution to the burst duration.

Contribution of BK, T-type calcium, and hyperpolarization-activated channels to spontaneous bursting. Extracellular recordings were made from Purkinje neurons under control conditions and in the presence of specific ion channel blockers. Cumulative probability plots of burst parameters similar to those shown in Figure 8 B were made. The average of the ratio of the median value in blocker to the median value in control conditions was determined for each burst parameter. These ratios accurately reflected the changes seen in the cumulative probability plots and are shown in this figure for each blocker. The horizontal bars indicate a value of 1. Ratios that significantly differ from 1 are indicated by asterisks. A, Recordings were made under control conditions and in the presence of iberiotoxin (100 nm) to block BK channels (*p < 0.05; *p < 0.01; determined by one-way ANOVA). Error bars represent ±SEM, (n = 11). The number of bursts analyzed was 11,172 under control conditions and 8517 in iberiotoxin. B, Burst firing was recorded under control conditions and in the presence of mibefradil (1 μm) to block T-type calcium channels (*p < 0.02; *p < 0.002; determined by one-way ANOVA). Error bars represent ±SEM, (n = 5). The number of bursts analyzed was 2184 under control conditions and 3567 in mibefradil. C, The effect of mibefradil (1 μm) was examined in the continuous presence of iberiotoxin (100 nm). (*p < 0.002; **p < 0.001; determined by one-way ANOVA). Error bars represent ±SEM (n = 5). The number of bursts analyzed was 3479 in iberiotoxin alone and 2100 in iberiotoxin plus mibefradil. D, The effect of iberiotoxin (100 nm) was examined in the continuous presence of mibefradil (1 μm) (*p < 0.05; **p < 0.005; determined by one-way ANOVA). Error bars represent ±SEM (n = 6). The number of bursts analyzed was 2639 in mibefradil alone and 2329 in mibefradil plus iberiotoxin. E, Cesium (1 mm) was used to block IH. Ratios significantly different from 1 are indicated by asterisks (*p < 0.002; determined by one-way ANOVA). Error bars represent ±SEM, (n = 5). The number of bursts analyzed was 2783 under control conditions and 2864 in cesium. For each cell, all bursts from at least two cycles of the trimodal pattern of activity were analyzed under control conditions and at least two cycles in the presence of the channel blocker.
One problem with interpreting the effect of iberiotoxin on Purkinje neurons is that block of BK channels is likely to affect the voltage trajectory of the cell and alter activation of other voltage-gated channels. BK channels have been shown to contribute to afterhyperpolarization after each action potential (Womack and Khodakhah, 2002b; Edgerton and Reinhart, 2003).
T-type voltage-gated calcium channels contribute to interspike and interburst intervals
T-type voltage-gated calcium channels, because of their low threshold for activation and inactivation, are well suited to contribute to oscillations in membrane potential that can cause bursting. In dissociated Purkinje neurons, together with resurgent sodium current, T-type calcium channels provide the dominant inward current between spikes during bursts (Swensen and Bean, 2003). In Purkinje neurons in slice culture, T-type calcium channels cause small regenerative dendritic potentials when P/Q-type calcium channels are blocked (Pouille et al., 2000; Cavelier et al., 2002). To determine whether T-type calcium channels contribute to spontaneous bursts in intact Purkinje neurons, recordings were made in the presence of mibefradil, a blocker of T-type calcium channels (McDonough and Bean, 1998). Block of mibefradil-sensitive calcium channels resulted in a modest and statistically significant decrease in the firing rate in all five cells tested (Fig. 9B). Although there was a slight increase in interburst interval, burst duration, and FR B/E, these results were not statistically significant. In addition to blocking T-type calcium channels, mibefradil also potently blocks R-type calcium channels (Randall and Tsien, 1997), which are thought to be present in Purkinje cells (Yokoyama et al., 1995; Meacham et al., 2003). In the absence of selective T-type calcium channel antagonists, we are unable to verify the extent to which the changes seen are the consequence of block of T-type calcium channels specifically.
In dissociated Purkinje neurons, T-type calcium currents (mibefradil-sensitive currents) are the major calcium current active between spikes (Swensen and Bean, 2003). Interpretation of the results obtained with mibefradil is complicated because the T-type calcium current not only contributes to the voltage trajectory between spikes but also is likely to activate calcium-dependent channels. Our unpublished observations suggest that in Purkinje neurons, SK channels are selectively coupled to P/Q-type calcium channels (Womack, Chevez, and Khodakhah, unpublished data). Experiments described in the next section were performed to evaluate the contribution of BK and T-type calcium channels to burst firing in isolation from each other.
BK and T channels interact to determine firing rate burst duration and interburst interval
To study the role of T-type calcium channels in isolation from the calcium-dependent BK channels, mibefradil was applied after block of BK channels with iberiotoxin. Average data from five cells are shown in Figure 9C. As in the absence of iberiotoxin, mibefradil significantly decreased the firing rate throughout the bursts. This finding suggests that T-type calcium channels make a contribution to depolarization of the membrane potential between spikes, consistent with the results of Swensen and Bean (2003). Furthermore, the contribution of T-type calcium channels at the end of the bursts is as great as at the beginning. The lack of pronounced inactivation of T-type calcium channels throughout each burst, as suggested by these findings, is also consistent with the observations of Swensen and Bean (2003).
Block of T-type calcium channels significantly increased the interburst interval by an average of 53% (p < 0.01). Therefore, T-type calcium channels contribute to membrane depolarization between bursts and help initiate the next burst. A paradoxical finding was that block of T-type calcium channels also increased the burst duration. One interpretation of this finding is that the T-type calcium current contributes to the initiation of dendritic calcium spikes that terminate the bursts. Block of T-type calcium channels will therefore prolong the time taken to reach threshold for generation of a calcium spike and increase the burst duration. An alternative explanation is that the threshold for initiation of dendritic calcium spikes is higher because at some voltages mibefradil blocks a small fraction of P/Q current (McDonough and Bean, 1998). No significant changes were observed in any of the other parameters measured. In the parameters where significant changes were observed, the variability in the average values was much smaller in these experiments than when BK channels were not blocked, suggesting that much of the cell-to-cell variability was a consequence of the interaction between T-type calcium and BK channels. As discussed, mibefradil also potently blocks R-type calcium channels. The finding that in the presence of mibefradil Purkinje cells continue to have regular spontaneous bursts suggests that neither T-type nor R-type calcium channels are required for the generation of dendritic calcium spikes.
To determine the contribution of BK channels to bursting in the absence of T-type calcium current, the effect of iberiotoxin on burst parameters was studied after block of T-type calcium channels with mibefradil. Figure 9D summarizes the results from six cells. In the absence of T-type calcium channels, block of BK channels significantly increased the beginning and average firing rates by 42 and 40%, respectively (p < 0.05). A smaller and statistically insignificant change was seen in the end firing rate, consistent with observations in dissociated Purkinje neurons, where the contribution of BK channels to the firing rate at the end of bursts is less than at the beginning (Swensen and Bean, 2003).
As in the absence of mibefradil, block of BK channels significantly decreased both interburst interval (75%) and burst duration (67%) (p < 0.05 for each). The decrease in interburst interval suggests that BK channels contribute a hyperpolarizing current that helps control the duration of the interburst interval. This is surprising because at hyperpolarized membrane potentials BK channels have a low affinity for calcium and, given the resting calcium concentration of the cell, are expected to have a very low open probability (Womack and Khodakhah, 2002b). It may be that after the calcium spikes that terminate bursts, the intracellular calcium remains sufficiently high to activate BK channels. Alternatively, in Purkinje cells, BK channels may be coupled to calcium channels that are active during the interburst intervals. Block of BK channels also significantly decreases the burst duration, most likely by permitting dendritic membrane potential to reach threshold for generation of a calcium spike sooner. No significant changes were observed in any of the other parameters measured. In the parameters where significant changes were observed, the variability in the average values was much smaller than in the presence of T-type calcium channels, again suggesting that much of the cell-to-cell variability was a consequence of the interaction between T-type calcium and BK channels.
IH does not contribute to burst firing
In many types of neurons, the IH also contributes to bursting (Pape, 1996). IH is present in Purkinje neurons (Crepel and Penit-Soria, 1986; Williams et al., 2002). The contribution of IH to bursting in Purkinje neurons was examined by blocking the channels with a low concentration of cesium (1 mm). Cesium had little effect on burst firing. Average results obtained from five cells in cesium show no significant effect of IH block on burst firing other than a decrease in interburst interval (Fig. 9E). Block of IH is expected to increase, not decrease, the interburst interval. It seems likely, therefore, that the decrease in interburst interval is caused by block of potassium channels by cesium rather than block of IH. Our observation that IH does not contribute to burst firing is consistent with the findings of Swensen and Bean (2003) in dissociated Purkinje cells.
Discussion
We have studied spontaneous bursting in cerebellar Purkinje neurons. In the absence of rapid synaptic input, Purkinje neurons exhibit long trains of regular bursts as part of a trimodal pattern of activity. The bursts exhibit a stereotypical waveform. Each burst develops with an increase in firing rate. The final few spikes in each burst show a more rapid increase in firing rate and a decrease in spike height, the result of a dendritic calcium spike that terminates the burst. We investigated the contribution of T- and P/Q-type calcium channels, hyperpolarization-activated channels, and SK and BK calcium-activated potassium channels to spontaneous bursting in Purkinje neurons. Surprisingly, none of the channels studied, with the exception of the P/Q-type calcium channels, are required for regular spontaneous bursts.
The bursts appear to result from the interplay between a progressively depolarizing membrane potential, on which somatic sodium-dependent action potentials ride, and the dendritic calcium spikes that terminate the bursts. Two mechanisms can account for this interaction. The first is that bursts are initiated by somatic conductances and terminated by dendritic calcium spikes; the second is that bursts are both initiated and terminated by the dendritic calcium spikes. In cerebellar Purkinje cells, somatic injection of depolarizing current can evoke dendritic calcium spikes (Hounsgaard and Yamamoto, 1979; Llinás and Sugimori, 1980; Lev-Ram et al., 1992). It is plausible, therefore, that calcium spikes that terminate bursts only occur when the progressive depolarization of the somatic membrane potential by somatic conductances has reached the threshold for activating a dendritic calcium spike. Therefore, activation of conductances that contribute an inward current during the bursts would shorten the burst duration, whereas activation of those that contribute an outward current would lengthen it. In the context of this “somatic” mechanism, the finding that block of T-type calcium current prolongs the burst duration suggests that these channels contribute to the depolarizing phase of the bursts. Conversely, block of BK channels decreases burst duration, suggesting that these channels slow the depolarizing phase; however, the fact that the depolarizing phase remains when both T-type calcium and BK channels are blocked indicates that additional factors contribute. These could include an increase in the fraction of sodium channels that recover from inactivation through the resurgent state, or slow inactivation of potassium channels.
If, in contrast to the somatic mechanism discussed, bursts are controlled entirely by the dendritic calcium spikes, it is possible that the foot of the calcium spikes causes the progressive depolarization of the somatic membrane seen during the bursts. Under this scenario, the fact that block of both BK and T-type calcium channels alters burst duration is consistent with the hypothesis that both contribute significantly to the slope of the depolarizing foot of the dendritic calcium spikes. Our data do not allow us to discriminate between somatic or dendritic mechanisms for initiation of spontaneous bursts; however, three observations provide indirect support for the dendritic mechanism. First, removal of the electrical contribution of the dendrites results in loss of the regular spontaneous bursts described here (Womack and Khodakhah, 2002a). Second, partial block of P/Q-type calcium channels concurrently eliminates both regular bursting and dendritic calcium spikes, indicating a critical role for these channels in generating the bursts. Third, cyclical generation of dendritic calcium spikes, which is associated with bursting, remains (albeit with a different rate) after elimination of somatic sodium-dependent action potentials. It is important to note that the rate and pattern of generation of calcium spikes are regulated by the somatic sodium spikes. Therefore, even in the case of a dendritic mechanism for initiation and termination of bursts, a clear and complex interaction between somatic and dendritic conductances is important to maintain normal spontaneous bursting in Purkinje cells.
It is clear that bursts are terminated by P/Q channel-dependent dendritic calcium spikes. Two mechanisms can account for termination of somatic bursts by calcium spikes. It is possible that the calcium spike inactivates such a large fraction of the somatic sodium channels that the cell is incapable of generating further action potentials. Alternatively, it may be that the large influx of calcium mediated by the calcium spike activates a substantial potassium conductance that abruptly terminates the somatic bursts. We favor the former possibility because although calcium-activated potassium channels contribute to the interburst interval, regular spontaneous bursting continues when both SK and BK channels are blocked simultaneously (Womack and Khodakhah, unpublished observations). The inward T-type calcium current contributes to depolarizing the membrane toward threshold and initiation of the next burst. Additional factors that contribute to the duration of the interburst interval may be the rate of recovery of sodium channels from inactivation or the gating kinetics of voltage-gated potassium channels.
Here we find that in some Purkinje neurons partial block of P/Q channels, which prevents generation of dendritic calcium spikes, produces irregular bursts similar to those seen in dissociated Purkinje neurons (Swensen and Bean, 2003). Such irregular bursting is also seen when the electrical contribution of dendrites to spontaneous firing is eliminated in intact Purkinje neurons (Womack and Khodakhah, 2002a). Although dendrites dominate spontaneous burst waveforms in intact Purkinje neurons, clearly Purkinje neurons are capable of burst firing with somatic conductances alone.
A surprising finding is that in Purkinje neurons, regular periodic generation of dendritic calcium spikes is retained in the absence of tetrodotoxin-sensitive sodium channels and fast synaptic input. This finding is in contrast to hippocampal and neocortical pyramidal neurons, where generation of dendritic calcium spikes requires intense synaptic stimulation and backpropagation of sodium-dependent action potentials (Golding et al., 1999; Larkum et al., 1999).
P/Q-type calcium channels play an essential role in regulating burst firing in Purkinje neurons. High- and low-threshold voltage-gated calcium channels also contribute to bursting in other neurons (Huguenard, 1996; Del Negro et al., 1999). In each of these cases, activation of the calcium channels contributes a depolarizing current that helps bring the cell to threshold during the bursts. Bursts are terminated by inactivation of the calcium channels (Kramer and Zucker, 1985; Huguenard and Prince, 1992; Bal and McCormick, 1997) or by activation of calcium-activated potassium channels after calcium entry (Wong and Prince, 1978; Del Negro et al., 1999; Golding et al., 1999). In Purkinje neurons, however, P/Q-type calcium channels are responsible for the abrupt termination of each burst by generating a dendritic calcium spike. The dendritic calcium spikes terminate bursts most likely by causing inactivation of sodium channels rather than by activation of calcium-dependent conductances. To our knowledge, such a mechanism for burst termination has not been described previously. Therefore, in Purkinje neurons the duration of the bursts is tightly regulated by the excitability of the dendrites and the timing of the dendritic calcium spikes rather than solely by somatic conductances. The interplay between intrinsic dendritic and somatic conductances is essential in controlling the pattern of activity in Purkinje neurons and emphasizes the importance of considering dendrites in the regulation of spontaneous neuronal activity, as suggested by Swensen and Bean (2003).
Footnotes
This work was supported by a grant from the National Institutes of Health (DA 14058).
Correspondence should be addressed to Kamran Khodakhah, Department of Neuroscience, Albert Einstein College of Medicine, 506 Kennedy Center, 1410 Pelham Parkway South, Bronx, NY 10461. E-mail: kkhodakh{at}aecom.yu.edu.
DOI:10.1523/JNEUROSCI.0290-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243511-11$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}