

Abstract
Spinocerebellar ataxia type 2 (SCA2) is an autosomal dominantly inherited, neurodegenerative disease caused by an expansion of polyglutamine tracts in the cytosolic protein ataxin-2 (Atx2). Cerebellar Purkinje cells (PCs) are predominantly affected in SCA2. The cause of PC degeneration in SCA2 is unknown. Here we demonstrate that mutant Atx2–58Q, but not wild-type (WT) Atx2–22Q, specifically associates with the cytosolic C-terminal region of type 1 inositol 1,4,5-trisphosphate receptor (InsP3R1), an intracellular calcium (Ca2+) release channel. Association with Atx2–58Q increased the sensitivity of InsP3R1 to activation by InsP3 in planar lipid bilayer reconstitution experiments. To validate physiological significance of these findings, we performed a series of experiments with an SCA2–58Q transgenic mouse model that expresses human full-length Atx2–58Q protein under the control of a PC-specific promoter. In Ca2+ imaging experiments, we demonstrated that stimulation with 3,5-dihydroxyphenylglycine (DHPG) resulted in higher Ca2+ responses in 58Q PC cultures than in WT PC cultures. DHPG-induced Ca2+ responses in 58Q PC cultures were blocked by the addition of ryanodine, an inhibitor of the ryanodine receptor (RyanR). We further demonstrated that application of glutamate induced more pronounced cell death in 58Q PC cultures than in WT PC cultures. Glutamate-induced cell death of 58Q PC cultures was attenuated by dantrolene, a clinically relevant RyanR inhibitor and Ca2+ stabilizer. In whole animal experiments, we demonstrated that long-term feeding of SCA1–58Q mice with dantrolene alleviated age-dependent motor deficits (quantified in beam-walk and rotarod assays) and reduced PC loss observed in untreated SCA2–58Q mice by 12 months of age (quantified by stereology). Results of our studies indicate that disturbed neuronal Ca2+ signaling may play an important role in SCA2 pathology and also suggest that the RyanR constitutes a potential therapeutic target for treatment of SCA2 patients.
Introduction
Spinocerebellar ataxia type 2 (SCA2) is an autosomal dominant genetic neurodegenerative disorder (Lastres-Becker et al., 2008). SCA2 patients usually present with progressive ataxia accompanied by a variety of additional symptoms, such as dysarthria, ophthalmoplegia, extrapyramidal or pyramidal signs, slow eye movements, and peripheral neuropathy (Filla et al., 1999; Schöls et al., 2004; Lastres-Becker et al., 2008). Purkinje cells (PCs) of the cerebellum are the primary locus of pathology in SCA2 patients (Geschwind et al., 1997; Lastres-Becker et al., 2008). At the molecular level, the cause of SCA2 is the expansion of an unstable trinucleotide CAG repeat in the ATXN2 gene, which encodes a polyglutamine (polyQ) track in the ataxin-2 protein (Atx2). In normal individuals, ATXN2 alleles contain between 14 and 31 CAG repeats, with the 22 CAG repeat allele predominant in the normal population (Imbert et al., 1996; Pulst et al., 1996; Sanpei et al., 1996). The pathological alleles of ATXN2 found in SCA2 patients contain ≥32 CAG repeats (Imbert et al., 1996; Pulst et al., 1996, 2005; Sanpei et al., 1996). SCA2 is a member of the family of polyglutamine expansion neurodegenerative disorders that include SCA1, Machado-Joseph disease (SCA3), SCA6, SCA7, Huntington's disease (HD), spinal bulbar muscular atrophy, and dentatorubral pallidoluysian atrophy (DRPLA).
Atx2 is composed of 1312 amino acid residues with a molecular mass of ∼140 kDa (Albrecht et al., 2004). Atx2 is a cytosolic ubiquitously expressed protein that contains an RNA-binding Lsm domain (Kozlov et al., 2001; Ralser et al., 2005a). Potential functions of Atx2 in RNA translation and splicing (Shibata et al., 2000; Ralser et al., 2005a; Satterfield and Pallanck, 2006), endocytosis (Ralser et al., 2005b), and actin-cytoskeleton organization (Satterfield et al., 2002) were postulated. Genetic knock-outs of ATXN2 orthologs in fly and worm resulted in embryonic lethality (Kiehl et al., 2000; Satterfield et al., 2002). Atxn2 knock-out mice were viable but displayed a late-onset obesity phenotype (Kiehl et al., 2006). Mice deficient in Atx2 did not show Purkinje cell loss or marked changes in the Purkinje cell dendritic tree (Kiehl et al., 2006). Nonessential role of Atx2 in rodents is most likely related to the presence of orthologs and redundancy in its function (Kiehl et al., 2006). Despite all these studies, the cause of neurodegeneration in SCA2 remains unknown.
It is generally agreed that the mechanism of toxicity in polyglutamine expansion disorders involves a “pathological gain of function” acquired by polyQ-expanded protein. It is also commonly assumed that many polyQ-expansion disorders share a common pathogenic mechanism (Cummings and Zoghbi, 2000; Gusella and MacDonald, 2000; Zoghbi and Orr, 2000). In our previous studies of HD, we discovered that mutant Huntingtin protein (Httexp) specifically bound to and activated type 1 inositol 1,4,5-trisphosphate receptor (InsP3R1), an intracellular calcium (Ca2+) release channel (Tang et al., 2003). Based on these results, we proposed that deranged Ca2+ signaling plays an important role in HD pathology (Bezprozvanny and Hayden, 2004). In our recent studies of SCA3, we found that mutant ataxin-3 protein (Atx3exp) also specifically binds to and activates InsP3R1 (Chen et al., 2008). We further demonstrated that long-term feeding of SCA3 mice with the Ca2+ stabilizer dantrolene alleviated motor deficits and prevented loss of pontine nuclei and substantia nigra neurons in these mice (Chen et al., 2008). The main aim of the present study was to determine whether deranged Ca2+ signaling mechanisms that we uncovered for HD and SCA3 may also be involved in SCA2 pathogenesis.
Materials and Methods
SCA2–58Q mouse colony.
Generation of SCA2–58Q transgenic mice (Pcp2–atxn2[Q58]5B9, B6D2F1 strain, a C57BL/6J×DBA/2J hybrid) has been described previously (Huynh et al., 2000). The expression of Atx2–58Q transgene in these mice is driven by Pcp2/L7 Purkinje cell-specific promoter (Huynh et al., 2000). To establish a colony at University of Texas Southwestern Medical Center at Dallas, the SCA2–58Q transgenic male mice were bred to the wild-type (WT) B6D2F1/J female mice (stock #100006; The Jackson Laboratory). The mice were housed in a temperature-controlled room at 22–24°C with a 12 h light/dark cycle and were fed a standard laboratory chow diet and double-distilled water ad libitum. All procedures involving mice were approved by Institutional Animal Care and Use Committee (IACUC) of the University of Texas Southwestern Medical Center at Dallas, in accord with the National Institutes of Health Guidelines for the Care and Use of Experimental Animals.
In vitro binding experiments.
Atx2–22Q and Atx2–58Q expression plasmids have been described previously (Huynh et al., 2007). The Atx2 expression constructs were transfected into COS7 cells by DEAE-dextran transfection. At 48 h after transfection, COS7 cells were collected with ice-cold PBS and solubilized for 30 min at 4°C in extraction buffer A (1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.4 mm KH2PO4, pH 7.2, 5 mm EDTA, 5 mm EGTA, and protease inhibitors). Extracts were clarified by centrifugation for 20 min at 100,000 × g. Efficient expression of Atx2–22Q and Atx2–58Q proteins was confirmed by Western blotting of COS7 lysates with anti-Atx2 monoclonal antibodies (mAb) (catalog #611378; BD Biosciences). Glutathione S-transferase (GST)–IC10 expression construct, encoding F2627–A2749 fragment of rat InsP3R1, was expressed in the BL21 Escherichia coli strain and purified on glutathione-agarose beads as described previously (Tang et al., 2003). GST, set as negative control, was expressed and purified in the same way. Lysates containing Atx2–22Q/58Q proteins were incubated with GST or GST–IC10 proteins attached to glutathione-agarose beads for 1 h at 4°C. The beads were washed three times with the extraction buffer A, and attached proteins were analyzed by Western blotting with anti-Atx2 mAb.
Full-length rat InsP3R1 (RT1) was expressed in Sf9 cells by baculoviral infection as described previously (Tu et al., 2002). The RT1-infected Sf9 cells were solubilized in extraction buffer A, cleared by centrifugation (100,000 × g), and mixed with the equal volume of Atx2–22Q/58Q COS7 lysates for 2 h at 4°C. The mixture was precipitated with anti-InsP3R1 polyclonal antibody (pAb) T443 (Kaznacheyeva et al., 1998) attached to protein A-Sepharose beads and analyzed by Western blotting with anti-Atx2 mAb. Preimmune sera (P/S) were used in the control immunoprecipitation experiments.
Brain immunoprecipitations.
The brain immunoprecipitation experiments were performed by modification of a procedure that we used previously in studies of InsP3R1 association with AKAP9 (Tu et al., 2004). Briefly, brains of adult wild-type and SCA2–58Q mice were collected out of skull, and only the cerebella were taken. The cerebella samples were homogenized and solubilized at 4°C for 2 h in extraction buffer A (1% CHAPS, 137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.4 mm KH2PO4, pH 7.2, 5 mm EDTA, 5 mm EGTA, and protease inhibitors). The homogenates were cleared by 30 min centrifugation at 50,000 rpm in TL-100 and incubated with anti-atx2 rabbit polyclonal antibody (SP714, raised against 714CEAKDSRLQDQRQNSPAG730 peptide derived from mouse Atxn2 sequence) attached to protein A-Sepharose CL-4B beads (GE Healthcare) at 4°C for 2 h. The resulting beads were washed three times with extraction buffer A containing protease inhibitors. Beads were pelleted by centrifugation, washed three times with the extraction buffer A, incubated for 30 min with 5 nm [3H]InsP3 (PerkinElmer) on ice, pelleted by centrifugation, and rinsed quickly with extraction buffer A. The amount of [3H]InsP3 in the final samples was quantified by β-counting. The nonspecific background signal was estimated by performing immunoprecipitation with empty protein A beads. The precipitated [3H]InsP3 counts were normalized to nonspecific background.
Functional reconstitution of Atx2–RT1 to black lipid membranes.
RT1 baculovirus encoding full-length rat InsP3R1 was described previously (Tu et al., 2005b). Full-length Atx2–22Q and Atx2–58Q coding sequences were amplified by PCR using mammalian expression constructs as templates and subcloned into the pFastBac1 vector (GE Healthcare). The Atx2–22Q and Atx2–58Q baculoviruses were generated and amplified using Bac-to-Bac system according to the instructions of the manufacturer (GE Healthcare). Expression of Atx2–22Q and Atx2–58Q proteins in Sf9 cells was confirmed by Western blotting with anti-Atx2 mAb. Sf9 cells were coinfected with RT1 and Atx2–22Q or Atx2–58Q baculoviruses. The endoplasmic reticulum (ER) microsomes from coinfected Sf9 cells were isolated 48 h after infection as described previously (Tang et al., 2003; Tu et al., 2005b; Chen et al., 2008) and stored at −80°C in the storage buffer. The microsomes were fused to the planar lipid bilayers as described previously (Tang et al., 2003; Tu et al., 2005b; Chen et al., 2008), and recordings of InsP3R1 activity were performed at 0 mV transmembrane potential using 50 mm Ba2+ in the trans (lumenal) chamber as a charge carrier. The cis (cytosolic) chamber contained 110 mm Tris dissolved in HEPES [pH 7.35, 0.5 mm Na2ATP, pCa 6.7 (0.2 mm EGTA plus 0.14 mm CaCl2)] (Bezprozvanny et al., 1991). InsP3R1 were activated by the addition of 100 nm or 2 μm InsP3 (Alexis) to the cis chamber as indicated in text. The InsP3R1 single-channel currents were amplified (OC-725; Warner Instruments), filtered at 1 kHz by a low-pass eight-pole Bessel filter, digitized at 5 kHz (Digidata 1200; Molecular Devices), and stored on computer hard drive and recordable optical discs. For offline computer analysis (pClamp 6; Molecular Devices), currents were filtered digitally at 500 Hz. For presentation of the current traces, data were filtered at 200 Hz.
Primary Purkinje cell culture.
The cerebellar dissociated cell culture was prepared as described previously (Tabata et al., 2000; Gimenez-Cassina et al., 2007) with some modifications. Male SCA2–58Q transgenic mice (58Q) were crossed with female WT mice of the same strain (B6D2F1 mouse strain, a C57BL/6J×DBA/2J hybrid). The postnatal day 0–1 neonatal pups were collected and genotyped by PCR with primers specific for the human Atx2 transgene. The 58Q pups and WT littermates were separated, the cerebella were dissected on ice from the whole brain, and meninges were removed. After a single wash with ice-cold Ca2+ and Mg2+-free HBSS (Invitrogen), the intact isolated cerebella were incubated in the papain solution (Papain Dissociation Kit System; Worthington Biochemical Corporation) prepared according to the guidelines of the manufacturer for 25 min at 37°C with gentle shaking every 3–4 min. The digestion was terminated by the ovomucoid solution (Papain Dissociation Kit System; Worthington Biochemical Corporation). The digested tissue was washed twice in DMEM/F-12 solution (Invitrogen) and triturated gently by fire-polished Pasteur pipettes with three different pore sizes. The resulting suspension was spun at 1000 rpm for 4 min. The pellet was resuspended in 10% FBS in DMEM/F-12. To remove fibroblasts and glial cells, the cell suspension was plated onto a 10 μg/ml poly-d-lysine-coated culture dish (10 cm diameter) for 10 min at 37°C and then recovered and seeded onto coverslips coated with 500 μg/ml poly-d-lysine at a density of 5 × 106 cells/ml. The next morning, PC culture media (DMEM/F-12 with addition of 100 mm putrescine, 30 nm sodium selenite, 3.9 mm l-glutamine, 20 nm progesterone, 10 ng/ml insulin, 100 mg/ml transferrin, 0.5 ng/ml tri-iodothyronine) was added with a final FBS concentration at 1%. At 5 and 12 d in vitro (DIV), half of the medium was replaced with the fresh PC culture medium containing 1% FBS and AraC (4 μm).
Immunostaining.
Immunostaining of cultured PCs was performed as described previously (Gimenez-Cassina et al., 2007). Briefly, PCs cultured on glass coverslips were fixed with paraformaldehyde and washed with PBS. After incubation in 0.25% Triton X-100 in PBS for 5 min, PCs were blocked in blocking solution (5% BSA and 0.1% Triton X-100 in PBS) for 1 h at room temperature, followed by the exposure to anti-InsP3R1 polyclonal antibody (T443) diluted in blocking solution overnight at 4°C. After three extensive washes with PBS, PCs were incubated with FITC-conjugated mouse anti-rabbit mAb for 1 h at room temperature. After PCs were stained with 4′,6′-diamidino-2-phenylindole (DAPI), the coverslips were immediately mounted and stored in darkness at 4°C. An Olympus IX70 fluorescence microscope was used for observations using blue (DAPI) and green (FITC) filter cubes (Chroma Technology). The pictures were taken using Cascade 650 ICCD camera and MetaFluor software (Nikon).
Ca2+ imaging.
Ca2+ imaging experiments with PC cultures were performed as described previously for rat medium spiny neuron (MSN) cultures (Tang et al., 2003) with minor modifications. Briefly, the 14–15 DIV 58Q and WT PC cultures were loaded with 5 μm fura-2 AM (Invitrogen) in PC culture media for 50 min at 37°C and then washed in prewarmed artificial CSF (ACSF) (in mm: 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, and 10 HEPES, pH 7.3) for 5 min at 37°C. The cells were maintained in ACSF at 37°C during imaging experiments (PH1 heater; Warner Instruments). For Ca2+ imaging experiments, the PC cells were intermittently excited by 340 and 380 nm UV light using DeltaRAM illuminator (Photon Technology International), and the 510 nm emitted light was collected by IC-300 camera (Photon Technology International). Images were digitized and analyzed by ImageMaster Pro software (Photon Technology International). The PCs were identified by the large size and oval shape of their soma and abundant dendrites (Southan and Robertson, 2000). Baseline (1–2 min) measurements were obtained before the bath application of 3,5-dihydroxyphenylglycine (DHPG), which was dissolved in ACSF and prewarmed at 37°C. In experiments with ryanodine, PCs were maintained in culture medium containing 100 μm ryanodine overnight before Ca2+ imaging was performed. Ryanodine was included at 100 μm in all solutions during Ca2+ imaging steps.
In vitro apoptosis cell death assays.
The in vitro cell death assays with cultured PCs were performed as described previously for MSN cultures (Tang et al., 2005, 2009) with minor modifications. Briefly, WT and 58Q PC cells at 14 DIV were exposed to 200 μm glutamate for 7 h in cell culture incubator (humidified 5% CO2, 37°C). Immediately after the exposure to glutamate, the cells were fixed for 25 min in 4% paraformaldehyde plus 4% sucrose in PBS, pH 7.4, permeabilized for 5 min in 0.25% Triton X-100 in PBS, and stained by in situ cell death terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) detection kit (TMR red; Roche) according to the instructions of the manufacturer. To identify the PCs, immunostaining was also performed using anti-InsP3R1 polyclonal antibody (T443) and FITC-conjugated mouse anti-rabbit mAb as described above. To evaluate the effects of glutamate treatment on the total PC number and on the fraction of apoptotic PCs, TUNEL-positive PCs and TUNEL-negative PCs were counted separately in 10–15 randomly chosen microscopic fields. Cell numbers of TUNEL-positive PCs and whole number of PCs for each microscopic field were averaged and presented as means ± SE. In addition, the number of TUNEL-positive PCs was calculated as a fraction of total PCs in each microscopic field, the fractions of TUNEL-positive PCs determined for each microscopic field were averaged, and the results are presented as means ± SE. Dantrolene was added to the cell culture medium (in final concentration of 10–50 μm) 1 h before the addition of glutamate as indicated in the text. In experiments with 1 μm dantrolene, PC cells were incubated with dantrolene for 24 h before addition of glutamate.
Dantrolene feeding.
Dantrolene feeding to SCA2–58Q mice was performed by following the same procedures that were used for tetrabenazine feeding to HD–YAC128 mice (Tang et al., 2007) and for dantrolene feeding to SCA3–YAC–84Q mice (Chen et al., 2008). Briefly, groups of age-matched female WT and SCA2–58Q mice were fed with dantrolene, which was suspended in 50 μl of PBS at the dosage of 5 mg/kg body weight. The control groups of WT and SCA2–58Q mice were fed with PBS. All mice were fed orally twice a week from 2 months of age until 11 months of age. Between 11 and 12 months of age, all mice were fed with PBS only (washout).
Motor coordination assessments in mice.
The motor coordination assays with SCA2–58Q mice were performed as described previously for HD–YAC128 and SCA3–YAC–84Q mice (Tang et al., 2007, 2009; Chen et al., 2008). The “beam-walk” assay was performed using a home-made experimental setup. The 17 mm round plastic beam, 11 mm round plastic beam, and 5 mm square wood beam were used in our studies. At each time point, the mice were trained on beams for 3 consecutive days (four training sessions per day) to traverse the beam to the enclosed box. Once the stable baseline of performance was obtained, the mice were tested in three consecutive trials on 17 and 11 mm round plastic beams and 5 mm square wood beam, in each case progressing from the widest to the narrowest beam. The latency to traverse the middle 80 cm of each beam and the number of times the hindpaws slipped off each beam were recorded for each trial. For those mice with “crawling behavior,” their every crawling step was counted as one foot slip. For each measurement, the mean scores of the three trials for each beam were used in the analysis.
Three days after completion of beam-walking assay the rotarod assay was performed using Economex rotarod apparatus (Columbus Instruments). Before the baseline collection, mice were screened on the rotarod rotated at a constant speed of 5 rpm. The mice that fell off the rotarod within 5 min were dismissed. At each age, the mice were trained on the accelerating rotarod (accelerated from 0 to 40 rpm over 200 s) with three training session per day for 3 consecutive days, by which time a steady baseline of performance was attained. The testing was executed over 1 d with 1.5 h of rest between tests. The mean latency to fall off the rotarod recorded in the three trials was used in the analysis.
Stereological analysis.
After the motor assays at the last time point (12 months of age), the SCA2–58Q mice were perfused and processed as described previously for HD–YAC128 and SCA3–YAC–84Q mice (Tang et al., 2007, 2009; Chen et al., 2008). The cerebella were frozen on dry ice and cut sagittally to 50-μm-thick sections using a Leica SM2000R sliding microtome. The cerebellar slices from every 20th section were picked and stained with anti-Calbindin D 28K (CBD28) monoclonal antibody (1:500 dilution, clone CB-955; Sigma). Biotinylated anti-mouse IgG reagent was used as secondary antibody (1:250 dilution; M.O.M kit; Vector Laboratories). Signal was amplified with an ABC Elite kit (Vector Laboratories) and detected with diaminobenzidine (Vector Laboratories).
An optical fractionator stereological design (West et al., 1991) was used to make unbiased estimates of total PC number. All stereological analyses were performed blindly with respect to the nature of slices (genotype and drug feeding) using Stereoinvestigator setup and software (MicroBrightField). The 40× objective was used for cell counting. For PC cell counting, the grid size of 500 × 500 μm and the counting frame size of 150 × 150 μm were applied to a systematic random sample of sites in the cerebellar cortex over the entire cerebellum (Sajdel-Sulkowska et al., 2005; Woodruff-Pak, 2006). PC cells with weak anti-CBD28 staining were counted as 0.5 of a cell.
Measurements of molecular layer thickness.
The molecular layer (ML) thickness was determined by measuring the distance between the PC body layers using Stereoinvestigator setup and dividing in half as described previously (Zu et al., 2004).
Pathological analysis.
After removal of the brains, the mouse carcasses were stored at −20°C until dissected. Representative sections of the heart, liver, and skeletal muscle from a hindleg of half of the animals from each group were obtained and fixed in 10% buffered Formalin. These samples were processed into paraffin blocks, sectioned at 4 μm thickness, stained with hematoxylin and eosin, and coverslipped for light microscopic evaluation. The presence and degree of lymphocytic infiltrates in the three tissue types was semiquantitatively graded using the following schema: 0, no inflammation; 1, rare, scattered, small collections of lymphocytes; 2, occasional scattered collections; and 3, frequent large collections.
Statistical analysis.
Differences between groups were judged by a two-tailed Student's unpaired t test using a significance level of 0.05. For comparison between more than two groups, ANOVA followed by post hoc Fisher's PLSD test was used.
Results
Mutant ataxin-2 specifically binds to InsP3R1 in vitro and in vivo
In previous studies, we demonstrated that polyQ-expanded Huntingtin (Httexp) and ataxin-3 (Atx3exp) specifically bound to the InsP3R1 C-terminal region (Tang et al., 2003; Chen et al., 2008). To determine whether Atx2exp also binds to InsP3R1 C-terminal region, we expressed and purified InsP 3 R1 C-terminal fragment (IC10; amino acids F2627–A2749 of rat InsP3R1) as GST–IC10 fusion protein in the BL21 E. coli strain. GST protein was expressed and purified as a negative control. Wild-type Atx2–22Q and mutant Atx2–58Q human proteins were expressed in COS7 cells by transient transfection (Huynh et al., 2007). The lysates from transfected COS7 cells were mixed with recombinant GST–IC10 or GST proteins and glutathione bead pull-down experiments were performed. The precipitated proteins were detected by Western blotting with anti-Atx2 mAb. As a result of these experiments, we found that GST–IC10 specifically associated with Atx2–58Q protein (Fig. 1 A), whereas GST did not. There was no interaction between Atx2–22Q and GST–IC10 or GST detected in our experiments (Fig. 1 A). Thus, we concluded that mutant Atx2exp specifically associated with the InsP3R1 C-terminal region, similar to Httexp and Atx3exp (Tang et al., 2003; Chen et al., 2008).

InsP3R1 specifically associates with Atx2exp. A , Pull-down of Atx2–22Q/58Q from COS7 cell lysates with GST–IC10 protein. GST was used as a negative control. B , Precipitation of Atx2–22Q/58Q from COS7 cell lysates by recombinant full-length InsP3R1 (RT1). Anti-InsP3R1 polyclonal antibodies (T443) and corresponding control P/S were used for the precipitation. The precipitated fractions in A and B were analyzed by Western blotting (WB) with anti-Atx2 monoclonal antibody. The input lanes in A and B contain 1:20 of the COS7 cell lysates used in GST and InsP3R1 pull-down experiments. C , Cerebellar homogenates prepared from adult WT and 58Q mice were extracted in 1% CHAPS and immunoprecipitated with rabbit polyclonal antibodies against Atx2. The amount of precipitated InsP3R1 was quantified in an [3H]InsP3 binding assay. The data were normalized to the results obtained using empty protein A beads and presented as mean ± SE (n = 3). The number of [3H]InsP3 binding sites precipitated from SCA2–58Q mice cerebellar lysates was significantly (**p < 0.01) higher than the number of [3H]InsP3 binding sites precipitated from the WT mice cerebellar lysates.
Does the full-length InsP3R1 bind to Atx2exp? To address this question, we expressed full-length rat InsP 3 R1 (RT1) in Sf9 cells by baculoviral infection (Tu et al., 2002) and solubilized recombinant InsP3R1 in 1% CHAPS. Atx2–22Q and Atx2–58Q proteins were expressed in COS7 cells as described above. The mixture of RT1-infected Sf9 cell lysates and Atx2–22Q/58Q COS7 lysates was precipitated with anti-InsP3R1 polyclonal antibody (T443) (Kaznacheyeva et al., 1998) attached to protein A-Sepharose beads. The P/S was used in control immunoprecipitation experiments. The precipitated fractions were analyzed by Western blotting with anti-Atx2 mAb. As a result of these experiments, we found that full-length InsP3R1 strongly associated with mutant Atx2–58Q protein but not with wild-type Atx2–22Q protein (Fig. 1 B). Thus, the association of Atx2exp with full-length InsP3R1 is similar to Atx3exp (Chen et al., 2008). In contrast to Atx2 and Atx3, not only mutant Httexp but also wild-type Htt binds to the full-length InsP3R1 (Tang et al., 2003).
To determine whether Atx2exp formed a complex with InsP3R1 in vivo, we took advantage of a transgenic SCA2 mouse model that expresses a human Atx2–58Q transgene under the control of the PC cell-specific Pcp2 promoter (Huynh et al., 2000). In our experiments, cerebellar lysates were prepared from WT and 58Q mice and precipitated with anti-Atx2 rabbit polyclonal antibody. The amount of precipitated InsP3R1 was quantified by [3H]InsP3 binding as we described previously in studies of InsP3R1 association with AKAP9 (Tu et al., 2004). The background signal was determined by performing control experiments with empty beads. We found that the amount of [3H]InsP3 binding sites precipitated by anti-Atx2 pAb from WT cerebellar lysates was not significantly different from background (Fig. 1 C). In contrast, anti-Atx2 pAb precipitated significantly more [3H]InsP3 binding sites from the 58Q mice cerebellar lysates (p = 0.0019 compared with WT) (Fig. 1 C). Thus, we concluded that mutant Atx2exp, but not wild-type Atx2, binds to cerebellar InsP3R1 in vivo.
Mutant ataxin-2 activates InsP3R1 in vitro
In previous studies, we demonstrated that the association of InsP3R1 with Httexp and Atx3exp increased sensitivity of InsP3R1 to activation by InsP3 (Tang et al., 2003; Chen et al., 2008). Does Atx2exp have a similar effect on InsP3R1 activity? To answer this question, we generated baculoviruses expressing full-length Atx2–22Q and Atx2–58Q proteins. The expression of Atx2–22Q/58Q proteins in baculovirus-infected Sf9 cells was confirmed by Western blotting with anti-Atx2 mAb (data not shown). The Sf9 cells were coinfected by Atx2–22Q or Atx2–58Q baculoviruses together with InsP3R1-encoding RT1 baculovirus. The ER microsomes from coinfected Sf9 cells were purified by gradient centrifugation and fused to planar lipid bilayers [black lipid membranes (BLM)] as we described previously (Tu et al., 2005a). No channel activity was observed before addition of InsP3 in our experiments (Fig. 2 A,B). Addition of 100 nm InsP3 resulted in low levels of channel activity of InsP3R1 coexpressed with Atx2–22Q (Fig. 2 A, top traces, B) but resulted in a dramatic activation of InsP3R1 coexpressed with Atx2–58Q (Fig. 2 C, top traces, D). Increase in InsP3 concentration to 2 μm maximally activated InsP3R1 coexpressed with Atx2–22Q (Fig. 2 A, bottom traces, B) but had no additional effect on the activity of InsP3R1 coexpressed with Atx2–58Q (Fig. 2 C, bottom traces, D). Similar changes in InsP3R1 functional properties were observed in our previous bilayer reconstitution studies as a result of coexpression with full-length Htt–82Q or full-length Atx3–84Q (Tang et al., 2003; Chen et al., 2008). From these results, we concluded that the association with Atx2exp increases apparent affinity of InsP3R1 to activation by InsP3, similar to the effects of Httexp and Atx3exp (Tang et al., 2003; Chen et al., 2008).

Effects of Atx2 exp on InsP3R1 in BLM. A , C , Single-channel recordings of InsP3R1 activity in planar lipid bilayers (BLM). InsP3R1 (RT1) was coexpressed in Sf9 cells together with Atx2–22Q ( A ) or Atx2–58Q ( C ). The channels were recorded in the presence of 100 nm and 2 μm InsP3. Each current trace corresponds to 10 s (2 s for expanded traces) of current recording for the same experiment. B , D , The average open probability (P o) of InsP3R1 in BLM at 100 nm and 2 μm InsP3 in the presence of Atx2–22Q ( B ) and Atx2–58Q ( D ). The average P o was calculated for a 5 s window of time and plotted for the duration of an experiment. The times of InsP3 addition are shown above the P o plot. Similar results were obtained in three experiments with Atx2–22Q and three experiments with Atx2–58Q.
Mutant ataxin-2 facilitates InsP3-induced Ca2+ release in cultured Purkinje cells
Cerebellar PCs are affected early and severely in SCA2 (Geschwind et al., 1997; Lastres-Becker et al., 2008). PCs abundantly express InsP3R1 (Furuichi et al., 1989; Mignery et al., 1989). Do interactions of InsP3R1 with Atx2exp described above (Figs. 1, 2) result in abnormal Ca2+ signals in SCA2 PCs? With the modification of previously described methods (Tabata et al., 2000; Gimenez-Cassina et al., 2007), we established primary PC cultures from the WT and 58Q neonatal mouse pups. The established cultures contained large numbers of cerebellar granule and glial cells of which the nuclei could be visualized by DAPI staining (Fig. 3 A, shown by pseudocolor red). Because InsP3R1 is very highly enriched in PCs (Furuichi et al., 1989; Mignery et al., 1989), PCs in these cultures could be identified by immunostaining with T443 pAb directed against InsP3R1 C termini (Fig. 3 A, shown by green). Cultured PCs undergo maturation with well differentiated dendrites (Tabata et al., 2000; Gimenez-Cassina et al., 2007). The shape and morphology of PC dendritic trees from WT and SCA2–58Q mice were indistinguishable at 14 DIV in our cultures (Fig. 3 A).

Effects of Atx2 exp on DHPG-induced Ca2+ release in Purkinje cells. A , Primary cultures of WT and 58Q PCs. PCs cultures were prepared as described in Materials and Methods. At 14 DIV, PCs were fixed and stained with anti-InsP3R1 polyclonal antibody (T443, green) and DAPI (pseudocolor, red). PC pictures under 20× and 40× magnification are shown as indicated. For 40× magnification, several fields of view of a single PC were taken and combined into one image. B , Representative images of Ca2+ responses in WT and 58Q PCs cultures. Fura-2 ratio recordings are shown for 10 μm DHPG-induced Ca2+ transients in WT PCs (first row), WT PCs in the presence of 100 μm ryanodine (Ry; second row), 58Q PCs (third row), and 58Q PCs in the presence of 100 μm ryanodine (fourth row). Fura-2 340/380 ratio images are shown at 1 min before (first column) and 10 s, 25 s, 1 min, and 3 min after the application of 10 μm DHPG as indicated. The pseudocolor calibration scale for 340/380 ratios is shown on the right.
To compare InsP3R1-mediated Ca2+ signals in WT and 58Q PCs, the cells at 14 DIV were loaded with fluorescent Ca2+ imaging indicator fura-2 and challenged with DHPG, a specific mGluR1/5 receptor agonist. Cytosolic Ca2+ concentration in the experiments was estimated from the ratio of fura-2 signals at 340 and 380 nm excitation wavelengths as shown by pseudocolor images (Fig. 3 B). On average, basal Ca2+ levels before DHPG application were similar in WT and 58Q PC cultures (Figs. 3 B, left column, 4 I). The application of 10 μm DHPG triggered strong Ca2+ responses in 58Q PCs (Figs. 3 B, third row, 4 E,F) but much smaller Ca2+ responses in WT PCs (Figs. 3 B, first row, 4 A,B). On average, the amplitude of Ca2+ responses elicited by 10 μm DHPG was significantly (p < 0.01) higher in 58Q PC cultures than in WT PC cultures (Fig. 4 I).

Atx2 exp facilitates DHPG-induced Ca2+ release in Purkinje cells. A , C , E , G , DHPG-induced Ca2+ responses are plotted as basal and peak 340/380 ratios for individual PCs from WT ( A ), WT with ryanodine (Ry) ( C ), 58Q ( E ), and 58Q with ryanodine ( G ) groups. Basal ratios were determined 1 min before DHPG application (−1 min), and peak ratios were measured from maximal signals observed within 30 s after DHPG application. Fura-2 340/380 ratio traces for representative cells (marked +) are shown in B , D , F , and H . The time of DHPG application is shown by a bar. Similar results were obtained in three different culture batches of PCs cells. I , Summary of Ca2+ imaging experiments with WT and 58Q PC cultures. Average basal and DHPG-evoked peak fura-2 340/380 ratios are shown for WT and 58Q PCs cells. The results are combined from three different batches of cells and shown as mean ± SE (n = number of cells). Fura-2 peak 340/380 ratios in 58Q PCs are significantly (***p < 0.001) higher than in WT PCs. With addition of ryanodine, the peak ratios in 58Q PCs are dramatically decreased (**p < 0.01).
Both InsP3R1 (Furuichi et al., 1989; Mignery et al., 1989) and the ryanodine receptor RyanR1 (Kuwajima et al., 1992; Mori et al., 2000) are abundantly expressed in PCs. There is significant functional overlap between these two intracellular Ca2+ release channels (Simpson et al., 1996; Turner et al., 2001). Furthermore, it has been demonstrated that RyanR1 and InsP3R1 use a common Ca2+ pool in PCs and that inhibition of RyanR by ryanodine significantly reduces InsP3R1-induced Ca2+ release (Khodakhah and Armstrong, 1997). Consistent with these observation, we found that preincubation of 58Q PCs with 100 μm ryanodine significantly reduced DHPG-induced Ca2+ responses in 58Q PC cells (Figs. 3 B, fourth row, 4 G,H). The Ca2+ responses in WT PCs were not significantly affected by the application of ryanodine (Figs. 3 B, second row, 4 C,D).
Mutant ataxin-2 sensitizes Purkinje cells to glutamate-induced apoptosis
Do enhanced Ca2+ signals observed in 58Q PCs (Figs. 3, 4) contribute to cell death of PC cells in SCA2? To answer this question, we performed in vitro cell death experiments with WT and 58Q PC cultures. In these experiments, 14 DIV WT and 58Q PC cultures were challenged by a 7 h application of 200 μm glutamate as we described previously for WT and HD–YAC128 striatal MSN cultures (Tang et al., 2005, 2009). After exposure to glutamate, the PCs were stained with T443 pAb to visualize cell shape. As mentioned above (Fig. 3 A), the shape and size of dendritic trees were similar in WT and 58Q PC cultures at 14 DIV before exposure to glutamate (Fig. 5, first column). The shape of both WT and 58Q PCs was affected by glutamate exposure. When compared with WT cells, 58Q cells exhibited less dendritic arborization and rougher shape (Fig. 5, second column, first and third rows). We reasoned that the changes in PC shape reflected apoptotic processes induced by exposure to glutamate. To test this hypothesis, we performed TUNEL staining assay to identify apoptotic nuclei (Fig. 5, third column, red). The DAPI staining was used to label all nuclei in the field of view (Fig. 5, third column, blue). Overlay of TUNEL/DAPI and T443-stained images enabled us to score apoptotic cell death specifically for PCs (Fig. 5, fourth column, merge). From this analysis, we concluded that PCs with the worst overall shape indeed corresponded to apoptotic (TUNEL-positive) cells (Fig. 5, second and fourth columns). Because changes in PC shape are difficult to quantify, we selected the TUNEL assay for quantitative scoring of PC death in the following experiments.

Glutamate-induced cell death of cultured Purkinje cells. Purkinje cells from WT and 58Q mice were treated with 200 μm glutamate (Glu) for 7 h at 37°C at 14 DIV and then fixed and stained with anti-InsP3R1 polyclonal antibody (T443). The representative images of four PCs from each group are shown for groups without glutamate treatment (first column) and after glutamate treatment (second column). PCs treated with glutamate were further stained using an in situ cell death TUNEL detection kit (red), and nuclei were counterstained with DAPI (blue). TUNEL/DAPI images (third column) and merged T443/TUNEL/DAPI images (fourth column) are shown. PC images are shown for WT (first row), 58Q (third row), WT plus dantrolene (Dan) (second row), and 58Q plus dantrolene (fourth row) groups of cells. The pictures of individual PC cells were taken with 40× objective and combined together digitally. Ctrl, Control.
The WT and 58Q cultures contained similar number of apoptotic PCs in the absence of glutamate (Fig. 6 A, first row). On average, the WT and 58Q PC cultures contained ∼12 PCs in the field of view (Fig. 6 B), of which on average 10% were TUNEL positive (Fig. 6 B,C). After exposure to 200 μm glutamate, an average number of PCs remaining in 58Q cultures was reduced (Fig. 6 B, p = 0.0002), and ∼80% of 58Q PCs were apoptotic (Fig. 6 A, second row, right, B,C). The number of WT PCs was also reduced after exposure to glutamate (Fig. 6 B), but only 40% of WT PCs were apoptotic in the same conditions (Fig. 6 A, second row, left, B,C). Thus, we concluded that expression of Atx2–58Q sensitizes glutamate-induced apoptosis of PCs.

Purkinje cells from SCA2–58Q mice sensitized to glutamate-induced apoptosis. A , Representative images of 14 DIV PCs from WT (left) and 58Q (right) mice. Merged images of T443 (green) and TUNEL (red) stainings are shown for each group of PCs. The images are shown for PCs from untreated group of cells (Ctrl; first row), cells treated with glutamate (Glu; second row), and cells treated with glutamate in the presence of dantrolene (Dan; third row). The images were taken from several adjacent fields using 20× objective and digitally assembled. B , Average number of PCs (open bars) and average number of TUNEL-positive PCs (hatched bars) in the field of view are shown. Results are shown for untreated cells and for cells treated with 200 μm glutamate as indicated. Data for WT and 58Q PCs are compared in the presence and in the absence of 10 μm dantrolene. C , Fraction of TUNEL-positive PCs is shown for untreated cells and cells treated with 200 μm glutamate. The average fraction of TUNEL-positive cells is shown for WT PCs (open black circles), 58Q PCs (open red circles), WT PC cells in the presence of 10 μm dantrolene (filled black circles), and 58Q PC cells in the presence of 10 μm dantrolene (filled red circles). Data for each groups of cells are shown as mean ± SE (n = 10–15 microscopic fields, 100–200 PCs in total). Similar results were obtained with two independent PC culture preparations. Glutamate-induced apoptosis of 58Q PCs was significantly (*p < 0.05) higher than apoptosis of WT. Addition of dantrolene resulted in significant (*p < 0.05) reduction in apoptotic cell death of 58Q PC cells.
Ca2+ imaging experiments suggested that inhibition of RyanR1 could efficiently attenuate Ca2+ signals in 58Q PCs (Figs. 3, 4). To determine whether inhibition of RyanR1 protected 58Q PCs cells from glutamate-induced cell death, we performed a series of experiments with dantrolene, a clinically relevant RyanR1 inhibitor and stabilizer of intracellular Ca2+ signaling. Dantrolene is used clinically for the treatment of malignant hyperthermia and muscle spasticity (Krause et al., 2004). Dantrolene has also been reported to protect neuronal cells from a variety of excitotoxic paradigms in animal models (Frandsen and Schousboe, 1991; Berg et al., 1995; Wei and Perry, 1996; Guo et al., 1999; Niebauer and Gruenthal, 1999; Schneider et al., 2001; Popescu et al., 2002; Makarewicz et al., 2003). In our experiments, we found that application of 10 μm dantrolene reduced glutamate-induced cell shape changes in 58Q PC cultures (Fig. 5, second column, fourth row) and significantly reduced the number of apoptotic 58Q PCs (Fig. 6 A, third row, right). Application of 10 μm dantrolene to WT cultures had no significant effect on changes in PCs shape (Fig. 5, second column, second row) or the number of apoptotic PCs (Fig. 6 A, third row, left). On average, the fraction of apoptotic 58Q PCs was reduced to 55% in the presence of 10 μm dantrolene (Fig. 6 B,C, p = 0.0065), whereas the fraction of apoptotic WT PCs was unchanged at 40% (Fig. 6 B,C). With higher concentrations of dantrolene (25 and 50 μm), the number of apoptotic 58Q PCs was reduced even further, nearly to the level of WT PCs (data not shown). Overnight incubation of 58Q PCs cultures with low concentration of dantrolene (1 μm) also resulted in similar protective effects (data not shown). These results suggested that blockage of RyanR1 with dantrolene protected 58Q PCs from glutamate-induced apoptosis.
Dantrolene alleviates motor coordination deficits in SCA2–58Q mice
The experiments with 58Q PC cultures suggested that deranged Ca2+ signaling (Figs. 3, 4) may facilitate excitotoxic cell death of these cells (Figs. 5, 6). Furthermore, pharmacological inhibition of RyanR1 attenuated Ca2+ signals in 58Q PCs (Figs. 3, 4) and protected these cells from glutamate-induced cell death (Figs. 5, 6). These results indicated that RyanR1 may constitute a potential therapeutic target for the treatment of SCA2. To test these ideas in vivo, we initiated a dantrolene trial in 58Q mice. The design of this trial was inspired by our recently completed trial of dantrolene in SCA3–YAC–84Q mice (Chen et al., 2008). Starting at 2 months of age, WT and 58Q mice were orally fed with dantrolene, which was resuspended in PBS, twice a week at the dosage of 5 mg/kg weight until the age of 11 months (Table 1, groups 2 and 4). Control groups of WT and 58Q mice were fed with PBS alone at the same time (Table 1, groups 1 and 3). Consistent with a potent muscle relaxant and sedative activity of dantrolene, mice fed with dantrolene were less active immediately after dantrolene intake. However, all mice could recover within several hours after dantrolene feeding and started to behave normally. The drug feeding was stopped after the 11 month time point and was followed by a 1 month washout period during which all four groups of mice were fed with PBS. The washout period was included to enable better discrimination between long-term and acute effects of dantrolene on mouse behavior.
Dantrolene trial in SCA2–58Q mice
Motor coordination abilities of the four groups of mice were assessed every 2 months by the beam-walk assay. The beam-walking test was performed on 17 mm round plastic, 11 mm round plastic, and 5 mm square wood beams. Motor performance of mice on each beam was scored every 2 months as the “latency” and the “number of foot slips” as we described previously (Tang et al., 2007, 2009; Chen et al., 2008). Before initiation of drug feeding, basal beam-walking performance was determined when the mice were 2 months old. Beam-walking analysis revealed age-dependent differences in motor coordination between WT control and 58Q control groups (Fig. 7). Starting from the age of 6 months, 58Q control mice (fed with PBS) exhibited progressive motor impairment (longer beam traverse latencies and increased number of foot slips) with the increment of age and beam difficulty when compared with the WT control group mice (Fig. 7 A–F). Significant differences between WT control and 58Q control groups were observed starting at 8 months of age for latency on all three beams (Fig. 7 A,C,E, all at p < 0.05 by one-way ANOVA), at 10 months of age for foot slips on the 11 mm round and the 5 mm square beams (Fig. 7 D,F), and at 12 months of age for foot slips on 17 mm round beam (Fig. 7 B, all p < 0.001 by one-way ANOVA). Importantly, significant differences between 58Q and 58Q control groups were observed for both measures after 1 month washout period on all three beams (Fig. 7 A–F, **p < 0.01, ***p < 0.001, one-way ANOVA).

Beam-walk performance of WT and SCA2–58Q mice during the dantrolene trial. A , C , E , Average times to traverse the beams are shown as latency against age of the mice for 17 mm round plastic beam ( A ), 11 mm round plastic beam ( C ), and 5 mm square wooden beam ( E ). B , D , F , Average numbers of foot slips are plotted against age of the mice on 17 mm round plastic beam ( B ), 11 mm round plastic beam ( D ), and 5 mm square wooden beam ( F ). In each panel, the data for WT control mice (WT Ctrl; open black circles), 58Q control mice (58Q Ctrl; open red circles), WT mice fed with dantrolene (WT Dan; filled black circles), and 58Q mice fed with dantrolene (58Q Dan; filled red circles) are shown as mean ± SE (for the number of mice in each group, see Table 1). The results are shown at 2, 4, 6, 8, 10, and 12 months time points. Significant differences between 58Q Ctrl and WT control groups were observed at 8, 10, and 12 months as indicated (*p < 0.05, **p < 0.01, ***p < 0.001). WO, Wash out.
Feeding dantrolene to WT mice had little effect on motor performance of these mice. Slight elevation in latencies on all three beams was observed for the WT dantrolene group when compared with the WT control group (Fig. 7 A,C,E), but the difference was not statistically significant. Most likely, the difference in latency was related to effects of dantrolene as muscle relaxant (Patrono et al., 1985; Ward et al., 1986). There was no difference between WT control and WT dantrolene groups in a number of foot slips (Fig. 7 B,D,F). In contrast, feeding dantrolene to 58Q mice dramatically improved the motor performance of these mice by shortening the latencies and decreasing the number of foot slips. Beginning at 6 months of age, the mice in the 58Q dantrolene group showed better beam traverse abilities than the 58Q control group (Fig. 7 A–F). The performance of 58Q dantrolene mice was similar to the performance of WT mice on all three beams at all ages tested (Fig. 7 A–F). The improvement in behavioral performance of 58Q mice was not attributable to acute effects of dantrolene, and it was maintained after a 1 month dantrolene washout (Fig. 7 A–F).
While conducting beam-walking assays, we observed that, with older age, some 58Q control mice developed a “crawling behavior” on the beams, an extreme motor deficit in mice. Similar behavior was also observed for some of the aging HD–YAC128 mice and SCA3–YAC–84Q mice evaluated in our previous studies (Tang et al., 2007, 2009; Chen et al., 2008). At the age of 8 months, two mice in the 58Q control group crawled on the 11 mm round beam. At the 10 month time point, the number of mice in the 58Q control group that crawled on 11 mm beam increased to three mice, and there was one mouse that crawled on the 5 mm square wood beam. Two months later (12 months time point), the number of crawling mice in the 58Q control group increased to two mice on 5 mm beam. In contrast, none of the mice in WT control group, WT dantrolene group, or 58Q dantrolene group exhibited crawling behavior.
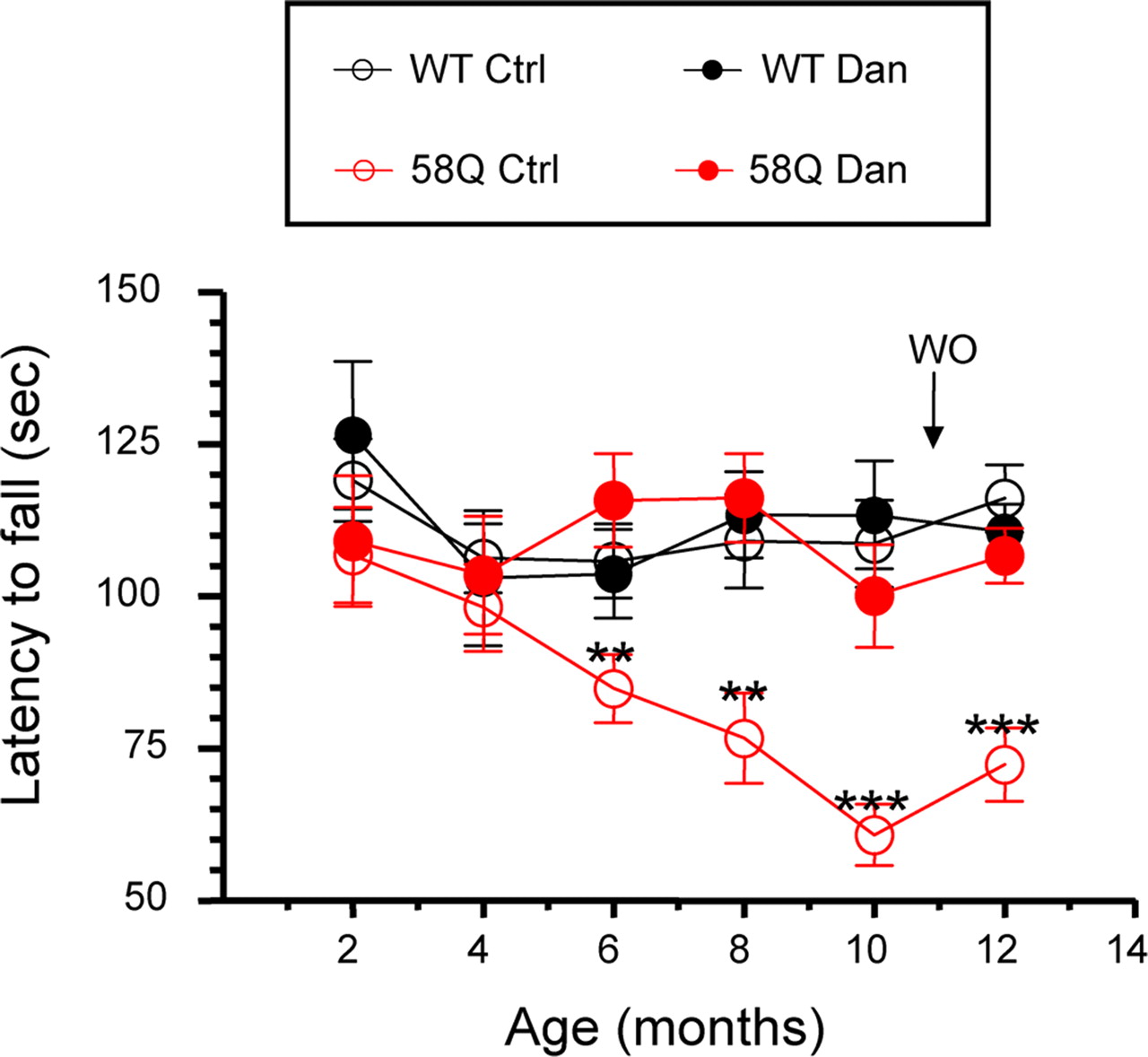
As an independent evaluation of motor performance of these mice, we also performed “accelerated rotarod” testing every 2 months. These experiments were performed as we described previously (Tang et al., 2007). The latency to fall is the readout used in rotarod testing. The rotarod assays showed that WT and 58Q mice performed similarly at 2 months of age, but, with increasing age, performance of 58Q control mice became impaired (Fig. 8). Significant differences (p < 0.01, one-way ANOVA) between 58Q control mice and WT control mice were detected starting at 6 months of age and became more pronounced with increasing age (Fig. 8). There was no apparent effect on the rotarod performance by feeding dantrolene to WT mice (Fig. 8). Feeding dantrolene to 58Q mice, however, alleviated the poor performance of these mice. At all ages, these mice performed as well on the rotarod as WT mice (Fig. 8). Consistent with results in the beam-walk assay, the improvement in rotarod performance of 58Q mice was not attributable to acute effects of dantrolene because it was maintained after a 1 month washout of dantrolene (Fig. 8).

Rotarod performance of WT and SCA2–58Q mice in dantrolene trial. Average latency to fall from the accelerating rotarod is shown for the WT control mice (WT Ctrl; open black circles), the 58Q control mice (58Q Ctrl; open red circles), WT mice fed with dantrolene (WT Dan; filled black circles), and 58Q mice fed with dantrolene (58Q Dan; filled red circles) as mean ± SE (for the number of mice in each group, see Table 1). Data are shown at 2, 4, 6, 8, 10, and 12 months time points. At 6, 8, 10, and 12 months of age, WT control mice performed significantly better (**p < 0.01, ***p < 0.001) than 58Q control mice. WO, Wash out.
Pathological analysis of mice fed with dantrolene
To determine potential toxic side effects resulting from long-term dantrolene treatment, we performed pathological analysis of several mouse carcasses from each group (10 mice from dantrolene-treated groups and 11 mice from control groups). Pathological evaluation of tissue samples from skeletal muscle, liver, and heart was performed. Only 2 of 21 mice analyzed (one mouse in the dantrolene-treated group and one mouse in the control group) had inflammation in the skeletal muscle (supplemental Table 1, available at www.jneurosci.org as supplemental material). Only one animal had inflammation in the heart (untreated group), and it was a small superficial focus, probably in the epicardium or even outside of the heart altogether. Inflammation in the liver was observed in 6 of 10 dantrolene-fed animals (60%) and in 4 of 11 untreated animals (36%) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Thus, long-term feeding with dantrolene appears to potentiate liver damage. However, when only higher degrees of inflammation, i.e., occasional or frequent foci of lymphocytes, were used to compare the two groups, the difference between them was not significant. Only 1 of 10 dantrolene-fed animals (10%) had this greater degree of inflammation in the liver versus 1 of 11 animals in the untreated group (9%) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Overall, we concluded that 9 months of treatment with dantrolene resulted in minimal pathological effects in skeletal muscle, liver, and heart tissues.
Dantrolene protects against Purkinje cell neuronal loss in SCA2–58Q mice
Specific degeneration of PCs in the cerebellum is a neuropathological hallmark of SCA2 patients (Geschwind et al., 1997; Lastres-Becker et al., 2008). To determine the role played by Ca2+ signaling in loss of PCs in SCA2–58Q mice (Huynh et al., 2000), we performed anatomical analyses of the cerebellum of WT and 58Q mice that had been studied in the dantrolene trial. Twelve-month-old mice from all four groups used in the trial (Table 1) were terminally anesthetized and transcardially perfused with fixation solution. The cerebella were extracted, frozen on dry ice, and cut sagittally to 50-μm-thick sections. The cerebellar slices were stained with anti-CBD28 monoclonal antibody to visualize PCs (Fig. 9 A).

Neuroanatomical analysis of WT and SCA2–58Q mice after dantrolene treatment. A , CBD immunoreactivity in the sagittal sections of WT mice cerebellum. The images were taken under 2×, 10×, and 40× objectives as indicated. B , Representative CBD staining of cerebella sections from 12-month-old WT and 58Q mice after dantrolene treatment. CBD is abundantly expressed in the PCs of WT mice (first column) but lost in both cell bodies and dendrites of PCs from 58Q control group mice (third column). Feeding dantrolene to SCA2–58Q mice (58Q + Dan) restored CBD staining of PCs in these mice (fourth column). Feeding of dantrolene to WT mice had no significant effect on CBD staining (WT + Dan; second column). Magnified images of representative cells (framed by white squares) are shown as insets. Scale bar, 30 μm. C , Average thickness of ML of 12-month-old WT and 58Q mice after dantrolene trial. The results are shown for WT mice, WT mice fed with dantrolene, 58Q mice, and 58Q mice fed with dantrolene as indicated. Data are presented as mean ± SE (for the number of mice in each group, see Table 1). Thickness of the molecular layer was determined by measuring the distance between the Purkinje cell body layers and dividing in half. ns, Not significant. D , Average number of PCs in WT and SCA2–58Q mice at the conclusion of the dantrolene trial. PCs counts from each group of mice were estimated with unbiased stereological techniques. The results are shown for WT mice, WT mice fed with dantrolene, SCA2–58Q mice, and SCA2–58Q mice fed with dantrolene as indicated. Data are presented as mean ± SE (for the number of mice in each group, see Table 1). 58Q control mice showed significant PC loss (**p < 0.01) when compared with control WT mice. Feeding dantrolene to 58Q mice significantly increased PC counts when compared with control 58Q mice (*p < 0.05). There was no significant change in PC counts in WT mice fed with dantrolene.
Consistent with the previous description of SCA2–58Q mice (Huynh et al., 2000), loss of calbindin immunoreactivity in PC bodies and dendrites was observed in cerebella from 12-month-old 58Q control mice (Fig. 9 B). This was in contrast to strong calbindin labeling of PCs in 12-month-old WT mice (Fig. 9 B). The calbindin staining of PCs was essentially restored in the 58Q dantrolene group (Fig. 9 B). There was no difference in calbindin staining between WT dantrolene and WT control PCs (Fig. 9 B). To quantify morphological changes in the cerebellum of aging 58Q mice, we performed measurements of ML thickness. We found that the ML was 10 μm thinner in 12-month-old 58Q control mice (166 ± 3 μm) than in WT control mice (176 ± 5 μm), but the difference did not reach a level of statistical significance (p = 0.078) (Fig. 9 C; Table 1). The ML thickness in the 58Q dantrolene group was very similar to WT control and WT dantrolene groups (Fig. 9 C; Table 1).
To further quantify the extent of PC loss in 58Q mice, we performed stereological analysis of PCs in all four groups of mice. As a result of this analysis, we discovered that the number of PCs of mice in the 12-month-old 58Q control group was reduced by 14% when compared with WT control group (Fig. 9 D; Table 1). The difference was statistically significant (p = 0.0013). Feeding dantrolene to 58Q mice significantly (p = 0.0122) reduced the death of PCs (Fig. 9 D; Table 1). The number of PCs in the 58Q dantrolene group was similar to the number of PCs in the WT control group (Fig. 9 D; Table 1, p = 0.7518). Feeding of dantrolene had no effect on PC number in WT mice (Fig. 9 D, p = 0.7237; Table 1). Thus, we concluded that feeding of dantrolene protected PCs from degeneration in aging 58Q mice.
Discussion
The causes of neurodegeneration in SCA2 and other polyQ-expansion disorders are poorly understood. In our previous studies, we demonstrated that abnormal neuronal Ca2+ signaling plays a significant role in the pathogenesis of HD and SCA3 polyQ-expansion disorders (Tang et al., 2003, 2005, 2007, 2009; Wu et al., 2006; Chen et al., 2008; Zhang et al., 2008). The data in the present study suggest that abnormal Ca2+ signaling plays a role in the pathogenesis of SCA2 as well. Similar to previous findings with Htt and Atx3, we found that polyQ-expanded Atx2 specifically binds to the C-terminal region of InsP3R1 (Fig. 1), increases apparent affinity of InsP3R1 to activation by InsP3 in an in vitro reconstitution system (Fig. 2), and enhances InsP3R1-mediated Ca2+ responses in cultured Purkinje neurons (Figs. 3, 4). Similar to our previous results with cultured striatal neurons from the transgenic HD–YAC128 mouse model (Tang et al., 2005), we here demonstrate that cultured PCs from transgenic SCA2–58Q mice are sensitized to glutamate-induced apoptosis (Figs. 5, 6). RyanR1 Ca2+ release channels are abundantly expressed in PCs (Kuwajima et al., 1992; Mori et al., 2000) and play an important role in PC Ca2+ signaling (Khodakhah and Armstrong, 1997). In our experiments, we demonstrated that inhibition of RyanR1 with ryanodine reduced glutamate-induced Ca2+ release in PCs from SCA2–58Q mice (Figs. 3, 4) and that exposure to dantrolene, a RyanR1 inhibitor and Ca2+ stabilizer, protected SCA2 PCs from glutamate-induced apoptosis in in vitro experiments (Figs. 5, 6). Consistent with in vitro findings, we found that long-term feeding of SCA2–58Q mice with dantrolene alleviated age-dependent motor deficits (Figs. 7, 8) and PC loss (Fig. 9) in these mice. Most of these findings parallel our previous studies of Ca2+ signaling in HD and SCA3 mouse models (Tang et al., 2003, 2005, 2007, 2009; Wu et al., 2006; Chen et al., 2008; Zhang et al., 2008) and indicate that deranged Ca2+ signaling contributes to pathogenesis of at least these three polyglutamine-expansion disorders. The role of Ca2+ signaling in SCA2 pathogenesis is also supported by the previously reported genetic association between polymorphisms in the CACNA1A gene encoding P/Q-type voltage-gated Ca2+ channels and the age of disease onset in SCA2 patients (Pulst et al., 2005).
Cerebellar Purkinje cells are the primary locus of pathology in three polyglutamine expansion disorders: SCA1, SCA2, and SCA6. In the case of SCA1, the polyQ expansion occurs in the nuclear protein ataxin-1, for SCA2 it occurs in the cytosolic protein ataxin-2, and for SCA6 it occurs in the C-terminal tail of the CaV2.1 pore-forming subunit of P/Q-type Ca2+ channels. Cerebellar Purkinje cells have a very potent Ca2+ signaling system and express extremely high levels of Ca2+ channel, transporters, and Ca2+ binding proteins. Ca2+ signaling abnormalities appear to play a role in pathogenesis of all three disorders. The reduction of PC Ca2+ binding protein levels was observed early in SCA1 pathology in patients and in SCA1 mouse model (Vig et al., 2001). Genetic crosses of SCA1 transgenic mice with calbindin knock-out mice resulted in an accelerated phenotype (Vig et al., 2001). Dramatic changes in expression of a number of genes related to Ca2+ signaling have been uncovered as a result of microarray analysis in the SCA1 transgenic mouse model (Lin et al., 2000; Serra et al., 2004). The genes downregulated in SCA1 transgenic mice included InsP3R1, Homer-3, EAAT4 glutamate transporter, SERCA3 Ca2+ pump, trp3 Ca2+ influx channel, the type 1 inositol polyphosphate 5-phosphatase, and the T-type voltage-gated Ca2+ channel (Lin et al., 2000; Serra et al., 2004). Despite these molecular changes, similar Ca2+ responses were observed in functional studies of PCs from adult SCA1–82Q mice (Inoue et al., 2001). It is likely, however, that abnormalities in PC Ca2+ signaling play a significant role in SCA1 pathology.
The mutation that causes SCA6 is a polyQ expansion in the C-terminal region of the CaV2.1 pore-forming subunit of P/Q-type Ca2+ channel (Zhuchenko et al., 1997). In a heterologous expression system, the polyQ-expanded CaV2.1 channel has been reported to support reduced (Matsuyama et al., 1999; Toru et al., 2000) or increased (Restituito et al., 2000; Piedras-Renteria et al., 2001) voltage-dependent Ca2+ influx when compared with the wild-type channel. Recent analysis of an SCA6 knock-in mouse model indicated that pathology may be related to aggregation of mutant CaV2.1 subunits and reduction in the density of dendritic P/Q-type Ca2+ currents (Watase et al., 2008). The Ca2+ signaling abnormalities appear to play an important role in SCA6 pathology, but the exact role of abnormal Ca2+ signaling in SCA6 needs to be further investigated.
Previous analysis of the SCA2–58Q mouse model demonstrated that nuclear localization or inclusion body formation of ataxin-2 is not necessary for the development of motor phenotypes or loss of calbindin staining in the cerebellum(Huynh et al., 2000). Using the same mouse model, we further confirmed and extended these findings. We demonstrated that aging SCA2–58Q mice developed progressive motor deficits as quantified by beam-walk and rotarod assays (Figs. 7, 8). At 12 months of age, these mice had significant reductions in calbindin staining in the molecular layer of the cerebellum (Fig. 9 B), slight reduction (∼6%) in molecular layer thickness (Fig. 9 C), and significant loss of PC number (14%) as quantified by stereology (Fig. 9 D). It is interesting to compare these results with the studies of the SCA1–82Q mouse model (Burright et al., 1995; Clark et al., 1997). SCA1–82Q mice start to display an abnormal phenotype at 8–10 weeks of age and become severely ataxic in the cage by 20 weeks of age (Burright et al., 1995; Clark et al., 1997). The SCA1–82Q mice display impaired performance on the accelerated rotarod as early as 5 weeks of age (Clark et al., 1997). Pathological analysis revealed significant shrinkage of the PC dendritic tree and disorganization of the PC layer in SCA1–82Q mice as early as 15–16 weeks of age (Burright et al., 1995; Clark et al., 1997). The SCA1–82Q mice have 30% fewer PCs than wild-type mice at 24 weeks of age (Clark et al., 1997) and display 30% shrinkage of molecular layer thickness at 32 weeks of age (Zu et al., 2004). All these results indicate that the SCA1–82Q mice phenotype is much more severe and develops much faster than the phenotype of SCA2–58Q mice described previously (Huynh et al., 2000) and in the present study (Figs. 7 ⇑–9; Table 1). Although the expression of human Atx1–82Q protein in SCA1–82Q mice and expression of human Atx2–58 protein in SCA2–58Q mice is driven by the same Pcp2/L7 promoter, it is possible that transgenes are expressed at different levels attributable to differences in copy number of integrated transgenes or location of the integration site. It is also possible that the severity of the SCA1–82Q phenotype is related to changes in PC cell gene expression induced by Atx1–82Q and resulting abnormalities in PC development. Ataxin-1 is a nuclear protein, and nuclear localization of Atx1–82Q is necessary for expression of severe phenotype in SCA1–82Q mice (Klement et al., 1998). In the nucleus, Atx1 associates with the transcriptional repressor Capicua (Lam et al., 2006) and interacts with the retinoid-related orphan receptor α (RORα) transcription factor (Serra et al., 2006). The pathological interactions of Atx1exp with Capicua and RORα impair PC development and contribute to the phenotypic severity observed in SCA1–82Q mice (Lam et al., 2006; Serra et al., 2006). In contrast, ataxin-2 is a cytosolic protein, and polyQ expansion in Atx2exp does not appear to affect PC development to a significant degree in the SCA2–58Q mouse model (Huynh et al., 2000; present study).
In our study, we established the connection between deranged intracellular Ca2+ signaling and neurodegeneration in SCA2 mice. Together with our previous studies of HD (Tang et al., 2003, 2005, 2007, 2009; Wu et al., 2006; Zhang et al., 2008) and SCA3 (Chen et al., 2008), these results suggest that abnormal neuronal Ca2+ signaling may play a role in pathogenesis of many polyglutamine-expansion disorders. In our experiments, we discovered that Httexp, Atx3exp, and Atx2exp specifically bind to InsP3R1 C-terminal region and increase InsP3R1 activity in the presence of low amounts of InsP3 (Tang et al., 2003; Chen et al., 2008; present study). Because Htt, Atx2, and Atx3 have no significant sequence similarity besides polyQ expansion, it is most likely that the InsP3R1 C terminus binds directly to the expanded polyQ track. It is, however, important to show that InsP3R1 binds to the polyQ-expanded stretch in each mutated protein, because polyQ conformation may be influenced by protein context. In additional experiments, we recently found that InsP3R1 C-terminal specifically bound to the polyQ-expanded atrophin-1 (protein mutated in DRPLA) and the polyQ-expanded ataxin-1 (protein mutated in SCA1) (X. Chen and I. Bezprozvanny, unpublished observations). It will be necessary to validate importance of these findings for neuronal dysfunction and loss in DRPLA and SCA1, but it appears that deranged Ca2+ signaling may play a significant role in pathogenesis of many polyQ-expanded disorders.
Short-term delivery of dantrolene has been demonstrated previously to be neuroprotective in acute excitotoxicity paradigms, such as cerebral ischemia (Wei and Perry, 1996) and a kainic acid injection model (Berg et al., 1995; Niebauer and Gruenthal, 1999; Schneider et al., 2001; Popescu et al., 2002). In our previous study, we demonstrated beneficial effects of long-term dantrolene treatment in an SCA3 genetic mouse model (Chen et al., 2008). Now we demonstrate that dantrolene exerted neuroprotective effects in an SCA2 mouse model as well. Our results indicate that Ca2+ signaling stabilizers such as dantrolene should be considered as potential therapeutics for the treatment of SCA3 and SCA2 patients and possibly for other polyQ-expansion disorders.
Footnotes
-
I.B. is a holder of Carla Cocke Francis Professorship in Alzheimer's Research and supported by the McKnight Neuroscience of Brain Disorders Award, Robert A. Welch Foundation, the National Organization for Rare Disorders, and National Institute of Neurological Disorders and Stroke (NINDS) Grants R01NS38082 and R01NS056224. O.N. is supported by a Division of Basic Science training grant and National Institutes of Health Predoctoral Fellowship Award for Minority Students F31 AG031692. D.P.H. is supported by NINDS Grant K01NS047548, and S.M.P. is supported by a grant from the National Ataxia Foundation and NINDS Grants R01NS033123 and 5P50NS038367. We thank Professor Masato Okada (Department of Oncogene Research, Research Institute for Microbial Diseases, Osaka University, Osaka, Japan) and Dr. Takenori Kotani (Laboratory of Biosignal Sciences, Institute for Molecular and Cellular Regulation, Gunma University, Gunma, Japan) for their helpful suggestions on Purkinje cell culture methods. We thank Huarui Liu and Yuemei Li for help with maintaining the SCA2 mouse colony, Tianhua Lei for assistance with biochemical experiments, and Leah Benson for administrative assistance.
- Correspondence should be addressed to Dr. Ilya Bezprozvanny, Department of Physiology, University of Texas Southwestern Medical Center at Dallas, 5323 Harry Hines Boulevard, Dallas, TX 75390. ilya.bezprozvanny{at}UTSouthwestern.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}